

Background: The AGAPs are a subtype of ArfGAPs with a G-protein like domain (GLD) postulated to function as an allosteric binding site.
Results: The C terminus of RhoA binds to AGAP1 and stimulates GAP activity specifically for Arf1 and dependent on the GLD.
Conclusion: AGAP1 is allosterically regulated by specific proteins that bind to the GLD.
Significance: These results are the first example of protein-dependent allosteric regulation of an ArfGAP.
Keywords: Actin, ARF, Enzymes, GTPase, Membrane Trafficking, Small GTPases, AGAP1, GTPase-activating Protein
Abstract
AGAPs are a subtype of Arf GTPase-activating proteins (GAPs) with 11 members in humans. In addition to the Arf GAP domain, the proteins contain a G-protein-like domain (GLD) with homology to Ras superfamily proteins and a PH domain. AGAPs bind to clathrin adaptors, function in post Golgi membrane traffic, and have been implicated in glioblastoma. The regulation of AGAPs is largely unexplored. Other enzymes containing GTP binding domains are regulated by nucleotide binding. However, nucleotide binding to AGAPs has not been detected. Here, we found that neither nucleotides nor deleting the GLD of AGAP1 affected catalysis, which led us to hypothesize that the GLD is a protein binding site that regulates GAP activity. Two-hybrid screens identified RhoA, Rac1, and Cdc42 as potential binding partners. Coimmunoprecipitation confirmed that AGAP1 and AGAP2 can bind to RhoA. Binding was mediated by the C terminus of RhoA and was independent of nucleotide. RhoA and the C-terminal peptide from RhoA increased GAP activity specifically for the substrate Arf1. In contrast, a C-terminal peptide from Cdc42 neither bound nor activated AGAP1. Based on these results, we propose that AGAPs are allosterically regulated through protein binding to the GLD domain.
Introduction
The ArfGAPs are a family of proteins encoded by 31 genes in humans (1). The proteins have a common catalytic domain but are otherwise diverse and can be divided into 10 subgroups based on domain structure. The physiologic function of ArfGAPs is thought to be related to the biochemical activity of inducing hydrolysis of GTP bound to Arf family proteins (2), which are regulators of membrane traffic and actin. Consistent with this function, the ArfGAPs have been found to affect membrane traffic and actin (3–5). Mechanisms of regulation of the GAP activity and the relationship of GAP2 activity to biologic function have not been established.
The largest subgroup, the AGAPs, are encoded by 11 genes, and two, AGAP1 and AGAP2, were found to function in post-Golgi trafficking. AGAP1 binds through its PH domain to the clathrin adaptor protein AP-3 (6), which is important for the biogenesis of lysosomes and lysosomal-related organelles (7, 8). AGAP2, which has been implicated in glioblastoma (9), binds to the clathrin adaptor protein AP-1 and affects transferrin recycling (10) and retrograde transport from the early endosome to the trans-Golgi network (11). Both AGAP1 and AGAP2, as recombinant proteins, associate with endocytic structures containing AP-1 and AP-3. The potential role of clathrin adaptor binding on GAP activity was considered, but no effect of AP-1 on the activity of AGAP2 (10) or AP-3 on AGAP1 was detected. The GLD of AGAP1, which is related to Ras superfamily proteins with highest homology to Miro-family proteins (12–15), might have a regulatory role. The role of the GLD in regulating GAP activity has not been explored.
The molecular basis for the function of a G-protein domain within a multidomain protein has been well characterized for several proteins, including leucine-rich repeat kinase 1/2 (LRRK1/2) (16–21), which contains a G-protein domain called ROC, a dimerization domain called COR, and a kinase domain. GTP binding to the ROC domain induces dimerization that increases kinase activity (16, 20).
The GLD of AGAP2 was initially reported to bind nucleotides (22). However, in subsequent work no nucleotide was detected by chemical methods, and nucleotide binding was not detected either by methods that detect high affinity binding sites or by equilibrium binding methods that would detect low affinity binding sites (23). The GLD of AGAP2 was crystallized, and no nucleotide was found (24), although nucleotidase activity of the purified protein indicated the possibility of a low affinity (mm) binding site. With low affinity nucleotide binding, a plausible hypothesis is that AGAPs are regulated by a similar mechanism to LRRK with nucleotides driving dimerization and activation of the enzyme.
