

Background: Immunoglobulin E (IgE) antibodies play a role in allergic disease.
Results: IgE has a bent conformation in solution that becomes more bent upon binding to the FcϵRI receptor but less bent upon binding the anti-IgE omalizumab.
Conclusion: Conformational change is critical for FcϵRI-mediated IgE activity.
Significance: The bent structure provides a molecular rationale for the susceptibility of IgE-FcϵRI complexes to allergenic stimulation.
Keywords: Antibodies, Biophysics, Biosensors, Fluorescence Resonance Energy Transfer (FRET), Immunology, Protein Conformation, CD23, FRET Biosensor, IgE, Omalizumab
Abstract
IgE binding to its high affinity receptor FcϵRI on mast cells and basophils is a key step in the mechanism of allergic disease and a target for therapeutic intervention. Early indications that IgE adopts a bent structure in solution have been confirmed by recent x-ray crystallographic studies of IgEFc, which further showed that the bend, contrary to expectation, is enhanced in the crystal structure of the complex with receptor. To investigate the structure of IgEFc and its conformational changes that accompany receptor binding in solution, we created a Förster resonance energy transfer (FRET) biosensor using biologically encoded fluorescent proteins fused to the N- and C-terminal IgEFc domains (Cϵ2 and Cϵ4, respectively) together with the theoretical basis for quantitating its behavior. This revealed not only that the IgEFc exists in a bent conformation in solution but also that the bend is indeed enhanced upon FcϵRI binding. No change in the degree of bending was seen upon binding to the B cell receptor for IgE, CD23 (FcϵRII), but in contrast, binding of the anti-IgE therapeutic antibody omalizumab decreases the extent of the bend, implying a conformational change that opposes FcϵRI engagement. HomoFRET measurements further revealed that the (Cϵ2)2 and (Cϵ4)2 domain pairs behave as rigid units flanking the conformational change in the Cϵ3 domains. Finally, modeling of the accessible conformations of the two Fab arms in FcϵRI-bound IgE revealed a mutual exclusion not seen in IgG and Fab orientations relative to the membrane that may predispose receptor-bound IgE to cross-linking by allergens.
Introduction
The incidence of allergic disease is on the increase world-wide, and this includes a range of conditions from seasonal hay fever to asthma and fatal anaphylactic shock triggered by allergens such as peanuts. The key mediator between the allergen and the cells of the immune system is the antibody immunoglobulin E (IgE) (1). In contrast to the comparatively high levels of the other immunoglobulin isotypes (∼2 mg/ml for IgA and 13 mg/ml for IgG), IgE is only found at low levels (∼50–200 ng/ml) in serum. Nevertheless, IgE binds with high affinity (Ka ∼ 1010 m−1) through its Fc region to the receptor FcϵRI expressed on the surface of mast cells and basophils, and this high affinity and long lifetime of receptor-bound IgE in tissue (dissociation t½ ∼14 days) (2, 3) accounts for the allergic sensitization and rapid activation of these cells upon allergen binding. Degranulation of these cells and the release of inflammatory mediators, triggered by often innocuous allergens, leads to the physiological responses associated with allergic reactions (1, 4), although the primary function of IgE may be to respond to multicellular parasitic pathogens such as helminths (5).
In IgG, the two Fab (antigen binding) arms are connected to the Fc (a dimer of Cγ2 and Cγ3 domains) by a disulfide-linked hinge region. IgE, however, has the (Cϵ2)2 domain pair in place of the hinge, and thus IgEFc comprises six domains (a dimer of Cϵ2, Cϵ3, and Cϵ4, disulfide-linked between the Cϵ2 domains). The crystal structure of IgEFc revealed the (Cϵ2)2 domain pair bent back against the Cϵ3 domains and even contacted one of the Cϵ4 domains (6). This structure was even more acutely bent than that proposed in an earlier FRET study with a chimeric IgE (7) or an x-ray and neutron scattering study of IgEFc in solution (8). Comparative studies of the kinetics of binding to the soluble extracellular domains of the IgE-binding α-chain of FcϵRI (sFcϵRIα)3 of both IgEFc and a subfragment lacking the Cϵ2 domains (IgEFc3–4) demonstrated that the Cϵ2 domains were in part responsible for the high affinity and slow dissociation rate (9). Although an “unbending” of the IgEFc was proposed to allow engagement of the Cϵ2 domains with FcϵRI (4), IgEFc was found to remain bent (in fact the bend is enhanced) in the crystal structure of the IgEFc·sFcϵRIα complex (10).
To discover whether the conformations and conformational changes seen in the crystal structures occur in solution, we generated an IgEFc biosensor to monitor conformational change in this region of the molecule. Previous solution-based fluorescence depolarization studies of human and murine IgE provided evidence only of flexibility of the Fabs relative to Fc (11–13); this was also observed by electron microscopy (14). FRET studies using recombinant mouse chimeric IgE (with Cϵ4 replaced by Cγ3 of IgG) did reveal a small change in energy transfer on binding to rat FcϵRI (7, 15), but the donor fluorophore was placed in the Fab antigen binding site rather than in Fc, making it difficult to distinguish between movements within Fc from those of the Fabs relative to Fc. We report here a conformational change within human IgEFc monitored by FRET between biologically encoded probes attached to the N and C termini of the molecule. Both hetero- and homo-energy transfer were measured using a combination of steady-state and excited-state decay techniques. These measurements were made in the presence and absence of soluble forms of the “high affinity” mast cell receptor FcϵRIα and the “low affinity” B cell receptor CD23 (FcϵRII) as well as the Fab of the therapeutic anti-IgE antibody omalizumab (XolairTM). The results reveal not only that IgEFc is bent in solution but also that the bend is enhanced upon binding to FcϵRIα. CD23 binding has no effect upon the bend, whereas the anti-IgE omalizumab causes an unbending. Finally, we modeled the disposition of the Fabs relative to FcϵRI-bound IgEFc and discovered a significant difference compared with receptor-bound IgG, which may have important implications for cross-linking of receptor-bound IgE and the activation of mast cells and basophils by allergens.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of Recombinant Proteins
cDNA sources were as follows: pRY 24 (16) for IgEFc (including secretion tag); pEGFP-N1 C (Clontech) for the minimal eGFP residues 1–230 (17); pRSET mRFP1 (a kind gift of Roger Tsien (18) for mRFP). For expression, the cDNAs were inserted into the pCEP4 vector (Invitrogen). In total, four vectors were made: eGFP-IgEFc, an IgEFc labeled N-terminally with eGFP; mRFP-IgEFc, an IgEFc labeled N-terminally with mRFP; mRFP-IgEFc-eGFP, a dual labeled IgEFc with mRFP at the N terminus and eGFP at the C terminus; IgEFc-eGFP, an IgEFc C-terminally labeled with eGFP (see supplemental Fig. S1). Each construct was individually transfected into HEK 293E cells using polyethyleneimine (19). Transfection-positive cells were selected using hygromycin (according to instructions provided with the pCEP4 vector) and maintained in triple-layered tissue culture flasks (Nunc). Supernatants were harvested at regular intervals, and active IgE fusion protein was purified by affinity chromatography using a sFcϵRIα receptor column (sFcϵRIα-IgG4Fc fusion protein) (20). Monomeric protein was isolated by size-exclusion chromatography (21). Omalizumab Fabs were produced by papain digestion of whole antibody (a kind gift of Gerald Dubois, Novartis Pharmaceuticals) using the Pierce Fab preparation kit (Thermo Scientific) followed by anion-exchange chromatography. sFcϵRIα, the IgEFc3–4 fragment, whole IgE, IgEFc, derCD23, and eGFP were produced as described previously (16, 20, 22–24).
Biochemical and Biophysical Characterization of Fluorescently Labeled IgEFc
Analytical size exclusion chromatography was used to test the “bindability” of stored protein preparations as previously described (25) and also on material prior and subsequent to all FRET assays. All fluorescent fusion proteins were compared with whole IgE for their affinity and kinetics of binding to sFcϵRIα by surface plasmon resonance using a Biacore 3000 as described previously (21). Dual polarization interferometry measurements (26) were made using an Analight Instrument (Farfield). All experiments were carried out at 20 °C. An active FcϵRI surface was generated by amine coupling sFcϵRIα-IgG4Fc fusion protein (20) to the chip at 100 μg/ml in HEPES-buffered saline after pre-activation of the chip surface with sulfo-GMBS (N-[γ-maleimidobutyryloxy]sulfosuccinimide ester) cross-linker (Thermo Lifesciences). The surface was exposed to protein for ∼8 min before remaining reactive esters were blocked using 1 m ethanolamine, pH 8, for 2 min. All experiments were carried out and analyzed according to the manufacturer's instructions.
