

Background: Reactive oxygen species (ROS) produced by TNFα induce apoptosis, but how this occurs and the actual molecules that damage mitochondria are undefined.
Results: Molecular manipulation of phospholipid peroxidation and oxidatively truncated phospholipid degradation shows that oxidized phospholipids are essential for TNFα-induced cell death.
Conclusion: Oxidatively truncated phospholipids couple membrane cytokine stimulation to mitochondrial apoptosis.
Significance: Fragmented phospholipids are endogenous ROS products that cause cell death.
Keywords: Apoptosis, Glutathione Peroxidase, NADPH Oxidase, Oxidative Stress, Tumor Necrosis Factor (TNF), Oxidized Phospholipid, PAF Acetylhydrolase
Abstract
TNFα generates reactive oxygen species (ROS) at the cell surface that induce cell death, but how ROS communicate to mitochondria and their specific apoptotic action(s) are both undefined. ROS oxidize phospholipids to hydroperoxides that are friable and fragment adjacent to the (hydro)peroxide function, forming truncated phospholipids, such as azelaoyl phosphatidylcholine (Az-PC). Az-PC is relatively soluble, and exogenous Az-PC rapidly enters cells to damage mitochondrial integrity and initiate intrinsic apoptosis. We determined whether this toxic phospholipid is formed within cells during TNFα stimulation in sufficient quantities to induce apoptosis and if they are essential in TNFα-induced cytotoxicity. We found that TNFα induced ROS formation and phospholipid peroxidation in Jurkat cells, and either chemical interference with NADPH oxidase activity or siRNA suppression of the NADPH oxidase-4 subunit blocked ROS accumulation and phospholipid peroxidation. Mass spectrometry showed that phospholipid peroxides and then Az-PC increased after TNFα exposure, whereas ROS inhibition abolished Az-PC accumulation and TNFα-induced cell death. Glutathione peroxidase-4 (GPx4), which specifically metabolizes lipid hydroperoxides, fell in TNFα-stimulated cells prior to death. Ectopic GPx4 overcame this, reduced peroxidized phospholipid accumulation, blocked Az-PC accumulation, and prevented death. Conversely, GPx4 siRNA knockdown enhanced phospholipid peroxidation, increasing TNFα-stimulated Az-PC formation and apoptosis. Truncated phospholipids were essential elements of TNFα-induced apoptosis because overexpression of PAFAH2 (a phospholipase A2 that selectively hydrolyzes truncated phospholipids) blocked TNFα-induced Az-PC accumulation without affecting phospholipid peroxidation. PAFAH2 also abolished apoptosis. Thus, phospholipid oxidation and truncation to apoptotic phospholipids comprise an essential element connecting TNFα receptor signaling to mitochondrial damage and apoptotic death.
Introduction
Tumor necrosis factor α (TNFα) is a master regulator of inflammatory and immune signaling (1), where its complex signals vary over time (2), with prolonged stimulation promoting extrinsic apoptosis through caspase activation (3). This induces cleavage of the BH3 domain-only member of the Bcl2 family Bid that promotes intrinsic mitochondria-dependent apoptosis (4) through release of cytochrome c to form an activated apoptosome (5). TNFα-induced apoptosis involves oxidizing radicals as intermediaries (6, 7), but neither the precise way TNFα couples cytokine stimulation (either at the plasma membrane or after internalization (8)) to cell death nor the precise role of reactive oxygen species (ROS)3 in cell death is known.
ROS are involved in a myriad of inflammatory, immunologic, and cell signaling events. Participation of ROS in the cytotoxicity of TNFα is firmly established (7, 9, 10), but the transient nature of radicals and their interconversion, along with a general lack of specific tools to identify specific compounds, combine to obfuscate understanding of how ROS actually cause cell death in TNFα-exposed cells.
TNFα stimulates ROS production from NADPH oxidase (9) that, depending on cell type, employs the non-phagocytic NOx1 (6) or NOx4 (11) NADPH oxidase complex. Polyunsaturated fatty acyl residues of membrane phospholipids are energetically favored ROS targets (12), producing phospholipid (hydro)peroxides. These peroxidized phospholipids are also products of the 12/15-lipoxygenase of several cells (13, 14). Chemical (13) reduction or enzymatic (15) reduction of these unstable oxidatively modified phospholipids by glutathione peroxidase-4 (GPx4) protects against the toxic effects of oxidative stress (16, 17), so these lipids are components of TNFα-induced toxicity. What remains unexplained, however, is the role of these oxidatively modified phospholipids in the decision to undergo or in the process of regulated cell death.
Phospholipid hydroperoxides oxidatively fragment adjacent to the (hydro)peroxide to a plethora of oxidatively truncated phospholipids (18–20). Oxidatively truncated phospholipid products of these reactions accumulate in the circulation during oxidative stress (21, 22). These truncated phospholipids are internalized, migrate to mitochondria, and then disrupt mitochondrial function, in a way aided by Bid, to initiate intrinsic apoptosis (23, 24).
PAF acetylhydrolases uniquely distinguish oxidatively modified phospholipids from biosynthetic, unmodified phospholipids because they very strongly select substrates with short sn-2 residues (18, 25). Hydrolysis of oxidatively damaged phospholipids may be the original role of this family of phospholipases A2 (26), and overexpression of the family member PAFAH2 in mammalian cells reduces toxicity to exogenous ROS (27).
Toxic oxidatively truncated phospholipids are not known to be products of cellular metabolism, so their potential involvement in endogenous mediators of apoptosis is also unknown. Here we define a new path from TNFα receptor activation to apoptosis through ROS production, phospholipid hydroperoxide formation, and accumulation of endogenous proapoptotic truncated phospholipids. Oxidatively damaged phospholipids in this path are required components of cytokine-induced cell death.
MATERIALS AND METHODS
Human recombinant TNFα was purchased from R&D Systems, pCMV-AC-GFP GPx4 and pCMV-PAFAH2 plasmids were obtained from Origene, caspase-3 (Asp-175) antibody was from Cell Signaling, and NOx4 antibody was from Abcam. Supelclean LC-NH2 SPE amino columns were from Supelco Analytical (Sigma). All of the solvents were HPLC/mass spectrometry grade. Diphenylene iodinium (DPI) and apocynin were purchased from Sigma. The caspase-3 inhibitor Z-DEVD-fmk was from BIOMOL. [2H4]Platelet-activating factor and azelaoyl phosphatidylcholine were from Cayman Chemicals. Hydroperoxyoctadecadienoyl phosphatidylcholine (HpODE-PC) was from Robert Salomon (Case Western Reserve University).
Cell Culture and Cytotoxicity
Jurkat cells (ATCC) were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, streptomycin (100 units/ml), and penicillin (100 μg/ml) and treated with 40 ng/ml TNFα in serum-free medium for 24 h or the stated times. Supernatants were collected by centrifugation at 300 × g for 5 min, and cell death was assessed by using the CytoTox-ONE homogenous membrane integrity assay kit (Promega) that measures lactate dehydrogenase release.
Reactive Oxygen Species
ROS were measured with Amplex Red® hydrogen peroxide/peroxidase kits (Invitrogen) in Jurkat cells (1.5 × 104 cells/20 μl) containing 50 μm Amplex Red and 0.1 unit/ml horseradish peroxidase in Hanks' balanced salt solution by monitoring with 540-nm excitation and 590-nm emission for 60 min in the presence or absence of TNFα and DPI (30 min, 20 μm).