RhoA and RhoB are members of the Ras superfamily of GTP-binding proteins (25, 26). They have been studied mostly as regulators of actin cytoskeleton and cellular adhesions, but some evidence implicates these proteins in the endocytic pathway (27–30). RhoA activation has been reported to inhibit clathrin-mediated endocytosis by activating Rho kinase, resulting in the phosphorylation of endophilin (31, 32). Rho family proteins have been reported to function in similar endocytic traffic as AGAPs. RhoB, at least in part through PRK1, slows traffic from early and late endosomes to lysosomes (28, 29, 33–38). RhoA activation caused redistribution of lysosomes without affecting other endosomal compartments (39–42).
We tested the hypothesis that the GLD of AGAP1 has a role in regulating GAP activity of AGAP1. The GLD domain by itself did not affect activity. In a two-hybrid screen, Rho family GTP-binding proteins were identified as potential binding partners for the GLD domain. Characterization of the interaction revealed that the C-terminal 12 amino acids of RhoA were able to specifically interact with AGAP1 stimulating GAP activity. We conclude that AGAP family proteins can be allosterically regulated by proteins that bind to the GLD.
MATERIALS AND METHODS
Plasmids
Plasmids for bacterial expression of His10AGAP1, His10[347–804]AGAP1 (his10[ΔGLD]AGAP1), GST[69–317]AGAP1 (GST-GLD1), and FLAG-AGAP1have been described (23). His10 [S85N]AGAP1 was made using the QuikChangeTM mutagenesis kit (Agilent Technologies, Santa Clara, CA). AGAP1-HA was subcloned into the NotI and EcoRI sites of pCDNA3.1(−) (Invitrogen). HA-AGAP2 and AGAP2-His6 have been described (11). Briefly, HA-AGAP2 was constructed from the full-length MGC image clone (IMAGE id 5218187; GenBank accession number NM014770). BamHI and XbaI sites were inserted in the N terminus and the C terminus of AGAP2, respectively. The cDNA with a sequence encoding an N-terminal hemagglutinin epitope tag was subcloned into pcDNA3 (Invitrogen). For a plasmid for bacterial expression of the His-tagged protein, the C-terminal stop codon was deleted, and BamHI and SalI sites were inserted in the N terminus and the C terminus of the AGAP2 cDNA by PCR. The PCR product was inserted into pET21a (EMD Biosciences). Myc RhoA, Myc [T19N]RhoA, Myc Rac1 and Myc CDC42 were from Addgene (Cambridge, MA). Plasmids for bacterial recombinant protein for His-RhoA, His-Rac1, and His-CDC42 were generous gifts from Dr. Silvio Gutkind. The plasmid for bacterial expression of leukemia-associated Rho guanine nucleotide exchange factor DH/PH ([766–1138]LARG) was kindly provided by Dr. John Sondek (43).
Antibodies
Rat monoclonal anti-HA antibody was from Roche Applied Science. Rabbit polyclonal Anti-Myc antibody was from Abcam Inc. (Cambridge, MA). Mouse monoclonal antibodies against adaptin γ (for AP-1), adaptin α (for AP-2), and Adaptin δ (for AP-3) were from BD Biosciences.