Steady-state Energy Transfer Measurements of Fluorescently Labeled IgEFc
Spectra were collected using a Spex Fluoromax (Jobin-Yvon) fluorimeter with polarizers set to vertical on excitation and magic angle (54.7°) on emission, to remove any polarization bias in the collected data. Measurements were made in a 50-μl fluorescence cuvette maintained at 20 °C in a thermostated cell holder with concentration-matched (200 nm) samples. Comparisons were made on the basis of acceptor mRFP fluorescence at 610 nm (monochromator bandwidth of 2 nm) with excitation wavelengths from 350 to 600 nm (bandwidth 2 nm). All spectra were corrected for the intensity of the source. Relevant spectra were also corrected for bleed-through of emission seen at 610 nm as a result of excitation of eGFP (always minimal); this was detected using IgEFc constructs labeled with eGFP alone. FRET efficiency (E) was calculated according to Lakowicz (27) based on the published extinction coefficient (ϵ) for the acceptor, mRFP, at the peak excitation wavelength for the eGFP donor (ϵA(λDex)) and the extinction coefficient for eGFP at the same wavelength (ϵD(λDex)) (24) together with the emission intensities of the free acceptor at the peak emission wavelength for the acceptor (FA(λAem)) and donor in the presence of acceptor at this wavelength (FAD(λAem)) using Equation 1.
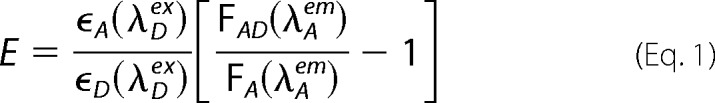 |
Fluorescence Lifetime Measurements of IgEFc Constructs
Measurements were made using an Edinburgh Instruments Lifespec spectrofluorometer fitted with a cooled micro channel plate-photomultiplier, emission polarizer, and monochromator set at 510 nm (bandwidth 4 nm) for eGFP fluorescence and a 468-nm polarized pulsed laser diode light source. The polarizer settings were the same as for the steady-state measurements. Sample preparation was identical to that used for steady-state measurements, with all experiments carried out in a 50-μl fluorescence cuvette at 20 °C maintained in a thermostated holder. Data were analyzed using TRI2 (28) updated with a single transient module. The photophysics of eGFP are complex, such that there are at least two electronic ground states and two excited states, for example see Refs. 29–32. Together with the biexponential decay kinetics, this precludes an entirely unambiguous measurement of the separation between the donor and acceptor fluorophores as the photophysical decay cannot be definitively modeled. Instead, the true average lifetimes (¯τ) were calculated using Equation 2, where αn were the relative amplitudes (normalized such that ∑nαn = 1), and τn are the lifetimes for the nth exponential.
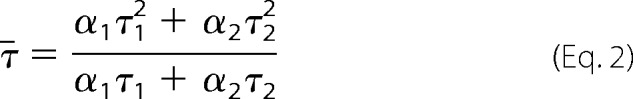 |
For the purpose of calculating FRET efficiencies, the excitation-weighted average lifetime (〈τ〉) was calculated using Equation 3.
The lifetime FRET efficiency (Eτ) was calculated using Equation 4 where 〈τDA〉 and 〈τ0D〉 are the excitation-weighted average lifetimes for the biosensor and control experiment where the acceptor was not on the same protein as the donor, respectively.
 |
Steady-state Anisotropy Measurements of Fluorescently Labeled IgEFc
Fluorescence anisotropy (r) measurements of eGFP-labeled IgEFc samples were made using the same Spex Fluoromax used for intensity FRET measurements. HomoFRET measurements were made according to a method adapted from Squire et al. (33); briefly, the emission monochromator was set at 560 nm (bandwidth 2 nm), and excitation was either performed at 488 nm (bandwidth 2 nm) and 514 nm (bandwidth 2 nm) or by carrying out an excitation scan from 470 to 510 nm (bandwidth 2 nm). Emission anisotropies (r) were then calculated according to Equation 5 using the orthogonally polarized emission intensities IVV and IVH taken with vertically polarized excitation together with polarized emission intensities IHV and IHH taken with horizontally polarized excitation to correct for instrumental polarization bias (G factor; Equation 6).
 |
 |
Anisotropy Decay Measurements of eGFP-labeled IgEFc
Anisotropy decay measurements were made using the same instrument used for the lifetime measurements. Polarized decays were collected at an emission wavelength of 510 nm (bandwidth 4 nm). G-factor normalization was accomplished by matching the summed counts in the VV-polarized and VH-polarized component decay curves to the separately measured steady-state anisotropies via Equation 7 (27, 34), where r is the steady-state anisotropy as defined by Equation 5.
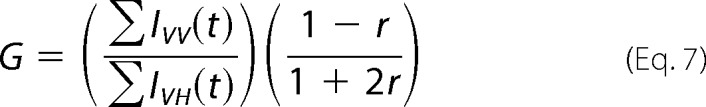 |
Generation of Fusion Protein Models
A model of the mRFP-IgEFc-eGFP biosensor was generated based on the crystal structures of IgEFc (PDB code 1O0V (6)) and GFP (1GFL chain A (35)). Although eGFP and mRFP show strong structural and sequence similarity in the barrel domains, the excited-state dipole transition moment vector geometry for mRFP is considerably less well defined relative to that in eGFP (36). Because this was a prerequisite for the use of the models in the calculation of theoretical FRET efficiencies (see below), eGFP was used in the model in place of mRFP. The models were constructed by fusing eGFP N- and C-terminally to the IgEFc (PDB code 1O0V) via rotational linker residues using the program FPMOD (37) to create a model of eGFP-IgEFc-eGFP simulating its genetically encoded form. The GFP units were allowed to move over 5 days computational time, and 1300 models with no clashes were generated. The procedure was repeated to produce 1300 models using the extended IgE (theoretical) model PDB 1IGE (38).
Theoretical Calculation of FRET Efficiencies from Fusion Protein Models
Inter-probe distances (R) for FRET biosensors are often related to FRET efficiency (E) using Equation 8.
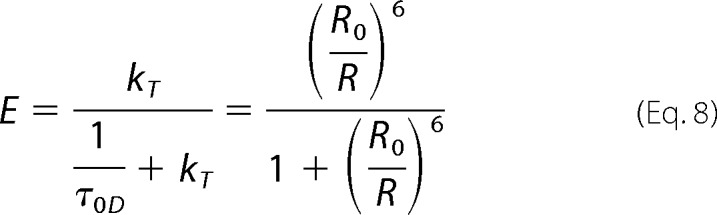 |
where kT is the rate constant for transfer, whereas R0 is the separation between the donor and the acceptor for which E = 50%, also known as the Förster distance, in the definition of which the orientation factor (κ2) is commonly assigned its dynamic random average value of 2/3. However, this expression is inadequate for the IgEFc biosensor, in which there are two acceptors for each donor (the sum of the rate constants to each acceptor replaces the single rate constant in Equation 8) and where averaging over the full multiplicity of conformers in the population exhibiting a wide range of probe separations and orientations is necessary. Similarly, the time-dependent depolarization due to rotation coupled with homoFRET between the two eGFP moieties in each of the conformers of the C-terminal construct and of the separate N-terminal construct is given by Equation 9.
where dT is the transfer depolarization factor (dT = cos2θT − ½), θT being the angle between the relevant transition moment vectors of the two eGFP fluorophores in excited-state exchange, kT is the one-way homoFRET transfer rate constant, and φ is the apparent rotational correlation time. Taking into account the substitution of r0 for its limiting value of 2/5 as well as the addition of the overall multiplier representing the rotational decay e−t/φ, Equation 9 (above) is essentially equivalent to Equation 16 of Ref. 39. With the additional condition of parallel absorption and emission transition moments (δ = 0), Equation 9 (above) is also equivalent to Equation 10 of Ref. 40, with both Refs. 39 and 40 deriving from the work of Tanaka and Mataga (41). Likewise with the relevant substitution r∞ = (r0 + r0dT)/2, which is the limiting anisotropy at very long (infinite) times when the excitation energy is equally shared between the initially excited fluorophore and initially unexcited one, Equation 9 (above) corresponds with the more recently presented Equation 11 in Ref. 42. Again, in the present cases, the measured depolarization will represent an ensemble average over the particular depolarization factors and homoFRET rate constants represented in the multiplicity of conformers.