Apoptotic DNA Fragmentation
Jurkat cells were incubated with DPI for 30 min and then with TNFα for 24 h before DNA was collected and separated with a Suicide-Track DNA ladder isolation kit (Calbiochem). DNA was resolved in a 1.5% agarose gel, stained with ethidium bromide, and detected during UV illumination.
Hydroperoxide and Truncated Phospholipids
Lipids were extracted (28) with [2H]PAF as an internal standard, purified over an aminopropyl column (29), and quantified by liquid chromatography/electrospray ionization/tandem mass spectrometry (LC/MS/MS). Sample in 85% methanol were injected onto a reverse phase C18 HPLC column (2 × 150 mm, 5-μm ODS(2) Phenomenex) equilibrated with 85% methanol containing 0.2% formic acid at a flow rate of 0.2 ml/min. Oxidized phospholipids were resolved with a linear gradient from 85 to 100% methanol for 17 min and then a linear gradient from 100 to 85% methanol in 0.5 min and held for 6.5 min. Analyses were performed with a Quattro Ultima triple-quadrupole mass spectrometer (Micromass, Wythenshawe, UK) configured with the capillary voltage at 5 kV, the cone voltage at 60 V, the source temperature at 120 °C, and a desolvation temperature at 250 °C. N2 and desolvation gas flow were 90 and 811 liters/h. Collision induced dissociation used argon gas. Analyses were performed using electrospray ionization in a positive ion mode with multiple reaction monitoring. The phosphocholine m/z 184 product ion is the dominant ion of both HpODE-PC and azelaoyl phosphatidylcholine (Az-PC), and the precursor to product ion transitions were m/z 791 → 184 and m/z 667 → 184, respectively. The identity of the phospholipids was confirmed in comparison with synthetic standards with the product ion scan performed in the positive mode for HpODE-PC because it produced few negative ions, whereas the product ion scan for Az-PC was performed in the negative mode.
Western Blot Analysis
Cells were washed with PBS, resuspended in 1× lysis buffer (Cell Signaling) containing protease inhibitor (Sigma), and incubated for 30 min on ice before centrifugation (14,000 × g, 30 min) and mixing with Laemmli gel loading buffer containing 10% SDS and 200 mm DTT, followed by boiling. SDS-PAGE used 10–12% gels that were blotted onto nitrocellulose membranes (Bio-Rad) and blocked with 5% nonfat dry milk (Amersham Biosciences). Detection used anti-caspase-3 (Cell Signaling), anti-GPx4 (R&D Systems), anti-12-lipoxygenase, anti-15-lipoxygenase (Abcam), or anti-PAFAH2 (Proteintech) for 1 h and then HRP-conjugated anti-rabbit or anti-mouse (1:5000) antibody before detection with Amersham Biosciences ECL Plus and reprobing with anti-β-actin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA).
Genetic Manipulations
Human NOx4 siGenome SMARTpool targeted NOx4, whereas GPx4 targeting used On-Target Plus SMARTpool human GPx4 siRNA (Thermo Scientific Dharmacon) and DhamaFECT4 transfection reagent. ALOX12 and ALOX15 were similarly targeted by OnTarget plus Smartpool. After 48 h, proteins were immunoblotted, or the siRNA post-transfected cells were then treated with TNFα for 24 h before the stated analyses. pCMV-PAFAH2, pCMV-GPx4, or control pCMV6-AC-GFP was expressed in Jurkat cells after Amaxa® electroporation with a nucleofector Kit V (Lonza) using 2 μg of vector with Nucleofector program X-05 and incubated for 48 h.
Data Analysis
All of the data were analyzed using one-way analysis of variance (multiple groups) or Student's t test for two groups by GraphPad Prism software, and data are expressed as mean ± S.E. Statistical significance was considered to be p < 0.05.
RESULTS
TNFα Stimulates Cytotoxic ROS
Reactive oxygen species, assessed as H2O2, were undetectable in quiescent Jurkat T lymphocytes, but the addition of TNFα immediately stimulated a burst of ROS formation (Fig. 1A). DPI, the small molecule inhibitor of flavoproteins, including NADPH oxidase, effectively reduced this ROS production in response to TNFα stimulation. ROS were essential components of cell death because DPI blockade of ROS production abolished TNFα-induced cell death (Fig. 1B).
FIGURE 1.
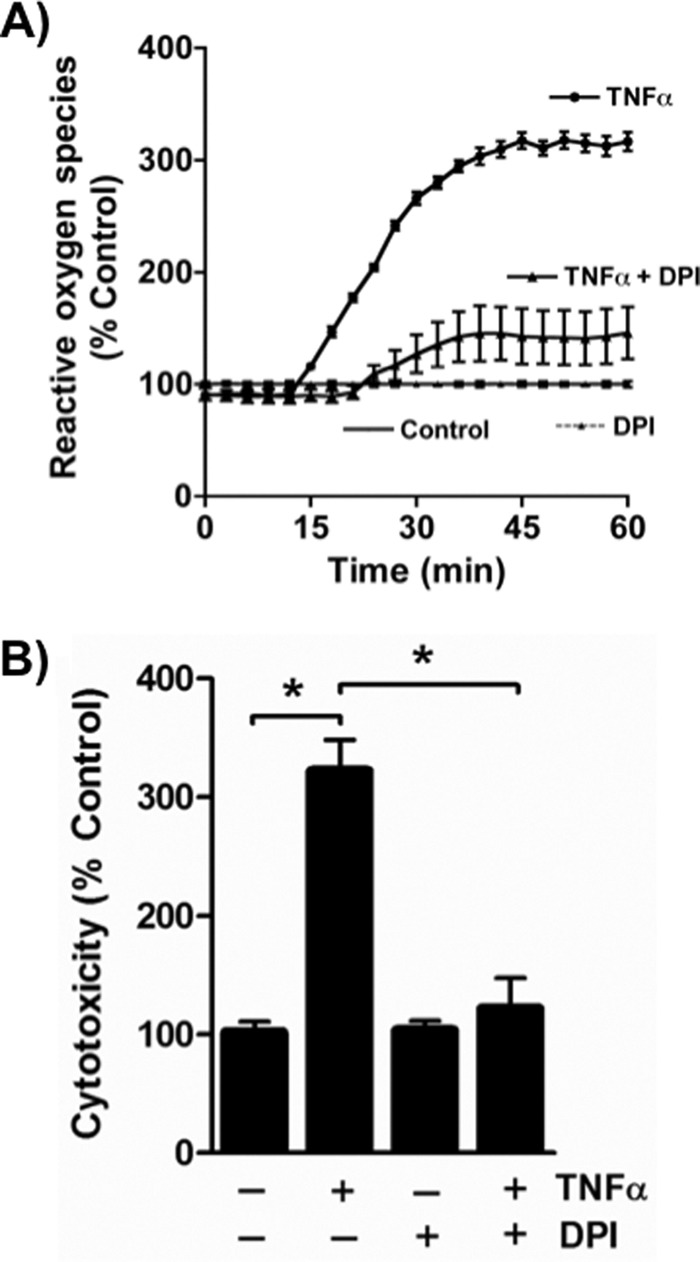
TNFα stimulates cytotoxic ROS formation in Jurkat cells. A, TNFα rapidly stimulates ROS formation in cultured Jurkat lymphoid cells. Amplex Red® fluorescence at 590 nm in the absence of or after the addition of 40 ng/ml TNFα with or without DPI addition. n = 3. B, DPI suppresses TNFα cytotoxicity. Jurkat cells were treated with TNFα for 24 h or not after a 30-min prior exposure to 20 μm DPI. Cell death was assessed as described under “Materials and Methods.” n = 3; *, p < 0.05. Error bars, S.E.