Protein Purification
Human myrArf1 and myrArf6 protein were expressed in and purified from bacteria as described (44–46). Recombinant His10AGAP1 and AGAP2-His6 were expressed as before (23). Proteins were purified by gradient chromatography on a 5-ml His Trap HPTM column (GE Healthcare) and eluted with imidazole followed by separation on a hydroxylapatite column as follows. Fractions from a His Trap HPTM column containing His10AGAP1 or AGAP2-His6 were pooled and diluted to a final buffer composition of 20 mm Tris, pH 8.0, 200 mm NaCl, 1 mm MgCl2, and 1 mm β-mercaptoethanol. The pool was applied to a 1-ml hydroxylapatite column that had been equilibrated with 50 mm potassium phosphate, pH 7.0, 100 mm NaCl, and 1 mm dithiothreitol (DTT). The column was then washed with 50 mm potassium phosphate, pH 7.0, 100 mm NaCl, and 1 mm DTT. The protein was eluted with 300 mm potassium phosphate, 100 mm NaCl, and 1 mm DTT and dialyzed into 20 mm Tris, pH 8.0, 200 mm NaCl, and 1 mm MgCl2. For recombinant His10[ΔGLD]AGAP1, recombinant protein was purified using a 5-ml Histrap HPTM column and further purified by gel filtration using a HiPrep 16/60 Sephacryl S-100 HRTM column (GE Healthcare) and developed in 20 mm Tris-HCl, pH 8.0, 200 mm NaCl, 1 mm MgCl2, and 5 mm β-mercaptoethanol. LARG DH/PH, His-RhoA, His-Rac1, and His-Cdc42 were expressed and purified as described (43, 47).
GAP Assay
The C50 (the concentration of ArfGAP that induces hydrolysis of 50% of the GTP bound to Arf in a fixed time) was determined as described in Luo et al. (44, 48). Lipids necessary for the reaction were provided as large unilamellar vesicles composed of 40% (mol %) phosphatidylcholine, 25% phosphatidylethanolamine, 15% phosphatidylserine, 9% phosphatidylinositol, 1% phosphatidylinositol 4,5-bisphosphate, and 10% cholesterol prepared as previously described (49). Total phospholipid concentration in the assays was 500 μm.
Synthesis of C-terminal Peptide of RhoA and CDC42
Peptides were assembled on Wang resin utilizing Fmoc (9-fluorenylmethoxycarbonyl)/tert-butyl chemistry. The coupling reactions were conducted by means of the 2(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate-hydroxybenzotriazole method. Biotinylation at the N terminus was accomplished by the N,N[prime]-diisopropylcarbodiimide/hydroxy succinamide method. Cleavage of the peptide from the resin was achieved with a trifluoroacetic acid/water/triisopropylsilane mixture (92.5/5/2.5, v/v) at room temperature. After the resin had been removed by filtration, the filtrate was concentrated by flushing with nitrogen gas, and crude peptides were precipitated with diethyl ether. Crude peptides were purified using reversed-phase high performance liquid chromatography (HPLC) on a preparative C4 column with a water/acetonitrile solvent system containing trifluoroacetic acid. Purified peptides were characterized by matrix-associated laser desorption ionization time-of-flight mass spectrometry and reversed-phase-HPLC on an analytical C18 column. The purity of all peptides was found to be >95%. Peptide from RhoA was biotin-GGGLQARRGKKKSG and from Cdc42 was biotin-GGGLEPPEPKKSRR.
Cell Culture and Immunoprecipitation (IP)
HeLa cell were maintained in Dulbecco's modified enriched media (DMEM) containing 10% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) at 37 °C with 5% CO2. Cells were transfected with plasmids directing expression of AGAP1-HA and or Myc-RhoA by Lipofectamine 2000 (Invitrogen). 24 h after transfection, HeLa cells were lysed in buffer containing 50 mm Hepes, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 10% glycerol, 1% Triton X-100, and protease inhibitor mixture (Roche Applied Science). Lysates were clarified by centrifugation at 14,000 rpm for 10 min at 4 °C. 1 mg of cell lysate were incubated with 2 μg of rat anti-HA Ab or polyclonal anti-myc Ab for 1 h at 4 °C. Immunocomplexes were precipitated by overnight incubation with γ BindTM beads (GE Healthcare). After washing the beads with lysis buffer 3 times, the immune complexes were eluted in a 2-fold concentrate of SDS sample buffer and analyzed by SDS-PAGE and immunoblotting using specific Abs.