The appropriate treatments for both cases are detailed in the supplemental information. For each of the 1300 donor-acceptor IgEFc models generated for both extended and bent forms, average intra- and cross-chain separation vectors R were calculated as the average of four vectors linking the coordinates of the donor and acceptor surrogate GFP benzylidene C1 and C2 and imidazolone N3 and C4 atoms within the fluorophores. In each case, the donor-acceptor separation was defined as the length of vector R (i.e. the modulus, |R|). The transition moments of the donor and acceptor fluorophores are represented by the vectors D and A, respectively, and were calculated for each conformation as the average of those obtained from the coordinates of the benzylidene C6 and imidazolone O5 and those of the benzylidene C3 and imidazolone N1 atoms (36, 43). The orientation factor, κ2, was calculated using Equation 10 (44, 45),
where θT is the angle between vectors D and A, θD is the angle between vectors D and R, and θA is the angle between vectors R and A. All calculations together with the FRET efficiencies and homoFRET depolarization courses and the distributions of separations and of orientation factors were carried out using Mathcad 13 (Parametric Technology Corp.) as described in the supplemental information.
Whole Antibody Modeling
The same process used to create the fluorescent IgEFc fusions was used to recreate whole IgE and IgG models based on the crystal structure of the Fabs bound to an allergen β-lactoglobulin (PDB code 2R56) (46). IgG models were based on crystal structures of available human IgG1-like molecules (PDB codes 1HZH (47) and 1IGT (48)) but with the PDB 2R56 Fabs.
RESULTS
Fusion of Fluorescent Proteins to IgEFc Does Not Perturb Binding to sFcϵRIα
Expressed protein was highly purified, free from aggregate and breakdown products as judged by analytical size exclusion gel filtration chromatography (see supplemental Fig. S2). IgE and FcϵRI interact with high affinity (KA ∼ 1010 m−1), and the integrity of stored preparations was confirmed by a complete shift in the gel filtration elution profile of fluorescently labeled IgEFc, detected by UV and fluorescence (see supplemental Fig. S3). In addition to confirming the bindability of the components, these analyses demonstrated that binding to sFcϵRIα did not lead to aggregation of labeled (homo-dimeric) IgEFc. The kinetics and affinities for the binding of all labeled IgEFc constructs to sFcϵRIα was compared with wild-type IgE by surface plasmon resonance (Biacore) (see supplemental Table S1); no noteworthy differences were detected.
As well as complementing the surface plasmon resonance affinity measurements, dual polarization interferometry provided a measure of the physical properties of the IgEFc·sFcϵRIα complex as it formed on the biosensor surface. Initial characterization (data not shown) indicated that all constructs had the same high affinity for sFcϵRIα as observed in surface plasmon resonance and gel filtration experiments. Upon the addition of IgEFc3–4, virtually no change in layer thickness was observed; however, the density increased, indicating that the fragment binds in the plane of the layer (see supplemental Table S2). An increase in thickness was seen upon the addition of IgEFc combined with a decrease in density, suggesting that the Cϵ2 domains present in IgEFc but not IgEFc3–4 point away from the surface. This was even more pronounced for both whole IgE and the FRET pair labeled IgEFc (mRFP-IgEFc-eGFP), which are expected to have similar dimensions. The thickness change for the two larger constructs also resulted in a substantial decrease in the apparent layer density. The marked increase in thickness seen when IgE (or mRFP-IgEFc-eGFP) is bound to receptor compared with IgEFc or IgEFc3–4 indicates that the Cϵ2 domains act to project the Fab region of IgE (or eGFP in the IgEFc biosensor) away from the surface (consistent with the models we derive). It appears that the biosensor mimics well the architecture and overall dimensions of the receptor-bound antibody.
Intramolecular FRET Occurs between eGFP and mRFP on IgEFc Biosensor
Excitation scans were collected at 610 nm (bandwidth 2 nm) for both concentration-matched (200 nm) samples of both dual-labeled (mRFP-IgEFc-eGFP) and individually labeled (IgEFc-eGFP and mRFP-IgEFc) proteins (Fig. 1A) and are shown corrected for residual fluorescence emission by eGFP (Fig. 1B). Comparison of the spectra for mRFP-IgEFc alone with dual-labeled mRFP-IgEFc-eGFP shows an increase in acceptor (mRFP) emission detected at the donor (eGFP) excitation maximum, revealing that intramolecular FRET indeed occurs with an efficiency of (9.6 ± 0.5)%. In contrast, in the control experiment in which the singly labeled mRFP-IgEFc and IgEFc-eGFP constructs were mixed together in the same cuvette, no FRET was observed (Fig. 1B).
FIGURE 1.

The mRFP-IgEFc-eGFP biosensor reveals IgEFc to be bent in solution and to undergo conformational change on binding to sFcϵRIα. A, schematics of the three fluorescent constructs are shown. B, shown is a steady-state fluorescence excitation scan (emission detected at 610 nm) of either the FRET biosensor mRFP-IgEFc-eGFP (blue line) or control molecules mRFP-IgEFc alone (red dashed line) or with IgEFc-eGFP (cyan line). The increase in fluorescence shown here is indicative of energy transfer. C, shown is a schematic of the proposed conformational change model to be tested using the mRFP-IgEFc-eGFP biosensor. D, shown is a steady-state fluorescence excitation scan (emission detected at 610 nm) of either the FRET biosensor mRFP-IgEFc-eGFP alone (blue line) or saturated with sFcϵRIα (green line) or control molecules mRFP-IgEFc and IgEFc-eGFP alone (cyan line) or with sFcϵRIα (black dashed line). The fluorescence increases when sFcϵRIα binds to the mRFP-IgEFc-GFP biosensor indicative of additional bending within IgEFc.
Fluorescence decay measurements of eGFP provide an additional way to quantify FRET in terms of the decrease it causes in the fluorescence lifetime of the donor. However, the complex photophysics of eGFP results in a biexponential decay even for a single eGFP in solution (see supplemental Fig. S4 and Table S3) as previously reported (29–32). We, therefore, compared the true average lifetimes ¯τ (Equation 2) for the FRET biosensor (mRFP-IgEFc-eGFP; 2.57 ± 0.01 ns), which were shorter than that in the control experiment (mRFP-IgEFc + IgEFc-eGFP; 2.76 ± 0.01 ns), providing further evidence that intramolecular FRET is indeed occurring (Fig. 2C). To calculate the FRET efficiencies Eτ (supplemental Table S3) from the lifetimes, the excitation-weighted average lifetimes 〈τ〉 (which are proportional to the relevant steady-state intensities) of the two exponential components (Equation 3) are employed (Equation 4) (see Fig. 2A, supplemental Fig. S5 and Table S3). These also qualitatively support the presence of FRET: 2.45 ± 0.01 ns for the FRET biosensor, shorter than the 2.69 ± 0.01 ns in the control, yielding a FRET efficiency of (8.9 ± 0.7)% (a summary of all the ¯τ, 〈τ〉 and Eτ determined in this study is presented in supplemental Table S3).
FIGURE 2.

Fluorescence lifetime measurements for biosensor and control molecules in the presence and absence of IgE ligands. A, shown are fluorescence decays (excitation 468 nm, emission 510 nm) and bi-exponential fits (line plots) and their photon-counting weighted residuals for the mRFP-IgEFc-eGFP biosensor (open black circles) or control molecules (open red circles). Green circles are the instrument response function (IRF). Non-overlap of the fits indicates a shorter fluorescence lifetime for the mRFP-IgEFc-eGFP biosensor and that it is, therefore, undergoing FRET. B, shown are fluorescence decays (excitation 468 nm, emission 510 nm) and bi-exponential fits (line plots) and their photon-counting weighted residuals for the mRFP-IgEFc-eGFP biosensor alone (open black circles) or saturated with sFcϵRIα (open red circles). Green circles are the instrument response function. Non-overlap of the fits indicates a shorter fluorescence lifetime for the mRFP-IgEFc-eGFP biosensor in the presence of sFcϵRIα. C, shown is mean values of the results of independent experiments (n = 5) for the true average lifetimes (¯τ) calculated using Equation 2. Significant values were determined using a one-way analysis of variance with the Tukey-Kramer test. Only the biosensor mRFP-IgEFc-eGFP shows FRET, with changes in true average lifetimes on binding to both sFcϵRIα and omalizumab Fab.