Cell death in response to TNFα exposure developed over time, with a significant increase in cell death by 12 h, which then increased over the subsequent 12 h (Fig. 2A). The loss of cellular viability from TNFα stimulation included extensive DNA fragmentation, which was absent in cells incubated in DPI (Fig. 2B). Cytokine stimulation also increased surface expression of phosphatidylserine (Fig. 2C), again indicative of apoptosis. Accordingly, activated caspase-3 fragments were present after TNFα treatment (Fig. 2D), and the caspase-3 inhibitor Z-DEVD-fmk abolished TNFα-induced cell death (Fig. 2E). TNFα therefore induced an apoptotic cell death in Jurkat cells.
FIGURE 2.

TNFα induces apoptotic cell death. A, TNFα induced a time-dependent increase in cell death. Jurkat cells were treated with TNFα (40 ng/ml) for the stated times before cytotoxicity was determined by the CytoTox One homogenous membrane integrity assay (Promega). n = 3; *, p < 0.05. B, DNA fragmentation is reduced by DPI. DNA was extracted after treatment or not with TNFα for 24 h with or without a 30-min preincubation with 20 μm DPI, which remained in the buffer, and resolved by electrophoresis before visualization after ethidium staining. C, phosphatidylserine appears on the outer aspect of TNFα-exposed cells. Jurkat cells were stained for Annexin V for 30 min after 24 h of TNFα treatment, and positive cells were analyzed using flow cytometry (FACScan). The data were analyzed using FlowJo software. n = 3; *, p < 0.05. D, TNFα induced cell death via activation of caspase-3. Jurkat cells were treated with TNFα for the indicated time, and then proteins were extracted and resolved in SDS-PAGE. Caspase 3 (Cell Signaling) fragments were detected by Western blot. E, caspase-3 inhibitor Z-DEVD-fmk abolished TNFα-induced cell death. Jurkat cells were preincubated with Z-DEVD-fmk for 30 min and then incubated with 40 ng/ml TNFα for 24 h, and cell death was assessed by LDH release assay as above. n = 3; *, p < 0.05. Error bars, S.E.
TNFα Stimulates ROS Production through NADPH Oxidase
Jurkat cells contain RNA encoding the non-phagocyte NADPH oxidase-1 subunit but also express mRNA for the related non-phagocytic NADPH oxidase-4 subunit (30). The NADPH oxidase-4 subunit was greatly suppressed with an appropriate siRNA (Fig. 3A), and this knockdown reduced ROS production by half after TNFα stimulation (Fig. 3B). The effect of reduced NADPH oxidase-4 on cell viability was far more dramatic because this knockdown fully suppressed TNFα-induced cell death (Fig. 3C).
FIGURE 3.

TNFα stimulated NADPH oxidase-4 generates cytotoxic ROS. A, NOx4 is reduced by targeted siRNA. Jurkat cells treated with nonspecific (NS) or NOx4 siRNA before NOx4 or β-actin were detected by Western blotting. n = 2. B, NOx4 produces half of TNFα-stimulated ROS. H2O2 was detected by Amplex Red® as in Fig. 1. n = 3. C, NOx4 knockdown reduces TNFα-stimulated cell death. Jurkat cells transfected with nonspecific or NOx4-directed siRNA were stimulated with TNFα before stimulation with TNFα, and the level of cytotoxicity was determined 24 h later. n = 3; *, p < 0.05. Error bars, S.E.
TNFα-stimulated ROS Generate Phospholipid Hydroperoxides
ROS rapidly interact with themselves, modify proteins, and modify nucleotides (31), but phospholipids are primary targets of ROS because radical attack is energetically favored between the olefinic bonds of polyunsaturated lipids abundantly esterified in membrane phospholipids. Linoleoyl (C18:2) sn-2 residues are the most abundant phospholipid polyunsaturated residues, and ROS attack forms phospholipids containing esterified HpODE residues. Lipid that co-eluted with a synthetic palmitoyl 9-HpODE-PC standard accumulated in TNFα-stimulated cells (Fig. 4A). Fragmentation in the positive mode of the peroxide function produced an m/z 773 fragment, an m/z 756 fragment from the loss of a hydroxyl function, and the m/z 184 phosphocholine fragment. Physical isolation and fragmentation and mass spectrometry in the positive mode (Fig. 4B) showed that palmitoyl linoleoyl HpODE-PCs are present in TNFα-treated Jurkat cells. These phospholipid hydroperoxides rapidly accumulated in response to TNFα stimulation (Fig. 4C), with the initial burst of HpODE-PC accumulation being reduced over the subsequent hours of stimulation. Still, HpODE-PC remained elevated throughout the entire duration of TNFα exposure.
FIGURE 4.

Phospholipid hydroperoxides and glutathione peroxidase-4 are inversely related after TNFα stimulation. A, phosphatidylcholine hydroperoxide is present in Jurkat cells after TNFα stimulation. Lipid extracted from Jurkat cells after treatment with TNFα (40 ng/ml) for 24 h or a synthetic HpODE-PC was analyzed by LC/MS/MS using the [M + H]+ m/z 791 → 184 transition as described under “Materials and Methods.” B, palmitoyl HpODE-PC is present after TNFα stimulation. Product ion scans in the positive mode were performed for both synthetic HpODE-PC and the resolved lipids from TNFα-treated cells that co-eluted with the standard. The fragmentation spectrum pattern establishes the presence of phosphocholine (m/z 184), whereas m/z 773 is produced from the loss of a hydroxyl moiety from palmitoyl HpODE-PC in both the standard and the co-eluting peak in TNFα-treated cells. C, TNFα induces a time-dependent increase in endogenous phosphatidylcholine hydroperoxide. HpODE-PC was determined as in B in relation to the standard as a function of the time of TNFα treatment of Jurkat cells. n = 6; *, p < 0.05 relative to time 0. D, TNFα stimulation depletes cellular GPx4. Western blot visualization of GPx4 and β-actin in control or TNFα-treated Jurkat cells. n = 2. E, NADPH oxidase, not lipoxygenase, promoted phospholipid peroxidation after TNFα activation. HpODE-PC was determined as in D using Jurkat cells previously transfected for 48 h with the stated siRNA or scrambled siRNA and then treated with TNFα for 3 h. n = 6; *, p < 0.05; NS, not significant. Error bars, S.E.
Reduction of phospholipid hydroperoxides to chemically stable hydroxyl phospholipid is the function of the type 4 family member (GPx4) because other members of this peroxidase family require water-soluble substrates. Jurkat cells constitutively expressed GPx4, but stimulation with TNFα reduced this protective cellular protein over time (Fig. 4D). Jurkat cells therefore lost a unique activity that reduces phospholipid hydroperoxides to non-fragmentable species just prior to their entry into apoptosis.
Phospholipid hydroperoxides may also accumulate through enhanced enzymatic synthesis in addition to decreased metabolism. 5-, 12-, and 15-lipoxygenase each oxidizes fatty acid to lipid hydroperoxides, but quantitative PCR of cellular mRNA and Western blotting for the encoded enzyme showed that Jurkat cells contained only the latter two mRNAs (not shown). We separately knocked down each lipoxygenase but found that loss of neither lipoxygenase reduced cellular phosphatidylcholine hydroperoxide accumulation after TNFα stimulation (Fig. 4E). In contrast, the complete loss of NOx4 by siRNA targeting did prevent accumulation of this phospholipid hydroperoxide. These results additionally suggest that H2O2 induced after TNFα stimulation of NADPH oxidase does not require either lipoxygenase to form highly reactive ROS to peroxidize membrane phospholipid.