For the binding of the C-terminal peptide of RhoA or Cdc42 to AGAP1, HeLa cells were transfected with plasmids directing expression of AGAP1-HA or FLAG-AGAP1. Cells were then lysed in the same buffer used in the IP experiments. Lysate were then incubated with 15 μm of each peptide overnight in the presence of streptavidin-coated beads (Sigma). Samples were collected by centrifugation at 750 × g for 5 min, washed in lysis buffer, and subjected to analysis by SDS-PAGE and immunoblot.
Yeast Two-hybrid Screening
Yeast two-hybrid screening was carried out at Myriad Genetics (Salt Lake City, UT) using the GLD domain of AGAP2 as bait with a mating-based method. The corresponding cDNA for AGAP2 GLD domain (amino acids 30–250) was cloned into pGBT.superB as fused to GAL4 DNA-binding domain. The bait plasmid was introduced into Myriad's ProNet yeast strain PNY200 (MATα ura3-52 ade2-101 trp1-901 his3-Δ200 leu2-3,112 gal4Δ gal80Δ). The bait yeast cells were allowed to mate with Myriad's ProNet MATa yeast cells, BK100 (MATa ura3-52 trp1-901 his3-Δ200 leu2-3,112 gal4Δ gal80Δ GAL2-ADE2 LYS2::GAL1-HIS3 met2::GAL7-lacZ) containing human brain, human spleen, or mouse embryo cDNA libraries. After mating, 5 million diploid yeast cells were obtained and selected on His- and Ade-lacking medium. The auxotrophy is suppressed if the bait and prey protein interact. The prey plasmid was isolated from the positive colonies, and the interaction was confirmed by expression of third reporter gene (lacZ). cDNA in the positive prey plasmid was sequenced.
Miscellaneous
All lipids were purchased from Avanti Polar Lipids (Alabaster, AL). BODIPY-GTPγS was purchased from Invitrogen. A fluorescence assay for nucleotide binding to Rho family proteins using BODIPY-labeled guanine nucleotide has been described (43). Data were collected using a FluorMax 3 spectrophotometer (Jobin Yvon Horiba, Edison, NJ). The reactions were conducted at 30 degree, and fluorescence was measured at an excitation wavelength of 502 nm and an emission wavelength of 511 nm. Adobe Photoshop and Illustrator (Adobe Systems Inc., San Jose, CA) were used to prepare composite figures.
RESULTS
GLD Domain Does Not Render AGAP1 Sensitive to Nucleotide
We determined GAP activity of full-length His10AGAP1 in the presence of GDP, GTP, ADP, or ATP (Fig. 1A, Table 1). In these experiments, AGAP1 was titrated into the reaction to determine the amount of GAP that induced hydrolysis of 50% of the GTP bound to myristoylated Arf1 in a fixed time, 3 min. The value, referred to as the C50, is inversely proportional to catalytic power. The C50 was not affected by the presence of nucleotide. We then compared the activity of full-length His10AGAP1 and AGAP1 with the GLD deleted (His10[ΔGLD]AGAP1) (Fig. 1B). The proteins had similar C50 values, and the C50 values were not affected by either guanine or adenine nucleotide (Table 1). We conclude that AGAP1 was not regulated by the same GTP-regulated homodimerization mechanism described for LRRK1/2 (20, 50).
FIGURE 1.