Modeling Reveals That FRET Can Only Occur If IgEFc Has a Bent Conformation
In contrast to the extended conformation of the six domains in the first published model for IgEFc (37), the crystal structure (6) revealed an acutely bent structure. Calculation of interprobe separations from FRET efficiencies for GFP-related proteins is complicated by uncertainty in the value of the orientation factor (κ2) (44, 46–51) and by the biexponential decays (49, 52). To discriminate between the two extreme sets of conformations of IgEFc, we simulated the flexible movements of fluorescent proteins at the N and C termini of the extended (PDB code 1IGE) and bent (PDB code 1O0V) structures and calculated their theoretical FRET efficiencies on a simplified basis, as described above and in the supplemental information, where the complications above are examined in more detail. The distributions of the κ2 values derived from the models are shown in supplemental Figs. S6 and S7 and are close to random, suggesting sufficient models were produced to sample the space available to the eGFP units.
The calculated efficiency based on the extended models (0.54%) would be experimentally indistinguishable from zero; in contrast, the value based on the bent model (9.2%) is in good agreement with the experimental values of (9.6 ± 0.5)% by steady-state FRET and (8.9 ± 0.7)% by lifetime FRET. This demonstrates first, that the IgEFc does indeed adopt a bent conformation free in solution, and second, that the mRFP-IgEFc-eGFP biosensor is a useful tool for analyzing IgEFc conformation in solution.
IgEFc Undergoes Conformational Change on FcϵRIα Binding
It had been proposed that IgEFc might unbend upon binding to FcϵRI (4, 9), whereas the crystal structure of the IgEFc·sFcϵRIα complex revealed an even more acute bend (10). Unbending (see the schematic in Fig. 1C) would result in a decrease in the FRET signal from the IgEFc biosensor, but the enhanced emission (Fig. 1D) and further decrease in 〈τ〉 to 2.38 ± 0.01 ns, (from 2.45 ± 0.01 ns) (Fig. 2B, and Supplemental Table S3) both point to increased intramolecular FRET; this is consistent with a greater degree of bending upon FcϵRIα binding, bringing the probes closer together (and/or, in principle, their relative orientations changing such that energy transfer between them is more favorable).
Omalizumab Anti-IgE, but Not CD23 (FcϵRII) Binding, Affects IgEFc Conformation
The soluble lectin “head” domain of CD23 (termed derCD23) binds to IgEFc with a 2:1 stoichiometry (20). Recombinant derCD23 was bound to mRFP-IgEFc-eGFP with no detectable change in 〈τ〉 (2.45 ± 0.01 ns; supplemental Table S3), implying no conformational change in IgEFc. In contrast, when omalizumab Fab was added to mRFP-IgEFc-eGFP, 〈τ〉 increased to (2.54 ± 0.01 ns; supplemental Table S3); the decreased FRET implies that the probes moved further apart upon omalizumab binding in an unbending of the IgEFc (and/or their relative orientations changed) such as to reduce FRET efficiency. No effect was seen on the addition of omalizumab Fab to a control mixture of donor-only (IgEFc-eGFP)- and acceptor-only (mRFP-IgEFc)-labeled molecules (Fig. 2C and supplemental Table S3).
HomoFRET Occurs between eGFPs Attached at Each Terminus
Because IgEFc is a dimer, it carries two copies of each biologically encoded label (see the schematics in Fig. 1, A and C, and Fig. 3A), and homoFRET may occur between these identical fluorescent molecules because their excitation and emission spectra overlap. GFP homoFRET is excitation wavelength-dependent and detected through a “red edge” effect in the anisotropy values, because the shape of the emission spectrum is sensitive to excitation wavelength (33, 53). Both N- and C-terminal IgEFc eGFP fusions had lower anisotropies than eGFP alone, and the measured anisotropy was excitation wavelength-dependent (Fig. 3B) indicative of homoFRET. The anisotropy values measured for the C-terminal fused IgEFc were substantially lower than those for the N-terminal construct (Fig. 3B), indicating that the eGFP molecules in this construct were closer (as expected from the IgEFc crystal structure) and/or the difference in their relative orientations more extreme on average.
FIGURE 3.

HomoFRET in eGFP-IgEFc and IgEFc-eGFP does not change on binding to sFcϵRIα. A, the schematic illustrates how homoFRET could be used to detect independent movements of polypeptide chains in the IgEFc homodimer on binding to sFcϵRIα. B, an anisotropy excitation scan of either eGFP alone (green circles), eGFP-IgEFc (blue circles), or IgEFc-eGFP (red circles) is shown. HomoFRET occurs in both constructs as shown by the significantly lower emission anisotropy (r) at lower excitation wavelengths and the increase in r with excitation wavelength. C, sFcϵRIα binding has no effect on the anisotropy excitation scans of either eGFP-IgEFc (blue circles) or IgEFc-eGFP (red circles). D, repeat measurements of steady-state anisotropies of eGFP-IgEFc at two wavelengths are shown. Data represent the average and S.D. for n = 9 independent measurements. Significant values were determined using a one-way analysis of variance with Tukey-Kramer test. The absence of any change on binding sFcϵRIα confirms that there is no significant change in orientation between the Cϵ2 domains. ns, not significant. E, a semi-log plot shows the anisotropy decay r(t) for eGFP alone, with raw data five-point-averaged for display purposes only (green circles). The fit line passing through the decay data is the best-fit impulse response for eGFP rotation (see the supplemental information for details). The overlaid line graph shows the instrument response function (IRF). The anisotropy decay of eGFP (in the absence of homoFRET) is monoexponential and thus appears linear in this semi-log plot. F, a semi-log plot shows the anisotropy decays for eGFP-IgEFc (blue circles) and IgEFc-eGFP (red circles). Raw data were five-point averaged for display purposes only with the lines passing through the data points being the theoretically calculated anisotropy decays based on the models (for a detailed description see supplemental information). The pronounced curvature of the decays in this semi-log plot demonstrate the expected deviation from monoexponential decay kinetics due to homoFRET.
For homoFRET pairs, the overlapping absorption and emission spectra are identical for each partner. Hence, given close enough separation and favorable relative orientations, there will be transfer of the excitation energy of one of them to the other and (because the same conditions apply to the newly excited partner) back again and so on. This leads to sharing of the initial excitation energy between them such that homoFRET is reversible, with identical transfer rate constants in both directions. The extent of completeness of this sharing is determined by the relative rates of transfer and deactivation. Because the excited-state behavior of the two partners is identical, homoFRET leads to no change in the overall emission spectrum nor, in contrast with heteroFRET, of the intensity (quantum yield) or decay kinetics of the exchanging pair as compared with an isolated partner (see Fig. 2 and supplemental Table S3). HomoFRET is only observed experimentally when the emission transition dipoles of the pair (only the case of parallel absorption and emission dipoles is considered here) are not aligned parallel to one another. This leads to depolarization of the overall emission relative to that observed for an isolated partner, the extent of depolarization depending on both the angle between the emission dipoles, and the fraction of emission originating from the initially unexcited partner. This depolarization occurs on top of any rotational depolarization, converting the monoexponential anisotropy decay due to rotational depolarization observed if there were no homoFRET to biexponential decay kinetics in its presence (see Equation 9 above and Ref. 54). Thus, whereas the anisotropy of monomeric eGFP decays monoexponentially as a result of depolarization due to rotational tumbling, with a harmonic mean correlation time 〈φ〉 = 19.8 ± 0.2 ns (see Fig. 3E and supplemental Fig. S8 and Table S4), in good agreement with other values in the literature (see Ref. 55 for example), the presence of homoFRET in the eGFP-IgEFc and IgEFc-eGFP constructs induces an additional anisotropy decay component (see Fig. 3F). The magnitude of this component of the biexponential decay depends on the angular separation of the transition dipole moments of the partners and has a rate constant [2kT + (1/φ)] (see Equation 9) involving the rate constant for homoFRET. It is thus always greater and may be very much greater than the rotational rate constant. In the present cases where many different conformers contribute differently, the overall average anisotropy decays are expected to be highly multiexponential with a wide range of correlation times. This is seen clearly for the C-terminal IgEFc-eGFP fusion (see Fig. 3F, supplemental Fig. S8C, 〈φ〉 = 54.5 ± 0.7 ns). It is less clearly seen for the N-terminal eGFP-IgEFc construct (see Fig. 3F, supplemental Fig. S8B, 〈φ〉 = 38.6 ± 0.3 ns), where the distribution of separations of the partner fluorophores in the various conformers is less favorable to FRET (see the supplemental information). Good fits to the experimental decays were obtained (see Fig. 3F and supplemental Fig. S8) by theoretical calculation of the homoFRET depolarization coupled to rotational depolarization for the 1300 bent-configuration conformers. Details of the calculations are described in the supplemental information, with derived rotational depolarization parameters shown in supplemental Table S4 together with an additional analysis of the effects of using excitation-averaged lifetimes and dynamic average orientation factors. The close to random distributions for the individual model homoFRET depolarization factors are shown in supplemental Figs. S9 and S10. The calculated decays were averaged across the 1300 models, weighted for their widely different extents of depolarization, which result in a large range of individual homoFRET anisotropy decays (supplemental Fig. S11). Despite the same approximations as for the heteroFRET modeling, this confirms the presence of homoFRET and strongly supports the validity of the modeling of heteroFRET efficiency.