Phospholipid Hydroperoxides Are Required for TNFα Cytotoxicity
We recreated cells lacking the phospholipid hydroperoxide reductase GPx4 using siRNA that targeted this enzyme, and this knockdown effectively abolished expression of this phospholipid hydroperoxide reductase (Fig. 5A). The non-targeted family member GPx1, which exclusively reduces aqueous substrates, did not increase to compensate for this loss of GPx4. We found that the viability of Jurkat cells lacking GPx4 was not different from that of cells transfected with an irrelevant siRNA in the absence of cytokine stimulation (Fig. 5A). However, TNFα was significantly more toxic to cells that lack GPx4 than to control cells.
FIGURE 5.

Glutathione peroxidase-4 blocks cell death by depleting cellular phospholipid hydroperoxides. A, GPx4 is reduced by targeted siRNA. GPx4 and β-actin were detected by immunoblotting Jurkat cells after transfection with nonspecific (NS) or GPx4-targeted siRNA. n = 2. Loss of endogenous GPx4 enhances TNFα-induced cytotoxicity determined by LDH release. n = 3; *, p < 0.05. B, ectopic GPx4 expression increased GPx4 protein. GPx4, GPx1, and β-actin were assessed by immunoblotting after transient transfection with pCMV-GPx4 or empty vector. n = 2. Overexpression of GPx4 blocked TNFα-induced cytotoxicity. Jurkat cells transiently expressing GPx4 were stimulated with 40 ng/ml TNFα or buffer, and 24 h later, the increase in released LDH was determined. n = 3; *, p < 0.05. C, loss of 12-lipoxygenase does not reduce TNFα cytotoxicity. siRNA directed to 12-lipoxygenase reduced cellular protein expression 48 h after transfection but did not suppress TNFα-induced cytotoxicity. n = 6; *, p < 0.05. D, loss of 15-lipoxygenase does not reduce TNFα cytotoxicity. siRNA directed to 15-lipoxygenase reduced cellular protein expression 48 h after transfection but did not suppress TNFα-induced cytotoxicity. n = 6; *, p < 0.05. Error bars, S.E.
We performed the converse experiment and increased cellular GPx4 protein content by overexpressing GPx4 under the control of a constitutive CMV promoter (Fig. 5B). Again, the viability of these cells did not differ from that of control cells in the absence of TNFα. GPx4-overexpressing cells, however, were fully protected from death when exposed to TNFα. We conclude the level of GPx4 is critical in to the apoptotic response to TNFα, so that the loss of GPx4 over time after exposure to TNFα increases the propensity to undergo apoptosis. These data also show that phospholipid hydroperoxides are critical elements in the pathway from the TNF receptor to apoptosis.
We separately reduced the content of 12-lipoxygenase (Fig. 5C) and 15-lipoxygenase (Fig. 5D) by siRNA knockdown, and as anticipated from the lack of effect on phospholipid peroxide formation, reduction of neither enzyme decreased TNFα induced cell death. We conclude that phospholipid hydroperoxides formed in TNFα-stimulated cells derive from NADPH oxidase-generated H2O2 and not from enzymatic oxidation of fatty acids followed by phospholipid remodeling.
Oxidatively Truncated Phospholipids Accumulate in TNFα-stimulated Cells
Phospholipid hydroperoxides themselves might be toxic to Jurkat cells, but these oxygenated phospholipids also are friable and fragment just proximal to the newly introduced oxygen, generating a range of truncated phospholipids (20, 32, 33). These oxidatively modified phospholipids would be relevant, if formed endogenously after cytokine stimulation, because exogenous truncated phospholipids, such as Az-PC, are transported into cells (34), selectively traffic to mitochondria (23), and initiate the intrinsic pathway to apoptotic cell death (24). The phospholipid pool of TNFα-treated cells contained material that co-migrated with a synthetic Az-PC standard (Fig. 6A), and mass spectrometry in the negative mode showed (Fig. 6B) that this lipid fragmented identically to the standard, including formation of a diagnostic m/z 201 fragment (35). This fragment is only formed in the gas phase by an intramolecular transfer of a methyl function from choline to the esterified azelaoyl carboxylate (36). We assessed the amount of Az-PC in cytokine-stimulated cells by mass spectrometry and discovered a time-dependent accumulation that became significant after 12 h of TNFα stimulation (Fig. 6C). Accumulation of Az-PC thus preceded cell death.
FIGURE 6.

TNFα stimulation generates oxidatively truncated phospholipid. A, Az-PC is present in Jurkat cells after TNFα stimulation. Lipids extracted from Jurkat cells after treatment with TNFα (40 ng/ml) for 24 h or a synthetic Az-PC were analyzed by LC/MS/MS using the [M + H]+ m/z 661 → m/z 184 transition. B, palmitoyl Az-PC is present after TNFα stimulation. Product ion scans in the negative mode were performed for both synthetic Az-PC standard and the co-eluting peak from TNFα-treated cells. The fragmentation spectrum pattern establishes the presence of the azelaoyl residue from the intramolecularly methylated azelaoyl fragment (m/z 201) in both the standard and co-eluting cellular peak. C, Az-PC increases over time of TNFα stimulation. Jurkat cells were treated with TNFα for the stated times before the cells were lysed, and Az-PC content was determined by mass spectrometry in relation to the synthetic standard. n = 3; *, p < 0.05 from time 0. Error bars, S.E.
TNFα-induced ROS were responsible for phospholipid fragmentation because DPI abolished Az-PC accumulation in TNFα-stimulated cells (Fig. 7A). In fact, the truncated phospholipid Az-PC primarily arose from stimulated NADPH oxidase action because its increase was strongly suppressed by siRNA directed to NADPH oxidase-4 (Fig. 7B).
FIGURE 7.

Phospholipid hydroperoxides are required precursors for TNFα-stimulated Az-PC accumulation. A, DPI abolishes TNFα-induced Az-PC accumulation. Jurkat cells were treated as stated with DPI and TNFα or not for 24 h before Az-PC was determined by mass spectrometry as in Fig. 6. n = 3; *, p < 0.05. B, NOx4 knockdown suppressed TNFα-initiated phospholipid fragmentation. Az-PC was quantified by mass spectrometry in cells with reduced NOx4 or cells expressing scrambled siRNA. n = 6; *, p < 0.05. C, siRNA knockdown of GPx4 enhances TNFα-initiated phospholipid truncation. GPx4 RNA and protein were reduced by siRNA before Az-PC was quantified. n = 6; *, p < 0.05. D, GPx4 overexpression abolishes TNFα-induced accumulation of Az-PC. Az-PC content after transient GPx4 or empty vector transfection with or without TNFα stimulation. n = 6; *, p < 0.05. Error bars, S.E.
Az-PC was the product of phospholipid hydroperoxide fragmentation because siRNA knockdown of GPx4, which chemically reduces the HpODE-PC precursor of Az-PC, augmented Az-PC accumulation (Fig. 7C). Conversely, GPx4 overexpression abolished the accumulation of Az-PC after stimulation (Fig. 7D). Thus, TNFα stimulation of NADPH oxidase-4 generates ROS sufficient to overwhelm normal cellular defenses, leading to oxidative truncation of cellular phospholipids.