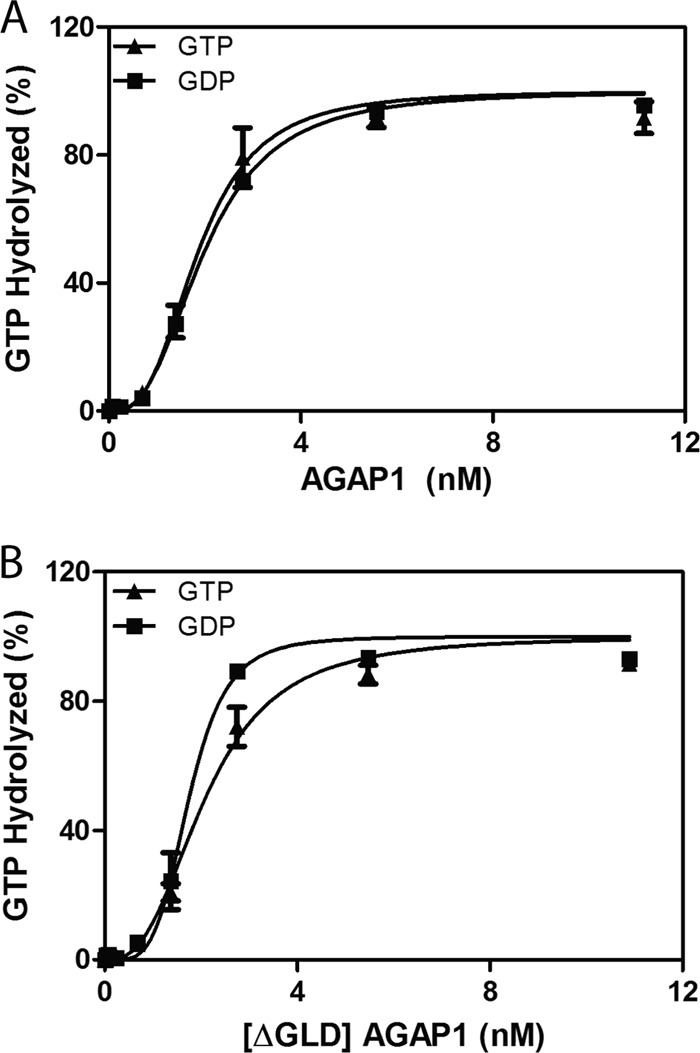
Effect of GLD on GAP activity of AGAP1. A, His10AGAP1 was titrated into a reaction containing 0.4 μm myrArf1·GTP, 500 μm large unilamellar vesicles, and either 1 mm GTP or 1 mm GDP. B, His10[ΔGLD]AGAP1 was titrated into the reaction as described in A.
TABLE 1.
Effect of nucleotides on GAP activity of AGAP1
His10AGAP1 was titrated into a reaction containing 0.4 μm myrArf1·GTP, 500 μm LUVs, and 1 mm concentrations of the indicated nucleotide. The results are the average and S.E. of three experiments. These experiments were independent of those presented in Fig. 1.
| Protein | C50 |
|||
|---|---|---|---|---|
| GTP | GDP | ATP | ADP | |
| nm | ||||
| His10AGAP1 | 2.0 ± 1.1 | 2.0 ± 1.3 | 3.1 ± 1.4 | 2.0 ± 1.0 |
| [ΔGLD]AGAP1 | 2.1 ± 1.2 | 1.8 ± 1.3 | ||
GLD Is Protein Binding Site
We considered an alternate hypothesis that AGAP1 and AGAP2 were regulated by proteins binding to the GLD. Two-hybrid screens for binding partners to the GLD of AGAP2 identified Rho family GTP binding proteins as candidates, including RhoA, Rac1, Rac2, and Cdc42 (Table 2). No other proteins were identified in the screen. The two-hybrid clones were missing critical elements for GTP binding (e.g. G1 motif GXXXXGK(T/S)) and the switch 1 region of the proteins but did contain at least part of switch 2 and the region immediately C-terminal to switch 2.
TABLE 2.
Clones from two-hybrid screen
Bait was [30–250]AGAP2.
AGAP1 and AGAP2 have significant sequence homology (Fig. 2A), which led us to determine if both bound to Rho family proteins. Interaction between RhoA and both AGAP1 and -2 was examined by coimmunoprecipitation (Fig. 2, B–D). We used epitope-tagged recombinant proteins because of limitations of antibodies to the native proteins. RhoA was tested first. The two-hybrid results indicated the interaction may be nucleotide-independent. Nevertheless, switch 2 was still present in the two-hybrid clones so we could not a priori assert that either GTP- or GDP-bound forms of Rho were involved. The dominant negative ([T19N]RhoA) and wild type forms of RhoA were expressed. The constitutively active form was toxic. Both wild type RhoA (not shown) and [T19N]RhoA (Fig. 2B) coimmunoprecipitated with AGAP1-HA, although [T19N]RhoA was more efficient. We examined [T17N]Rac1 by the same approach but did not detect binding to AGAP1 (not shown). HA-AGAP2 and AGAP2-GFP were also examined (Fig. 2, C and D). As with AGAP1, binding to [T19N]RhoA was detected by coimmunoprecipitation using AGAP2 with either an N-terminal HA tag or a C-terminal GFP fusion. Thus, [T19N]RhoA coprecipitated with AGAP1 and AGAP2 independent of the type and position of the epitope tag used to detect the protein. We focused on AGAP1 and RhoA because both have been implicated in lysosomal function.