Cϵ2 Domains Move as Unit upon FcϵRI Binding
The presence of two eGFP moieties in each of the N- and C-terminal IgEFc fusions enables the following question to be addressed; do the domains within the (Cϵ2)2 or (Cϵ4)2 pairs change their relative orientation upon FcϵRI binding? Gould et al. (4) speculated that the individual Cϵ2 domains might be displaced upon FcϵRI binding (see the schematic, Fig. 3A), although the subsequent IgEFc·sFcϵRIα crystal structure (10) showed no evidence of this. We, therefore, measured the homoFRET for both eGFP-IgEFc and IgEFc-eGFP before and after sFcϵRIα binding. No change was observed in steady-state anisotropy (Fig. 3C and D) or anisotropy decay for either protein measurements (see supplemental Fig. S8, B and C), indicating that even in solution the separation and relative orientation of the two probes at either terminus remained unchanged upon FcϵRIα binding. The (Cϵ2)2 pair thus moves as a rigid unit when the IgEFc becomes more bent upon receptor binding, and as expected, the (Cϵ4)2 pair also behaves as a unit.
The Bent IgEFc Allows Fabs to Move Freely When Bound to FcϵRI
The close agreement between the observed FRET efficiencies and those calculated from the modeled structures of the IgEFc FRET biosensor suggests that modeling could also produce a realistic approximation of the relative movements of the Fabs relative to the Fc. Because the Fabs, like the probes, are connected to IgEFc by flexible linkers, 2000 models were generated for both a whole IgE and a whole IgG to assess the impact of the bent IgEFc on the movement of the Fab arms (Fig. 4 and a supplemental video). When the IgE models (generated in the absence of sFcϵRIα) were overlaid with the crystal structure of the IgEFc·sFcϵRIα complex (PDB code 2Y7Q (10)) only one of the 2000 models clashed with the receptor. The models also show (Fig. 4A, right-hand panel) that the Fabs occupy two essentially independent hemispheres of space pointing away from each other. These conclusions, with the Fab arms pointing away from the cell membrane, are consistent with the dual polarization interferometry results that showed the thickness of the IgE·sFcϵRIα layer to be substantially greater than that of the IgEFc·sFcϵRIα layer. The models are also consistent with the 140° inter-Fab angle seen in EM (14), and the average distance between the antigen binding sites of the models (110–130 Å) agrees well with estimates from small molecule binding studies (56).
FIGURE 4.

Molecular modeling of the space occupied by Fab arms in whole IgE and IgG. Example models of whole IgE (A) and IgG (B) are overlaid with their respective receptor complexes (see also supplemental video). The receptor α-chains are shown in blue with the C-terminal β-strand leading into the cell membrane in red. Fc domains are shown in green, and the Fabs are in purple and orange. Plots showing the spatial distributions of the Fab paratope residue Arg-101 are shown for 2000 models. C, shown is the arrangement of Fabs and allergen dimer in the Fab-allergen-allergen-Fab crystal structures for Bos d 5 (PDB code 2R56 (46)) and Bla g 2 (PDB code 2NR6 (67)). Images of the structures were produced using PyMOL (68).
The IgG models show two distinct differences to those of IgE. First, the regions accessible to the two Fabs interpenetrate (Fig. 4B), and therefore, the Fabs cannot move totally independently. In IgE, the Cϵ2 domains direct the Fabs apart, whereas in IgG they are held closer together by the much smaller hinge region. Second, when the IgG models are overlaid onto the crystal structure of the IgG-Fc·sFcγRIIIα complex (PDB code 1T83 (57)), almost a fifth of the models (392 of 2000) clash with the receptor (Fig. 4B). It is unclear whether the bent hinge seen in whole IgG and some IgGFc crystal structures (and used in the modeling) reflects the natural arrangement in solution, but without the bend, far more IgG-receptor clashes would occur. Fig. 4B also shows that the addition of the D3 domain in FcγRIα prevents clashes with the cell membrane that would inevitably occur in FcγRIIIα.
The range of orientations that the Fabs can adopt relative to the receptor and cell membrane are very different for IgE and IgG (see Fig. 4), and the greater range of inter-Fab angles for IgG is consistent with its enhanced flexibility as seen spectroscopically (11) and by EM (14). In IgE, the models further show that whereas one of the Fabs points upwards, away from the cell membrane, the other lies predominantly parallel to the cell membrane (Fig. 4A); this may have consequences for allergen recognition.
DISCUSSION
Previous efforts to investigate IgE flexibility and conformation focused on the whole antibody. Anisotropy decay measurements on fluorescently labeled murine IgE revealed less flexibility than IgG (11). Steady-state fluorescence anisotropy data obtained using the same murine IgE showed no change in flexibility on FcϵRI binding (12), although later time-resolved analyses distinguished two components, one that was unaffected by receptor binding and abrogated when the Fab arms were cross-linked, and another, longer time-scale movement that was affected by receptor binding (13). Together with FRET measurements (58), this implied a rigidly bent IgE molecule bound to FcϵRI with “wagging” Fabs directed away from the cell surface (13). In further studies with a chemically FRET-labeled chimeric murine IgE/IgG, a bent conformation was confirmed that became less flexible and slightly more bent when bound to FcϵRI on cells (7, 15). This was also consistent with the “cusp-like” structure of rat myeloma IgE inferred from x-ray solution scattering (59). A compact structure for human IgEFc was also shown by solution scattering (8), and the crystal structures of both free (6) and receptor-bound (10) IgEFc reveal asymmetrically and acutely bent conformations, with a more acute bend in the complex.
In the present study we show that the asymmetrically bent IgEFc conformation exists in solution by quantitatively modeling heteroFRET and homoFRET energy transfer and depolarization processes within an ensemble of 1300 models. Rigid-body simulations that sample the conformational space of the fluorescent protein (FP) domains within the IgEFc biosensor allowed detailed calculation of distances between the FRET partners as well as their relative orientations. These factors enabled us to determine for each conformer the degree of depolarization in homoFRET and to avoid inappropriately assuming the problematic 2/3 value for orientation factors (Equation 10) needed in both heteroFRET and homoFRET calculations. This assures optimally reliable predictions of the population average heteroFRET efficiency in the mRFP-IgEFc-eGFP construct and of the population average homoFRET-induced fluorescence anisotropy decays of the C-terminal and the N-terminal eGFP constructs, to compare with the experimental measurements. A remarkable degree of correspondence is found.
Upon binding to sFcϵRIα, the FRET efficiency of the mRFP-IgEFc-eGFP biosensor increases, as measured by a decrease in the average eGFP fluorescence lifetime, suggesting that the bent structure of the IgEFc becomes more bent upon sFcϵRIα binding in solution (Equation 8). Increased bending of IgEFc within the IgEFc·sFcϵRIα complex was also observed by crystallography (10), and we show this is not an artifact of crystal packing. Additionally, the homoFRET results show that during this conformational change the (Cϵ2)2 and the (Cϵ4)2 domain pairs appear to behave as rigid units.