Oxidatively Truncated Phospholipids Are Required Effectors of TNFα Cytotoxicity
Phospholipid hydroperoxides were required for TNFα-induced cell death, but either these or their truncation products could be the essential element in TNFα cytotoxicity. Membranous phospholipids and HpODE-PC with its long sn-2 residue can be distinguished from its phospholipid fragmentation products by their sensitivity to PAF acetylhydrolase that specifically selects short sn-2 residues (18, 37). Jurkat cells normally express little intracellular PAF acetylhydrolase PAFAH2, but they do when transfected (Fig. 8A). PAFAH2, as expected, did not significantly decrease TNFα-induced phospholipid oxidation because HpODE-PC was unaffected by overexpression (Fig. 8B). This newly expressed enzyme, however, was active against oxidatively truncated phospholipids because the Az-PC that accumulated after TNFα treatment was abolished in cells expressing this oxidized phospholipid phospholipase (Fig. 8C). Moreover, these PAF acetylhydrolase substrates are the actual effectors of TNFα cytotoxicity because cells expressing PAFAH2 were completely resistant to the cell death induced by this cytokine (Fig. 8D). Thus, truncated phospholipids like Az-PC are apoptotic when presented exogenously (23, 24), and these data show that they are equally deleterious when generated after a burst of H2O2 from stimulated NADPH in cells responding to TNFα.
FIGURE 8.

Oxidatively truncated phospholipids are required for TNFα-induced cytotoxicity. A, transient transfection increases PAFAH2. Jurkat cells were transfected by electroporation with PAFAH2 under control of a CMV promoter or an empty construct for 48 h before control lysates were resolved, and PAFAH2 and β-actin were analyzed by Western blotting. B, PAFAH2 overexpression does not reduce cellular phospholipid hydroperoxide content. HpODE-PC was determined as in Fig. 4. n = 3. C, overexpression of PAFAH2 abolishes TNFα-stimulated Az-PC accumulation. Jurkat cells transiently overexpressing PAFAH2 or not were stimulated with TNFα as above before Az-PC was quantitated by mass spectrometry. n = 3; *, p < 0.05. D, transient expression of PAFAH2 prevents apoptosis. Viability of Jurkat cells expressing ectopic PAFAH2 or not was determined by LDH release as in Fig. 1. n = 3; *, p < 0.05. E, schematic representation of TNFα stimulation of phospholipid oxidation and fragmentation to species that damage mitochondria, allowing caspase activation that promotes cell death. Error bars, S.E.
DISCUSSION
We show ROS production in TNFα-stimulated cells is sufficient to overwhelm endogenous cellular antioxidant mechanisms, allowing peroxidation of membrane phospholipids. The cytokine-stimulated oxidizing potential was also sufficient to oxidatively truncate the newly accumulated phospholipid hydroperoxides. Exogenous truncated phospholipids induce apoptosis in isolated cells through mitochondrial dysfunction accompanied by release of cytochrome c, caspase activation, and initiation of the intrinsic apoptotic cascade (23, 24). We show that enzymatic metabolism of peroxidized phospholipids, and especially their phospholipid truncation products, abolished TNFα-induced cell death. Therefore, TNFα cytotoxicity is completely a reflection of this novel pathway to cell death, so endogenous truncated phospholipids formed after cytokine stimulation, like those presented exogenously, are apoptotic. These data also clarify why ROS comprise essential components of cytokine-induced apoptosis; ROS are required because they attack and fragment cellular polyunsaturated phospholipids to proapoptotic truncated phospholipids.
TNFα signaling proximal to TNFR1 and recruitment of receptor-interacting proteins has been described, but it includes multiple, poorly defined elements of redox signaling (7). It is apparent that ROS participate in TNFα-induced cytotoxicity (10) and that the membrane-permeable radical scavenger tempol restores TNFα-damaged tissue function (38). The amount of ROS is a critical determinant of cellular response to TNFα because high amounts of H2O2 are lethal to hepatocytes, whereas smaller amounts sensitize the cells to TNFα cytotoxicity (39).
We detected ROS in TNFα-stimulated cells as H2O2 with Amplex Red. Whereas this assay specifically responds to H2O2, the direct product of NOx4 (40), it requires the participation of peroxidase activity and is subject to interference by cellular reductants (41). TNFα, however, did increase cellular phospholipid hydroperoxides from NOx4-derived H2O2 because efficient siRNA knockdown of the NOx4 subunit of the complex saved the cells from truncated phospholipid accumulation and apoptotic cell death. The actual phospholipid oxidant will not be H2O2, but H2O2 with heme-containing proteins catalyzes formation of hydroxyl radicals (42) through Fenton-Haber-Weiss chemistry (43). Potentially, then, hydroxyl radicals may be the species attacking polyunsaturated fatty acyl residues esterified in membrane phospholipids. Molecular oxygen then adds to the newly formed lipoxyl radical, forming phospholipid peroxy radicals that can fragment (12) to truncated phospholipids.
Phospholipid hydroperoxides report excess cellular oxidative stress (44) because they are sequestered in the lipid phase of membranes, protecting them from glutathione peroxidases and reductases that reduce water-soluble hydroperoxides (45). Just two peroxidases, GPx4 (45) and peroxiredoxin-6 (46), effectively metabolize phospholipid hydroperoxides, but this depends on the nature of the oxidative stress. For example, loss of peroxiredoxin 6 does not prevent ethanol-mediated oxidative stress (47) that is accompanied by the presence of circulating peroxidized phospholipids (21).
Manipulation of GPx4 establishes that phospholipid hydroperoxides are essential components of TNFα-induced cytotoxicity, but either phospholipid hydroperoxides themselves or their truncated reaction products could connect TNF receptor signaling to apoptosis.
Two lines of evidence implicate oxidative truncation products as the essential distal mediators of TNFα cytotoxicity. First, oxidatively truncated phospholipids themselves are potent, receptor-independent agonists of apoptosis when presented as pure, exogenous phospholipid (23). Exogenous oxidatively truncated phospholipid affects intracellular events because these short chain, relatively water-soluble phospholipids are actively internalized into cells in part through a transport system conserved since the divergence of yeast (34). Internalized Az-PC preferentially traffics to mitochondria where it depolarizes these organelles, allows cytochrome c to escape with formation of an active apoptosome and caspase-9 and effector caspase-3 cleavage and activation (23). Presentation of Az-PC to mitochondria is aided by Bid (24), a proapoptotic Bcl-2 family member that, alone among this family, is a lipid transfer protein (48). Az-PC depolarization of mitochondria initially is reversed by introduction of an albumin sink (24), showing that continuous exposure to Az-PC is required for mitochondrial depolarization, so it is relevant that Az-PC was present for prolonged periods in Jurkat cells stimulated with TNFα. Conversely, intact phospholipid hydroperoxides, although thrombotic (49), are not known to directly induce cell death.
The second line of evidence implicating oxidatively truncated phospholipids in TNFα cytotoxicity employed the unusual specificity of PAFAH2 as an oxidized phospholipid phospholipase. Phospholipases A2, with a single exception (50), are without selectivity for the length, saturation, or regio- or stereoisomeric configuration of the esterified sn-2 residue that they accept as substrates. However, mammalian group VII phospholipases A2, the PAF acetylhydrolases, display remarkably distinct substrate recognition. These enzymes were purified (25, 51) and cloned (52–54) based on their hydrolysis of the short two-carbon acetyl sn-2 residue of PAF. Lengthening the sn-2 residue to six carbon atoms precludes catalysis by the plasma enzyme, but this remarkably sharp discrimination of residue length is overcome for the plasma and type II enzymes if the ω-end of the short sn-2 residue contains a polar function (18, 55), as is present in many truncated phospholipids (18–20).