FIGURE 2.

Interaction of AGAP1 and AGAP2 with RhoA. A, a schematic of AGAP1 and AGAP2 is shown. The sequence identity for each domain is indicated. Ank, ankryrin repeat. B–D, shown is detection of RhoA-AGAP interaction by coimmunoprecipitation. In panel B, HeLa cells expressing AGAP1-HA and myc[T19N]RhoA were lysed. In panel i proteins were precipitated using an antibody to the myc epitope, and the relative quantities of AGAP1-HA in the precipitates (ppt) were determined by immunoblotting for the HA epitope. Blots for proteins in the total lysates are labeled IB HA and IB Myc. The blot for the HA-tagged AGAP1 that was precipitated with mycRhoA is labeled IP Myc and IB HA. In panel ii, proteins were precipitated with an antibody to the HA epitope, and the amount of RhoA in the precipitate was detected with an antibody to the myc epitope. In panel C, HeLa cells expressing AGAP2-GFP and myc[T19N]RhoA were examined. In panel i, proteins were precipitated with an antibody to GFP. In panel ii, proteins were precipitated with an antibody to myc. In panel D, cells expressing HA-AGAP2 (the epitope was fused to the N terminus of AGAP, as opposed to the epitope on the C terminus of AGAPs in other experiments presented in this figure) and myc[T19N]RhoA were examined. Proteins were precipitated with an antibody to the myc epitope. For all experiments, the input blots are 6% of the lysate used for the IP.
Rho Family Proteins Increase Activity of AGAP1
We tested the hypothesis that Rho protein binding to AGAP1 regulates GAP activity by determining the effects of His-RhoA, His-Cdc42, and His-Rac1, bound to either GTP or GDP, on Arf GAP activity of purified full-length AGAP1or AGAP2 using Arf1·GTP as a substrate (Fig. 3, A and B). RhoA, bound to either GDP or GTP, was found to stimulate activity. Although Rac1 did not detectably interact with AGAP1 as measured by coimmunoprecipitation, we found it did activate GAP activity of both AGAP1 and AGAP2-His6 independent of the bound nucleotide. Titration revealed that Rac1 was less efficient than RhoA in activating AGAP1 (Table 3), consistent with a higher affinity for RhoA than for Rac. The difference in affinity could explain the ability to detect the interaction of RhoA and AGAPs, but not Rac with AGAPs, by IP. Cdc42 did not increase activity significantly.
FIGURE 3.