The IgEFc biosensor undergoes no measurable change in FRET efficiency upon binding to CD23 (FcϵRII). Two molecules of derCD23 (the IgE-binding lectin domain) can bind to IgEFc (20) at locations that are distinct from the FcϵRI binding site (as indicated by mutagenesis (61)). Clearly the binding mechanisms also differ, and IgEFc can bind to derCD23 in its naturally bent conformation without an apparent measurable change in the bend angle.
The binding of the Fab fragment of omalizumab (an anti-IgE therapeutic IgG) caused a decrease in the FRET signal (as measured by an increase in the average donor fluorescence lifetime), indicative of an unbending of the IgEFc. Again, the precise location of the epitope recognized by omalizumab is not known, but it acts by preventing the binding of free IgE molecules to FcϵRI (62). This mechanism may thus be indirect and allosteric, as a result of bending the IgE in the opposite direction to that required for receptor engagement.
Changes in FRET efficiency could potentially arise for reasons other than changes in inter-probe separation, but these can be discounted. (i) sFcϵRIα binding could distort the eGFP fluorophore and/or its close environment altering the distribution between its different photophysical states, so that the average lifetime changes for a reason other than FRET. This appears unlikely, however, as sFcϵRIα binding has no effect on the donor alone control (see supplemental Table 3). (ii) A structural change in the eGFP protein could lead to a change in the steady-state spectra (60), but this is not observed upon sFcϵRIα binding (Fig. 1D). Furthermore, the modeling also showed no steric clashes between sFcϵRIα and the FP domains. (iii) The change in FRET efficiency could be solely due to a change in the relative orientations of the FPs in the IgEFc without any appreciable change in distances between them. This can be discounted, first because an unfeasibly large (30–40%) change in the population average κ2 would be required, in contrast to only a 4–5% change in distance. Second, and perhaps more importantly, other experimental evidence indicates that the distribution of orientations of the fluorescent protein probes is unchanged upon sFcϵRIα binding. This is provided by steady-state fluorescence anisotropy (Fig. 3, C and D) and anisotropy decay measurements (supplemental Fig. 8, B and C), where no change is observed. Furthermore, analysis of the biosensor models showed that there was a close to random distribution of κ2 irrespective of the interprobe distance (see supplemental Figs. S6 and S7), suggesting that even if the space available to the probes is more constrained upon sFcϵRIα binding, the randomness of their orientations is not likely to be affected.
The good agreement between the observed FRET efficiencies and those calculated from the modeled IgEFc biosensor suggested that the flexibility of the Fab arms might also be modeled reliably. A comparison between the range of conformations available to the Fab arms in IgE and IgG revealed marked differences (Fig. 4, supplemental video). In IgG, not only do the regions accessible to the two Fab arms overlap extensively, but when mapped onto the crystal structure of an IgG·FcγRIII complex there is considerable clashing with the receptor and also with the membrane. Remarkably, the Fab arms of IgE occupy distinct regions (Fig. 4A) and show virtually no clashing with either the receptor or membrane. The rigid (Cϵ2)2 domain pair appears to act as a steric insulator and presents (and conformationally restricts) the Fab arms in a manner unique to IgE. In part, this difference between IgG and IgE may relate to a difference in biological function; IgG·antigen complexes generally form in solution and then interact with their low affinity receptors, whereas most IgE is receptor-bound before encountering antigen (allergen). If IgE had an IgG-like structure, the Fabs would be much closer to both the cell membrane and the receptor, which might inhibit antigen recognition. However, the manner in which the bent IgE molecule and its (Cϵ2)2 domains present the Fab arms may have important implications for allergen recognition and effector cell activation.
The crystal structure of the high affinity FcγRI, which has an additional domain compared with FcγRII and FcγRIII, is instructive in this context. The biological roles of the high affinity IgG·FcγRI and IgE·FcϵRI complexes have common features; 1) they are both found on dendritic cells where the high affinity results in their action as scavenger receptors that bind and internalize antigens; 2) they are both found on cells that combat infecting organisms; that is, macrophages and bacterial infections for IgG·FcγRI and mast cells and helminth infections for IgE·FcϵRI. The FcγRI crystal structure (PDB code 3RJD (63)) shows that the additional extracellular domain (D3), despite the predictions from mutagenesis (64), does not contact IgG. Instead, it acts as a spacer pushing the D1 and D2 domains (that do contact IgG) away from the membrane (Fig. 4B). Therefore, it is a similar adaptation to the Cϵ2 domains in IgE (only this time in the receptor) that allows the Fabs to move freely without clashing with the membrane, an important ability for a scavenging receptor.
As depicted in Fig. 4A, one of the IgE Fabs predominantly points away from the membrane, whereas the other is oriented parallel to the membrane. The optimal inter-IgE epitope distance for FcϵRI activation has been shown to be 40–60 Å using rigid dinitrophenol-labeled molecular spacers (65, 66). Distances shorter than this were not assayed, but the efficiency of activation decreased rapidly at greater separations. A key feature of allergens is the spatial clustering of more than one IgE epitope and for small allergens or those presenting only a single epitope such as β-lactoglobulin (Bos d 5), this frequently occurs through oligomerization. Two crystal structures of dimerized allergens with bound Fabs have been solved, Bos d 5 (2R56 (46) and Bla g 2 (PDB code 2NR6 (67)) and are both shown in Fig. 4C. These two complexes are particularly instructive for not only do they reveal that the allergen epitopes are optimally spaced for receptor activation (40 and 63 Å, respectively), but they also both display an approximately co-linear arrangement, consistent with IgE Fabs on adjacent receptors oriented parallel to the membrane. The acutely bent receptor-bound IgEFc and conformationally constrained Fab arms may, therefore, predispose IgE-sensitized cells to respond to small antigens with only a particular arrangement of epitopes; this may represent a structural determinant of protein allergenicity, the understanding of which has hitherto proved elusive.
Supplementary Material
Acknowledgments
We thank members of the Sutton and Beavil groups past and present for useful discussions. S.A-B acknowledges his collaborators in the development of the TRI2 software.
This work was supported by a grant from Asthma UK and the Medical Research Council (UK).

This article contains supplemental information, Tables S1–S4, Figs. S1–S11, Equations S1–S20, and a video.
- sFcϵRIα
- soluble fragment of the high affinity IgE receptor FcϵRI α-chain
- mRFP
- monomeric red fluorescent protein
- FP
- fluorescent protein
- eGFP
- enhanced green fluorescent protein.