PAF acetylhydrolases are an ancient form of protection against an oxidizing environment, with an ortholog present prior to divergence of yeast that protects these free living cells against phospholipid oxidation and oxidative death (26). Similarly, forced expression of PAFAH2 in cultured cells protects against oxidative stress (27), foam cell formation in vitro (56), neointima formation and atherosclerosis (57), and focal cerebral ischemia (58) and reduces glutamate-induced apoptosis in vitro (59). The basis for the wide protective effect of PAF acetylhydrolase overexpression has been assumed, but not proven, to result from the removal of their substrates. We show that cellular oxidatively truncated phospholipid content was inversely proportional to cellular PAFAH activity. Overexpression of PAFAH2 in Jurkat cells decreased the amount of Az-PC that accumulated after TNFα treatment to less than that of control cells, and PAFAH2 overexpression maintained this muted level of the truncated phospholipid AZ-PC throughout TNFα stimulation. Moreover, this ectopic enzyme fully protected cells from TNFα-induced cell death. Thus, oxidative truncation of polyunsaturated sn-2 residues generated substrates for PAF acetylhydrolase from intact membrane phospholipids.
This is the first documentation that oxidatively truncated phospholipids are formed within stimulated cells and are formed in sufficient quantities to affect mitochondrial integrity and function. Exogenous Az-PC circulates in animals as alcoholic steatohepatitis develops (21), and exogenous truncated phospholipid is rapidly internalized at least in part by a phospholipid transport system (34). Internalized truncated phospholipid readily traffics to mitochondria (23) to induce intrinsic apoptotic cell death (24). The amount of exogenous Az-PC added to cells (≥5 μm) necessary to induce apoptosis (23) is somewhat greater than the amount of endogenous Az-PC generated in response to TNFα examined here. We ascribe this to inefficient, rate-limiting internalization of truncated phospholipids. Potentially, the rate of internalization as well as the rate of intracellular degradation by intracellular PAF acetylhydrolase varies among cells, thereby modulating the apoptotic response to TNFα.
Az-PC depolarization of isolated mitochondria requires the continual presence of this disruptive phospholipid (24), and the prolonged presence of endogenously generated Az-PC relative to exposure to a bolus of exogenous Az-PC may additionally potentiate cellular responsiveness to endogenous oxidatively truncated phospholipid. It is also relevant that Az-PC, although abundant, is not the sole oxidatively truncated phospholipid to induce apoptosis (23), and Az-PC additionally marks the presence of numerous other proapoptotic phospholipid oxidation products.
Endogenous phospholipid hydroperoxide formation can be non-enzymatic, from NADPH oxidase O2˙̄ or H2O2 for instance, or enzymatic after esterification of peroxy fatty acids into phospholipids (49). The regio- and stereoisomeric identity of the reaction products differ between enzymatic and non-enzymatic formation, but lipoxygenases interconvert phospholipid hydroperoxide stereoisomers (60). This disguises the relationship between the identity of the recovered isomers and whether they are enzymatic products or not. We instead resolved the issue of whether phospholipids were formed chemically or enzymatically to find, at least in Jurkat cells, that phospholipid peroxide formation was non-enzymatic. We conclude this because suppression of NADPH oxidase reduced ROS production and effectively suppressed Az-PC formation and cell death, whereas siRNA knockdown of neither 12- or 15-liopxygenase was similarly effective.
Oxidatively truncated phospholipids intervene between TNF receptor signaling and enhanced mitochondrial permeability, so Bid (7), depolarizing agents (61), mitochondrial oxidative damage (62), prolonged JNK activation (6), thiol oxidation (63), tyrosine phosphorylation (64), and caspase activation (3, 65) either act below truncated phospholipids or, as for Bid, enhance the effect of membrane-disruptive phospholipids. Oxidatively fragmented phospholipids are proximal to TNF receptor signaling but also are distal effectors that reversibly damage mitochondria, allowing cytochrome c escape and formation of a functional apoptosome that amplifies cytotoxicity through caspase activation (Fig. 7E). This defines a previously unappreciated pathway connecting TNF receptor signaling to mitochondrial damage and apoptosis.
Acknowledgments
We thank Gang Zhou and Rui Chen for expertise and initial help with this project.
This work was supported, in whole or in part, by National Institutes of Health Grants AA017748 and HL092747.
- ROS
- reactive oxygen species
- GPx4
- glutathione peroxidase-4
- DPI
- diphenylene iodinium
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- HpODE
- hydroperoxyoctadecadienoyl
- PAF
- platelet-activating factor
- PC
- phosphatidylcholine
- Az-PC
- azelaoyl phosphatidylcholine.
REFERENCES
- 1. Parameswaran N., Patial S. (2010) Tumor necrosis factor-α signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 20, 87–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gordon J. W., Shaw J. A., Kirshenbaum L. A. (2011) Multiple facets of NF-κB in the heart. To be or not to NF-κB. Circ. Res. 108, 1122–1132 [DOI] [PubMed] [Google Scholar]
- 3. Shakibaei M., Sung B., Sethi G., Aggarwal B. B. (2010) TNF-α-induced mitochondrial alterations in human T cells requires FADD and caspase-8 activation but not RIP and caspase-3 activation. Antioxid. Redox Signal. 13, 821–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Falschlehner C., Emmerich C. H., Gerlach B., Walczak H. (2007) TRAIL signaling. Decisions between life and death. Int. J. Biochem. Cell Biol. 39, 1462–1475 [DOI] [PubMed] [Google Scholar]
- 5. Schafer Z. T., Kornbluth S. (2006) The apoptosome. Physiological, developmental, and pathological modes of regulation. Dev. Cell 10, 549–561 [DOI] [PubMed] [Google Scholar]
- 6. Kim Y. S., Morgan M. J., Choksi S., Liu Z. G. (2007) TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell 26, 675–687 [DOI] [PubMed] [Google Scholar]
- 7. Han D., Ybanez M. D., Ahmadi S., Yeh K., Kaplowitz N. (2009) Redox regulation of tumor necrosis factor signaling. Antioxid. Redox Signal. 11, 2245–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tchikov V., Bertsch U., Fritsch J., Edelmann B., Schütze S. (2011) Subcellular compartmentalization of TNF receptor-1 and CD95 signaling pathways. Eur. J. Cell Biol. 90, 467–475 [DOI] [PubMed] [Google Scholar]
- 9. Vanden Berghe T., Declercq W., Vandenabeele P. (2007) NADPH oxidases. New players in TNF-induced necrotic cell death. Mol. Cell 26, 769–771 [DOI] [PubMed] [Google Scholar]