Characterization of the effect of Rho family protein on GAP activity of AGAP1 and AGAP2. A, activation of AGAP1 is shown. GAP assays contained 0.2 μm [α-32P]GTP·myrArf1, 2.2 nm His10AGAP1, 500 μm large unilamellar vesicles, and either 1 mm GDP with 0.5 μm His-RhoA·GDP, His-Cdc42·GDP, and His-Rac1·GDP or 10 μm GTPγS with 0.5 μm His-RhoA·GTPγS, His-Cdc42·GTPγS, and His-Rac1·GTPγS. Activity is expressed as -fold increase over His10AGAP1 only control. Statistical analysis of the data included one way analysis of variance followed by the Dunnett multiple comparison test using AGAP1 only as the control. * p < 0.05; ***, p < 0.001; ns, not significant. B, activation of AGAP2 is shown. Experiment were performed essentially the same as in A, but 1.7 nm AGAP2-His6 was used instead of AGAP1. Statistical analysis was the same as for A. **, p < 0.01 compared with AGAP2 alone. C, heat sensitivity of RhoA effect is shown. The reactions were performed the same as in A, except 0.5 μm his-RhoA·GDP or his-Rac1·GDP was heated at 95 °C for 10 min where indicated. Data were analyzed by analysis of variance followed by Bonferroni's multiple comparison test. **, p < 0.01 compared with AGAP1 alone; ns, not significant compared with AGAP1 alone; #, p < 0.05 compared with AGAP1 + RhoA. D, the effect of RhoA on ArfGAP1 is shown. GAP activity for ArfGAP1 (20 nm) incubated with 0.5 μm RhoA·GDP or RhoA·GTP was determined. The results were analyzed by analysis of variance followed by the Bonferroni's multiple comparison test. ns, not significant compared with ArfGAP1 alone. E, shown is the effect of RhoA·GDP on [ΔGLD]AGAP1. Either 2.2 nm His10AGAP1 or 2.2 nm His10[ΔGLD]AGAP1 was used in the GAP assays. Statistical analysis was the same as for C. ***, p < 0.001 compared with AGAP1 alone; ns, not significant compared with [ΔGLD]AGAP1. F and G, shown is a comparison of substrates for RhoA-dependent AGAP1 catalytic activity. His10AGAP1 was titrated into a reaction containing myrArf1·GTP or myrArf6·GTP either in the absence (F) or presence (G) of 0.5 μm his-RhoA·GDP.
TABLE 3.
Comparison of RhoA and Rac1 as activators of AGAP1
RhoA and Rac1 were titrated into a reaction mixture contain 2.2 nm His10AGAP1 and 0.2 μm [α−32P]GTP·myrArf1. A plot of GAP activity vs. RhoA or Rac1 was fit to a hyperbola to determine the amount required to achieve one-half maximal effect (EC50). The results are the mean ± S.E. for three experiments. The means were compared by Student's t test.
| EC50 | |
|---|---|
| RhoA | Rac1 |
| nm | |
| 91 ± 24 | 197 ± 36a |
a, p < 0.05.
Stimulation of AGAP1 GAP activity was dependent on properly folded Rho protein (Fig. 3C); RhoA heated to 95 °C for 5 min did not stimulate activity. The stimulation was also dependent on the GLD of AGAP1 (Fig. 3E). The activity of a recombinant protein comprised of the PH, ArfGAP, and Ank repeat domains, but lacking the GLD domain of AGAP1 ([ΔGLD]AGAP1), was not stimulated by RhoA·GDP. In addition, the stimulation was specific for Arf isoforms (Fig. 3, F and G, Table 4). RhoA increased activity of AGAP1 toward Arf1 but decreased activity toward Arf6. The effect was specific for AGAP1. ArfGAP1 activity was not affected by RhoA (Fig. 3D).
TABLE 4.
Effect of RhoA on the Arf specificity of AGAP1
AGAP1 was titrated into a GAP reaction containing either 0.2 μm myrArf1· [α32P]GTP or myrArf6· [α32P]GTP and 0.5 μm RhoA·GDP as indicated. The concentration of AGAP1 required to achieve 50% hydrolysis was determined from curves. The means for three experiments are presented. The results were analyzed by Student's t test.
a, p < 0.001 compared to no RhoA.
b, p < 0.05 compared to no RhoA.
C-terminal 12 Amino Acids of RhoA Are Sufficient for Effect on AGAP1
The activation and binding of AGAP1 by RhoA is nucleotide-independent. This is similar to that described for other enzymes such as protein kinase C-related kinase/protein kinase N for RhoA and phosphatidylinositol phosphate kinase, Tor2, and CD2AP for Rac1. In these three instances, the Rho family proteins were found to bind through their C termini to targets (51–54). We determined whether RhoA may bind to AGAP1 by a similar mechanism. Biotin-conjugated peptides derived from the C termini of RhoA and Cdc42 were mixed with lysates of cells expressing AGAP1-HA or FLAG-AGAP1. The peptides were precipitated using streptavidin conjugated to agarose beads. AGAP1-HA and FLAG-AGAP1, detected by immunoblotting, were in precipitates with the RhoA peptide but not the Cdc42 peptide (Fig. 4A). The peptides were titrated into GAP assay reaction mixtures containing Arf1·GTP and His10AGAP1 (Fig. 4B). The peptide from RhoA increased activity by 44-fold with a half-maximal effect at 10 μm. The peptide from Cdc42 had no detectable effect. Unlike the full-length protein, the peptide was not heat-sensitive (Fig. 4C). The reason for the difference with full-length protein may be that the full-length may aggregate on heating, rendering the C terminus inaccessible, or the C terminus may be sequestered by another mechanism, e.g. oxidation of C-terminal cysteines. The short peptide would not be expected to aggregate on heating and does not contain cysteines.