REFERENCES
- 1. Gould H. J., Sutton B. J. (2008) IgE in allergy and asthma today. Nat. Rev. Immunol. 8, 205–217 [DOI] [PubMed] [Google Scholar]
- 2. Wank S. A., DeLisi C., Metzger H. (1983) Analysis of the rate-limiting step in a ligand-cell receptor interaction. The IgE system. Biochemistry 22, 954–959 [DOI] [PubMed] [Google Scholar]
- 3. Miller L., Blank U., Metzger H., Kinet J. (1989) Expression of high affinity binding of human immunoglobulin E by transfected cells. Science 244, 334–337 [DOI] [PubMed] [Google Scholar]
- 4. Gould H. J., Sutton B. J., Beavil A. J., Beavil R. L., McCloskey N., Coker H. A., Fear D., Smurthwaite L. (2003) The biology of IgE and the basis of allergic disease. Annu. Rev. Immunol. 21, 579–628 [DOI] [PubMed] [Google Scholar]
- 5. Gounni A. S., Lamkhioued B., Ochiai K., Tanaka Y., Delaporte E., Capron A., Kinet J. P., Capron M. (1994) High affinity IgE receptor on eosinophils is involved in defense against parasites. Nature 367, 183–186 [DOI] [PubMed] [Google Scholar]
- 6. Wan T., Beavil R. L., Fabiane S. M., Beavil A. J., Sohi M. K., Keown M., Young R. J., Henry A. J., Owens R. J., Gould H. J., Sutton B. J. (2002) The crystal structure of IgE Fc reveals an asymmetrically bent conformation. Nat. Immunol. 3, 681–686 [DOI] [PubMed] [Google Scholar]
- 7. Zheng Y., Shopes B., Holowka D., Baird B. (1991) Conformations of IgE bound to its receptor FcϵRI and in solution. Biochemistry 30, 9125–9132 [DOI] [PubMed] [Google Scholar]
- 8. Beavil A. J., Young R. J., Sutton B. J., Perkins S. J. (1995) Bent domain structure of recombinant human IgE-Fc in solution by X-ray and neutron scattering in conjunction with an automated curve-fitting procedure. Biochemistry 34, 14449–14461 [DOI] [PubMed] [Google Scholar]
- 9. McDonnell J. M., Calvert R., Beavil R. L., Beavil A. J., Henry A. J., Sutton B. J., Gould H. J., Cowburn D. (2001) The structure of the IgE Cϵ2 domain and its role in stabilizing the complex with its high affinity receptor FcϵRIα. Nat. Struct. Biol. 8, 437–441 [DOI] [PubMed] [Google Scholar]
- 10. Holdom M. D., Davies A. M., Nettleship J. E., Bagby S. C., Dhaliwal B., Girardi E., Hunt J., Gould H. J., Beavil A. J., McDonnell J. M., Owens R. J., Sutton B. J. (2011) Conformational changes in IgE contribute to its uniquely slow dissociation rate from receptor FcϵRI. Nat. Struct. Mol. Biol. 18, 571–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oi V. T., Vuong T. M., Hardy R., Reidler J., Dangle J., Herzenberg L. A., Stryer L. (1984) Correlation between segmental flexibility and effector function of antibodies. Nature 307, 136–140 [DOI] [PubMed] [Google Scholar]
- 12. Slattery J., Holowka D., Baird B. (1985) Segmental flexibility of receptor-bound immunoglobulin E. Biochemistry 24, 7810–7820 [DOI] [PubMed] [Google Scholar]
- 13. Holowka D., Wensel T., Baird B. (1990) A nanosecond fluorescence depolarization study on the segmental flexibility of receptor-bound immunoglobulin E. Biochemistry 29, 4607–4612 [DOI] [PubMed] [Google Scholar]
- 14. Roux K. H., Strelets L., Brekke O. H., Sandlie I., Michaelsen T. E. (1998) Comparisons of the ability of human IgG3 hinge mutants, IgM, IgE, and IgA2, to form small immune complexes. A role for flexibility and geometry. J. Immunol. 161, 4083–4090 [PubMed] [Google Scholar]
- 15. Zheng Y., Shopes B., Holowka D., Baird B. (1992) Dynamic conformations compared for IgE and IgG1 in solution and bound to receptors. Biochemistry 31, 7446–7456 [DOI] [PubMed] [Google Scholar]
- 16. Young R. J., Owens R. J., Mackay G. A., Chan C. M., Shi J., Hide M., Francis D. M., Henry A. J., Sutton B. J., Gould H. J. (1995) Secretion of recombinant human IgE-Fc by mammalian cells and biological activity of glycosylation site mutants. Protein Eng. 8, 193–199 [DOI] [PubMed] [Google Scholar]
- 17. Li X., Zhang G., Ngo N., Zhao X., Kain S. R., Huang C. C. (1997) Deletions of the Aequorea victoria green fluorescent protein define the minimal domain required for fluorescence. J. Biol. Chem. 272, 28545–28549 [DOI] [PubMed] [Google Scholar]
- 18. Campbell R. E., Tour O., Palmer A. E., Steinbach P. A., Baird G. S., Zacharias D. A., Tsien R. Y. (2002) A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. U.S.A. 99, 7877–7882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Durocher Y., Perret S., Kamen A. (2002) High level and high throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 30, E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shi J., Ghirlando R., Beavil R. L., Beavil A. J., Keown M. B., Young R. J., Owens R. J., Sutton B. J., Gould H. J. (1997) Interaction of the low affinity receptor CD23/FcϵRII lectin domain with the Fcϵ3-4 fragment of human immunoglobulin E. Biochemistry 36, 2112–2122 [DOI] [PubMed] [Google Scholar]
- 21. Hunt J., Bracher M. G., Shi J., Fleury S., Dombrowicz D., Gould H. J., Sutton B. J., Beavil A. J. (2008) Attenuation of IgE affinity for FcϵRI radically reduces the allergic response in vitro and in vivo. J. Biol. Chem. 283, 29882–29887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brüggemann M., Williams G. T., Bindon C. I., Clark M. R., Walker M. R., Jefferis R., Waldmann H., Neuberger M. S. (1987) Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J. Exp. Med. 166, 1351–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cook J. P., Henry A. J., McDonnell J. M., Owens R. J., Sutton B. J., Gould H. J. (1997) Identification of contact residues in the IgE binding site of human FcϵRIα. Biochemistry 36, 15579–15588 [DOI] [PubMed] [Google Scholar]
- 24. Peter M., Ameer-Beg S. M., Hughes M. K., Keppler M. D., Prag S., Marsh M., Vojnovic B., Ng T. (2005) Multiphoton-FLIM quantification of the EGFP-mRFP1 FRET pair for localization of membrane receptor-kinase interactions. Biophys. J. 88, 1224–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keown M. B., Ghirlando R., Young R. J., Beavil A. J., Owens R. J., Perkins S. J., Sutton B. J., Gould H. J. (1995) Hydrodynamic studies of a complex between the Fc fragment of human IgE and a soluble fragment of the FcϵRIα chain. Proc. Natl. Acad. Sci. U.S.A. 92, 1841–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Swann M. J., Peel L. L., Carrington S., Freeman N. J. (2004) Dual-polarization interferometry. An analytical technique to measure changes in protein structure in real time, to determine the stoichiometry of binding events, and to differentiate between specific and nonspecific interactions. Anal. Biochem. 329, 190–198 [DOI] [PubMed] [Google Scholar]
- 27. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd Ed., Springer-Verlag New York Inc [Google Scholar]
- 28. Barber P. R., Ameer-Beg S. M., Gilbey J., Carlin L. M., Keppler M., Ng T. C., Vojnovic B. (2009) Multiphoton time-domain fluorescence lifetime imaging microscopy. Practical application to protein-protein interactions using global analysis. J. R. Soc. Lond. Interface 6, S93-S105 [Google Scholar]
- 29. Suhling K., Siegel J., Phillips D., French P. M., Lévêque-Fort S., Webb S. E., Davis D. M. (2002) Imaging the environment of green fluorescent protein. Biophys. J. 83, 3589–3595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heikal A. A., Hess S. T., Webb W. W. (2001) Multiphoton molecular spectroscopy and excited-state dynamics of enhanced green fluorescent protein (EGFP). Acid-base specificity. Chem. Phys. 274, 37–55 [Google Scholar]
- 31. Uskova M. A., Borst J. W., Hink M. A., van Hoek A., Schots A., Klyachko N. L., Visser A. J. (2000) Fluorescence dynamics of green fluorescent protein in AOT reversed micelles. Biophys. Chem. 87, 73–84 [DOI] [PubMed] [Google Scholar]
- 32. Cotlet M., Hofkens J., Maus M., Gensch T., Van der Auweraer M., Michiels J., Dirix G., Van Guyse M., Vanderleyden J., Visser A. J. W. G., De Schryver F. C. (2001) Excited-state dynamics in the enhanced green fluorescent protein mutant probed by picosecond time-resolved single photon counting spectroscopy. J. Phys. Chem. B 105, 4999–5006 [Google Scholar]