- 10. Morgan M. J., Liu Z. G. (2010) Reactive oxygen species in TNFα-induced signaling and cell death. Mol. Cells 30, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Basuroy S., Bhattacharya S., Leffler C. W., Parfenova H. (2009) Nox4 NADPH oxidase mediates oxidative stress and apoptosis caused by TNF-α in cerebral vascular endothelial cells. Am. J. Physiol. Cell Physiol. 296, C422–C432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frankel E. N. (1984) Chemistry of free radical and singlet oxidation of lipids. Prog. Lipid. Res. 23, 197–221 [DOI] [PubMed] [Google Scholar]
- 13. O'Donnell V. B., Spycher S., Azzi A. (1995) Biochem. J. 310, 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seiler A., Schneider M., Förster H., Roth S., Wirth E. K., Culmsee C., Plesnila N., Kremmer E., Rådmark O., Wurst W., Bornkamm G. W., Schweizer U., Conrad M. (2008) Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase-dependent and AIF-mediated cell death. Cell Metab. 8, 237–248 [DOI] [PubMed] [Google Scholar]
- 15. Imai H., Nakagawa Y. (2003) Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. 34, 145–169 [DOI] [PubMed] [Google Scholar]
- 16. Yagi K., Komura S., Kojima H., Sun Q., Nagata N., Ohishi N., Nishikimi M. (1996) Expression of human phospholipid hydroperoxide glutathione peroxidase gene for protection of host cells from lipid hydroperoxide-mediated injury. Biochem. Biophys. Res. Commun. 219, 486–491 [DOI] [PubMed] [Google Scholar]
- 17. Ran Q., Liang H., Gu M., Qi W., Walter C. A., Roberts L. J., 2nd, Herman B., Richardson A., Van Remmen H. (2004) Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J. Biol. Chem. 279, 55137–55146 [DOI] [PubMed] [Google Scholar]
- 18. Stremler K. E., Stafforini D. M., Prescott S. M., McIntyre T. M. (1991) Human plasma platelet-activating factor acetylhydrolase. Oxidatively fragmented phospholipids as substrates. J. Biol. Chem. 266, 11095–11103 [PubMed] [Google Scholar]
- 19. Podrez E. A., Poliakov E., Shen Z., Zhang R., Deng Y., Sun M., Finton P. J., Shan L., Gugiu B., Fox P. L., Hoff H. F., Salomon R. G., Hazen S. L. (2002) Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J. Biol. Chem. 277, 38503–38516 [DOI] [PubMed] [Google Scholar]
- 20. Reis A., Domingues P., Ferrer-Correia A. J., Domingues M. R. (2004) Fragmentation study of short-chain products derived from oxidation of diacylphosphatidylcholines by electrospray tandem mass spectrometry. Identification of novel short-chain products. Rapid Commun. Mass Spectrom. 18, 2849–2858 [DOI] [PubMed] [Google Scholar]
- 21. Yang L., Latchoumycandane C., McMullen M. R., Pratt B. T., Zhang R., Papouchado B. G., Nagy L. E., Feldstein A. E., McIntyre T. M. (2010) Chronic alcohol exposure increases circulating bioactive oxidized phospholipids. J. Biol. Chem. 285, 22211–22220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Podrez E. A., Byzova T. V., Febbraio M., Salomon R. G., Ma Y., Valiyaveettil M., Poliakov E., Sun M., Finton P. J., Curtis B. R., Chen J., Zhang R., Silverstein R. L., Hazen S. L. (2007) Platelet CD36 links hyperlipidemia, oxidant stress, and a prothrombotic phenotype. Nat. Med. 13, 1086–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen R., Yang L., McIntyre T. M. (2007) Cytotoxic phospholipid oxidation products. Cell death from mitochondrial damage and the intrinsic caspase cascade. J. Biol. Chem. 282, 24842–24850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen R., Feldstein A. E., McIntyre T. M. (2009) Suppression of mitochondrial function by oxidatively truncated phospholipids is reversible, aided by Bid, and suppressed by Bcl-XL. J. Biol. Chem. 284, 26297–26308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hattori K., Hattori M., Adachi H., Tsujimoto M., Arai H., Inoue K. (1995) Purification and characterization of platelet-activating factor acetylhydrolase II from bovine liver cytosol. J. Biol. Chem. 270, 22308–22313 [DOI] [PubMed] [Google Scholar]
- 26. Foulks J. M., Weyrich A. S., Zimmerman G. A., McIntyre T. M. (2008) A yeast PAF acetylhydrolase ortholog suppresses oxidative death. Free Radic. Biol. Med. 45, 434–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matsuzawa A., Hattori K., Aoki J., Arai H., Inoue K. (1997) Protection against oxidative stress-induced cell death by intracellular platelet-activating factor-acetylhydrolase II. J. Biol. Chem. 272, 32315–32320 [DOI] [PubMed] [Google Scholar]
- 28. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 29. Marathe G. K., Davies S. S., Harrison K. A., Silva A. R., Murphy R. C., Castro-Faria-Neto H., Prescott S. M., Zimmerman G. A., McIntyre T. M. (1999) Inflammatory platelet-activating factor-like phospholipids in oxidized low density lipoproteins are fragmented alkyl phosphatidylcholines. J. Biol. Chem. 274, 28395–28404 [DOI] [PubMed] [Google Scholar]
- 30. Van Buul J. D., Fernandez-Borja M., Anthony E. C., Hordijk P. L. (2005) Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid. Redox Signal. 7, 308–317 [DOI] [PubMed] [Google Scholar]
- 31. Avery S. V. (2011) Molecular targets of oxidative stress. Biochem. J. 434, 201–210 [DOI] [PubMed] [Google Scholar]
- 32. Harrison K. A., Davies S. S., Marathe G. K., McIntyre T., Prescott S., Reddy K. M., Falck J. R., Murphy R. C. (2000) Analysis of oxidized glycerophosphocholine lipids using electrospray ionization mass spectrometry and microderivatization techniques. J. Mass Spectrom. 35, 224–236 [DOI] [PubMed] [Google Scholar]
- 33. Milne G. L., Seal J. R., Havrilla C. M., Wijtmans M., Porter N. A. (2005) Identification and analysis of products formed from phospholipids in the free radical oxidation of human low density lipoproteins. J. Lipid Res. 46, 307–319 [DOI] [PubMed] [Google Scholar]
- 34. Chen R., Brady E., McIntyre T. M. (2011) Human TMEM30a promotes uptake of antitumor and bioactive choline phospholipids into mammalian cells. J. Immunol. 186, 3215–3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davies S. S., Pontsler A. V., Marathe G. K., Harrison K. A., Murphy R. C., Hinshaw J. C., Prestwich G. D., Hilaire A. S., Prescott S. M., Zimmerman G. A., McIntyre T. M. (2001) Oxidized alkyl phospholipids are specific, high affinity peroxisome proliferator-activated receptor γ ligands and agonists. J. Biol. Chem. 276, 16015–16023 [DOI] [PubMed] [Google Scholar]
- 36. Kayganich-Harrison K. A., Murphy R. C. (1994) Characterization of chain-shortened oxidized glycerophosphocholine lipids using fast atom bombardment and tandem mass spectrometry. Anal. Biochem. 221, 16–24 [DOI] [PubMed] [Google Scholar]
- 37. Kono N., Inoue T., Yoshida Y., Sato H., Matsusue T., Itabe H., Niki E., Aoki J., Arai H. (2008) Protection against oxidative stress-induced hepatic injury by intracellular type II platelet-activating factor acetylhydrolase by metabolism of oxidized phospholipids in vivo. J. Biol. Chem. 283, 1628–1636 [DOI] [PubMed] [Google Scholar]
- 38. Mariappan N., Soorappan R. N., Haque M., Sriramula S., Francis J. (2007) TNF-α-induced mitochondrial oxidative stress and cardiac dysfunction. Restoration by superoxide dismutase mimetic Tempol. Am. J. Physiol. Heart Circ. Physiol. 293, H2726–H2737 [DOI] [PubMed] [Google Scholar]