- 33. Squire A., Verveer P. J., Rocks O., Bastiaens P. I. (2004) Red-edge anisotropy microscopy enables dynamic imaging of homo-FRET between green fluorescent proteins in cells. J. Struct. Biol. 147, 62–69 [DOI] [PubMed] [Google Scholar]
- 34. Blumberg W. E., Dale R. E., Eisinger J., Zuckerman D. M. (1974) Energy transfer in tRNAphe (yeast). The solution structure of transfer RNA. Biopolymers 13, 1607–1620 [DOI] [PubMed] [Google Scholar]
- 35. Yang F., Moss L. G., Phillips G. N. (1996) The molecular structure of green fluorescent protein. Nat. Biotechnol. 14, 1246–1251 [DOI] [PubMed] [Google Scholar]
- 36. Wan S., Liu S., Zhao G., Chen M., Han K., Sun M. (2007) Photoabsorption of green and red fluorescent protein chromophore anions in vacuo. Biophys. Chem. 129, 218–223 [DOI] [PubMed] [Google Scholar]
- 37. Pham E., Chiang J., Li I., Shum W., Truong K. (2007) A computational tool for designing FRET protein biosensors by rigid-body sampling of their conformational space. Structure 15, 515–523 [DOI] [PubMed] [Google Scholar]
- 38. Padlan E. A., Davies D. R. (1986) A model of the Fc of immunoglobulin E. Mol. Immunol. 23, 1063–1075 [DOI] [PubMed] [Google Scholar]
- 39. Gautier I., Tramier M., Durieux C., Coppey J., Pansu R. B., Nicolas J. C., Kemnitz K., Coppey-Moisan M. (2001) Homo-FRET microscopy in living cells to measure monomer-dimer transition of GFP-tagged proteins. Biophys. J. 80, 3000–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bastiaens P. I., van Hoek A., Benen J. A., Brochon J. C., Visser A. J. (1992) Conformational dynamics and intersubunit energy transfer in wild-type and mutant lipoamide dehydrogenase from Azotobacter vinelandii. A multidimensional time-resolved polarized fluorescence study. Biophys. J. 63, 839–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tanaka F., Mataga N. (1979) Theory of time-dependent photo-selection in interacting fixed systems. Photochem. Photobiol. 29, 1091–1097 [Google Scholar]
- 42. Yeow E. K., Clayton A. H. (2007) Enumeration of oligomerization states of membrane proteins in living cells by homo-FRET spectroscopy and microscopy. Theory and application. Biophys. J. 92, 3098–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shi X., Basran J., Seward H. E., Childs W., Bagshaw C. R., Boxer S. G. (2007) Anomalous negative fluorescence anisotropy in yellow fluorescent protein (YFP 10C). Quantitative analysis of FRET in YFP dimers. Biochemistry 46, 14403–14417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dale R. E., Eisinger J., Blumberg W. E. (1979) The orientational freedom of molecular probes. The orientation factor in intramolecular energy transfer. Biophys. J. 26, 161–193; Correction (1980) Biophys. J.30, 365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Förster Th. (1951) Fluoreszenz Organischer Verbindungen, Vandenhoeck & Ruprecht, Göttingen, Germany [Google Scholar]
- 46. Niemi M., Jylhä S., Laukkanen M. L., Söderlund H., Mäkinen-Kiljunen S., Kallio J. M., Hakulinen N., Haahtela T., Takkinen K., Rouvinen J. (2007) Molecular interactions between a recombinant IgE antibody and the β-lactoglobulin allergen. Structure 15, 1413–1421 [DOI] [PubMed] [Google Scholar]
- 47. Saphire E. O., Stanfield R. L., Crispin M. D., Parren P. W., Rudd P. M., Dwek R. A., Burton D. R., Wilson I. A. (2002) Contrasting IgG structures reveal extreme asymmetry and flexibility. J. Mol. Biol. 319, 9–18 [DOI] [PubMed] [Google Scholar]
- 48. Harris L. J., Larson S. B., Hasel K. W., McPherson A. (1997) Refined Structure of an intact IgG2a monoclonal antibody. Biochemistry 36, 1581–1597 [DOI] [PubMed] [Google Scholar]
- 49. Seward H. E., Bagshaw C. R. (2009) The photochemistry of fluorescent proteins. Implications for their biological applications. Chem. Soc. Rev. 38, 2842–2851 [DOI] [PubMed] [Google Scholar]
- 50. van der Meer B. W. (2002) Kappa squared. From nuisance to new sense. Rev. Mol. Biotechnol. 82, 181–196 [DOI] [PubMed] [Google Scholar]
- 51. Clegg R. M. (1992) Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 211, 353–388 [DOI] [PubMed] [Google Scholar]
- 52. Hink M. A., Griep R. A., Borst J. W., van Hoek A., Eppink M. H., Schots A., Visser A. J. (2000) Structural dynamics of green fluorescent protein alone and fused with a single chain Fv protein. J. Biol. Chem. 275, 17556–17560 [DOI] [PubMed] [Google Scholar]
- 53. Vogel S. S., Thaler C., Blank P. S., Koushik S. V. (2010) in FLIM Microscopy in Biology and Medicine (Periasamy A., Clegg R. M., eds) pp. 245–320, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 54. Thaler C., Koushik S. V., Puhl H. L., 3rd, Blank P. S., Vogel S. S. (2009) Structural rearrangement of CaMKIIα catalytic domains encodes activation. Proc. Natl. Acad. Sci. U.S.A. 106, 6369–6374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Suhling K., Davis D. M., Philips D. (2002) The influence of solvent viscosity on the fluorescence decay and time-resolved anisotropy of green fluorescent protein. J. Fluoresc. 12, 91–95 [Google Scholar]
- 56. Schweitzer-Stenner R., Licht A., Lüscher I., Pecht I. (1987) Oligomerization and ring closure of immunoglobulin E class antibodies by divalent haptens. Biochemistry 26, 3602–3612 [DOI] [PubMed] [Google Scholar]
- 57. Radaev S., Motyka S., Fridman W. H., Sautes-Fridman C., Sun P. D. (2001) The structure of a human type III Fcγ receptor in complex with Fc. J. Biol. Chem. 276, 16469–16477 [DOI] [PubMed] [Google Scholar]
- 58. Baird B., Holowka D. (1985) Structural mapping of Fc receptor-bound immunoglobulin E. Proximity to the membrane surface of the antibody combining site and another site in the Fab segments. Biochemistry 24, 6252–6259 [DOI] [PubMed] [Google Scholar]
- 59. Davis K. G., Glennie M., Harding S. E., Burton D. R. (1990) A model for the solution conformation of rat IgE. Biochem. Soc. Trans. 18, 935–936 [DOI] [PubMed] [Google Scholar]
- 60. Kirchhofer A., Helma J., Schmidthals K., Frauer C., Cui S., Karcher A., Pellis M., Muyldermans S., Casas-Delucchi C. S., Cardoso M. C., Leonhardt H., Hopfner K. P., Rothbauer U. (2010) Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 17, 133–138 [DOI] [PubMed] [Google Scholar]
- 61. Sayers I., Housden J. E., Spivey A. C., Helm B. A. (2004) The importance of Lys-352 of human immunoglobulin E in FcϵRII/CD23 recognition. J. Biol. Chem. 279, 35320–35325 [DOI] [PubMed] [Google Scholar]
- 62. Holgate S., Casale T., Wenzel S., Bousquet J., Deniz Y., Reisner C. (2005) The anti-inflammatory effects of omalizumab confirm the central role of IgE in allergic inflammation. J. Allergy Clin. Immunol. 115, 459–465 [DOI] [PubMed] [Google Scholar]
- 63. Lu J., Ellsworth J. L., Hamacher N., Oak S. W., Sun P. D. (2011) Crystal structure of Fcγ receptor I and its implication in high affinity γ-immunoglobulin binding. J. Biol. Chem. 286, 40608–40613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Harrison P. T., Allen J. M. (1998) High affinity IgG binding by FcγRI (CD64) is modulated by two distinct IgSF domains and the transmembrane domain of the receptor. Protein Eng. 11, 225–232 [DOI] [PubMed] [Google Scholar]
- 65. Paar J. M., Harris N. T., Holowka D., Baird B. (2002) Bivalent ligands with rigid double-stranded DNA spacers reveal structural constraints on signaling by FcϵRI. J. Immunol. 169, 856–864 [DOI] [PubMed] [Google Scholar]
- 66. Sil D., Lee J. B., Luo D., Holowka D., Baird B. (2007) Trivalent ligands with rigid DNA spacers reveal structural requirements for IgE receptor signaling in RBL mast cells. ACS Chem. Biol. 2, 674–68418041817 [Google Scholar]
- 67. Li M., Gustchina A., Alexandratos J., Wlodawer A., Wünschmann S., Kepley C. L., Chapman M. D., Pomés A. (2008) Crystal structure of a dimerized cockroach allergen Bla g 2 complexed with a monoclonal antibody. J. Biol. Chem. 283, 22806–22814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. DeLano W. L. (2011) The PyMOL Molecular Graphics System, Version 1.5.0, Schrödinger LLC, Cambridge, MA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.