- 39. Han D., Hanawa N., Saberi B., Kaplowitz N. (2006) Hydrogen peroxide and redox modulation sensitize primary mouse hepatocytes to TNF-induced apoptosis. Free Radic. Biol. Med. 41, 627–639 [DOI] [PubMed] [Google Scholar]
- 40. Takac I., Schröder K., Zhang L., Lardy B., Anilkumar N., Lambeth J. D., Shah A. M., Morel F., Brandes R. P. (2011) The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 286, 13304–13313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Votyakova T. V., Reynolds I. J. (2004) Detection of hydrogen peroxide with Amplex Red. Interference by NADH and reduced glutathione auto-oxidation. Arch. Biochem. Biophys. 431, 138–144 [DOI] [PubMed] [Google Scholar]
- 42. Sadrzadeh S. M., Graf E., Panter S. S., Hallaway P. E., Eaton J. W. (1984) Hemoglobin. A biologic Fenton reagent. J. Biol. Chem. 259, 14354–14356 [PubMed] [Google Scholar]
- 43. Kanner J., Harel S., Salan A. M. (1988) The generation of ferryl or hydroxyl radicals during interaction of hemeproteins with hydrogen peroxide. Basic Life Sci. 49, 145–148 [DOI] [PubMed] [Google Scholar]
- 44. Loscalzo J. (2008) Membrane redox state and apoptosis. Death by peroxide. Cell Metab. 8, 182–183 [DOI] [PubMed] [Google Scholar]
- 45. Ursini F., Bindoli A. (1987) The role of selenium peroxidases in the protection against oxidative damage of membranes. Chem. Phys. Lipids 44, 255–276 [DOI] [PubMed] [Google Scholar]
- 46. Fisher A. B., Dodia C., Manevich Y., Chen J. W., Feinstein S. I. (1999) Phospholipid hydroperoxides are substrates for non-selenium glutathione peroxidase. J. Biol. Chem. 274, 21326–21334 [DOI] [PubMed] [Google Scholar]
- 47. Roede J. R., Orlicky D. J., Fisher A. B., Petersen D. R. (2009) Overexpression of peroxiredoxin 6 does not prevent ethanol-mediated oxidative stress and may play a role in hepatic lipid accumulation. J. Pharmacol. Exp. Ther. 330, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Esposti M. D., Erler J. T., Hickman J. A., Dive C. (2001) Bid, a widely expressed proapoptotic protein of the Bcl-2 family, displays lipid transfer activity. Mol. Cell. Biol. 21, 7268–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thomas C. P., Morgan L. T., Maskrey B. H., Murphy R. C., Kühn H., Hazen S. L., Goodall A. H., Hamali H. A., Collins P. W., O'Donnell V. B. (2010) Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. J. Biol. Chem. 285, 6891–6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Six D. A., Dennis E. A. (2000) The expanding superfamily of phospholipase A2 enzymes. Classification and characterization. Biochim. Biophys. Acta 1488, 1–19 [DOI] [PubMed] [Google Scholar]
- 51. Stafforini D. M., Prescott S. M., McIntyre T. M. (1987) Human plasma platelet-activating factor acetylhydrolase. Purification and properties. J. Biol. Chem. 262, 4223–4230 [PubMed] [Google Scholar]
- 52. Tjoelker L. W., Eberhardt C., Unger J., Trong H. L., Zimmerman G. A., McIntyre T. M., Stafforini D. M., Prescott S. M., Gray P. W. (1995) Plasma platelet-activating factor acetylhydrolase is a secreted phospholipase A2 with a catalytic triad. J. Biol. Chem. 270, 25481–25487 [DOI] [PubMed] [Google Scholar]
- 53. Hattori K., Adachi H., Matsuzawa A., Yamamoto K., Tsujimoto M., Aoki J., Hattori M., Arai H., Inoue K. (1996) cDNA cloning and expression of intracellular platelet-activating factor (PAF) acetylhydrolase II. Its homology with plasma PAF acetylhydrolase. J. Biol. Chem. 271, 33032–33038 [DOI] [PubMed] [Google Scholar]
- 54. Hattori M., Adachi H., Tsujimoto M., Arai H., Inoue K. (1994) The catalytic subunit of bovine brain platelet-activating factor acetylhydrolase is a novel type of serine esterase. J. Biol. Chem. 269, 23150–23155 [PubMed] [Google Scholar]
- 55. Stremler K. E., Stafforini D. M., Prescott S. M., Zimmerman G. A., McIntyre T. M. (1989) An oxidized derivative of phosphatidylcholine is a substrate for the platelet-activating factor acetylhydrolase from human plasma. J. Biol. Chem. 264, 5331–5334 [PubMed] [Google Scholar]
- 56. Turunen P., Jalkanen J., Heikura T., Puhakka H., Karppi J., Nyyssönen K., Ylä-Herttuala S. (2004) Adenovirus-mediated gene transfer of Lp-PLA2 reduces LDL degradation and foam cell formation in vitro. J. Lipid Res. 45, 1633–1639 [DOI] [PubMed] [Google Scholar]
- 57. Quarck R., De Geest B., Stengel D., Mertens A., Lox M., Theilmeier G., Michiels C., Raes M., Bult H., Collen D., Van Veldhoven P., Ninio E., Holvoet P. (2001) Adenovirus-mediated gene transfer of human platelet-activating factor-acetylhydrolase prevents injury-induced neointima formation and reduces spontaneous atherosclerosis in apolipoprotein E-deficient mice. Circulation 103, 2495–2500 [DOI] [PubMed] [Google Scholar]
- 58. Umemura K., Kato I., Hirashima Y., Ishii Y., Inoue T., Aoki J., Kono N., Oya T., Hayashi N., Hamada H., Endo S., Oda M., Arai H., Kinouchi H., Hiraga K. (2007) Neuroprotective role of transgenic PAF-acetylhydrolase II in mouse models of focal cerebral ischemia. Stroke 38, 1063–1068 [DOI] [PubMed] [Google Scholar]
- 59. Hirashima Y., Ueno H., Karasawa K., Yokoyama K., Setaka M., Takaku A. (2000) Transfection of the plasma-type platelet-activating factor acetylhydrolase gene attenuates glutamate-induced apoptosis in cultured rat cortical neurons. Brain Res. 885, 128–132 [DOI] [PubMed] [Google Scholar]
- 60. Tokumura A., Sumida T., Toujima M., Kogure K., Fukuzawa K., Takahashi Y., Yamamoto S. (2000) Structural identification of phosphatidylcholines having an oxidatively shortened linoleate residue generated through its oxygenation with soybean or rabbit reticulocyte lipoxygenase. J. Lipid Res. 41, 953–962 [PubMed] [Google Scholar]
- 61. Bedard K., Krause K. H. (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313 [DOI] [PubMed] [Google Scholar]
- 62. Montero J., Mari M., Colell A., Morales A., Basañez G., Garcia-Ruiz C., Fernández-Checa J. C. (2010) Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochim. Biophys. Acta 1797, 1217–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Buettner G. R. (1993) The pecking order of free radicals and antioxidants. Lipid peroxidation, α-tocopherol, and ascorbate. Arch. Biochem. Biophys. 300, 535–543 [DOI] [PubMed] [Google Scholar]
- 64. Conrad M., Sandin A., Förster H., Seiler A., Frijhoff J., Dagnell M., Bornkamm G. W., Rådmark O., Hooft van Huijsduijnen R., Aspenström P., Böhmer F., Ostman A. (2010) 12/15-Lipoxygenase-derived lipid peroxides control receptor tyrosine kinase signaling through oxidation of protein-tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 107, 15774–15779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Holler N., Zaru R., Micheau O., Thome M., Attinger A., Valitutti S., Bodmer J. L., Schneider P., Seed B., Tschopp J. (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495 [DOI] [PubMed] [Google Scholar]