FIGURE 4.

C terminus of RhoA interacts with AGAP1. A, peptide from RhoA C terminus binds to AGAP1. Lysates of HeLa cells expressing FLAG-AGAP1 or AGAP1-HA were mixed with biotin-tagged-RhoA C-terminal peptide (biotin-GGGLQARRGKKKSG) or the biotin-tagged CDC42 C-terminal peptide (biotin-GGGLEPPEPKKSRR). The peptides were precipitated with streptavidin-agarose. The beads were washed three times with lysis buffer, and precipitates were analyzed by SDS-PAGE followed by immunoblotting for the FLAG or HA epitope. B, effect of RhoA C terminus on GAP activity is shown. RhoA or Cdc42 C-terminal peptides were titrated into reactions containing 1.1 nm His10AGAP1 and 0.5 μm myrArf·GTP. C, a lack of heat sensitivity of peptide activator is shown. The RhoA C-terminal peptide was heated for 10 min at 95 °C before being added to the GAP assay at the indicated concentration. D, the effect of peptide was observed with a nonmyristoylated Arf. RhoA or Cdc42 peptide were titrated into reactions containing 1.1 nm His10AGAP1 and 0.2 μm [α32P]GTP·[L8K]Arf1.
Effect of Peptide Does Not Depend on Myristoylation of Arf
Myristoylated Arf is restricted to membrane surfaces. Activation by RhoA could be achieved by driving recruitment to the membrane surface containing Arf1·GTP. Alternatively, RhoA binding to AGAP1 could be inducing a conformational change independent of membrane recruitment. To test this idea, the effect of the peptide from RhoA on AGAP1 activity using a soluble mutant of Arf1, [L8K]Arf1, was determined. As for myrArf1, peptide from RhoA stimulated activity, whereas the peptide from Cdc42 had no effect (Fig. 4D).
Nucleotides Do Not Have Detectable Effect on Allosteric Regulation
Although we did not detect an effect of nucleotide on activity when determining GAP activity of AGAP1 in the absence of a GLD protein binding partner, it is plausible that nucleotides bind to the GLD to affect binding to Rho. We reexamined nucleotide binding to the AGAP1 as a first step in examining this possibility. The change of fluorescence of Bodipy-GTPγS, whose fluorescence increases upon protein binding, was determined. AGAP1 did not affect fluorescence (Fig. 5A). As a positive control, the change in fluorescence due to binding to the same concentration of RhoA, catalyzed by the RhoA exchange factor leukemia-associated Rho guanine nucleotide exchange factor is shown. The result that AGAP1 does not bind nucleotide is consistent with previous work in which binding of radioisotopically labeled GTP could not be detected, nor could nucleotide bound to AGAP1 be detected chemically (23). However, nucleotide binding could not be excluded on the basis of these negative results. To determine if the ability to bind nucleotide was necessary for RhoA-dependent activation, serine 85 on AGAP1 was mutated to asparagine ([S85N]AGAP1). Serine 85 is a residue in the P-loop that is critical for nucleotide binding in other G-proteins and is often mutated to asparagine to disrupt GTP binding. The effect of RhoA on [S85N]AGAP1 was determined. The mutant was activated by RhoA·GDP (not shown) and the C-terminal peptide from RhoA to a similar extent as the wild type protein (Fig. 5, B and C). Related to this finding is a recent report that, in contrast to previous reports, the ROC domain of LRRK2 affected kinase activity independently of nucleotide binding (21).
FIGURE 5.
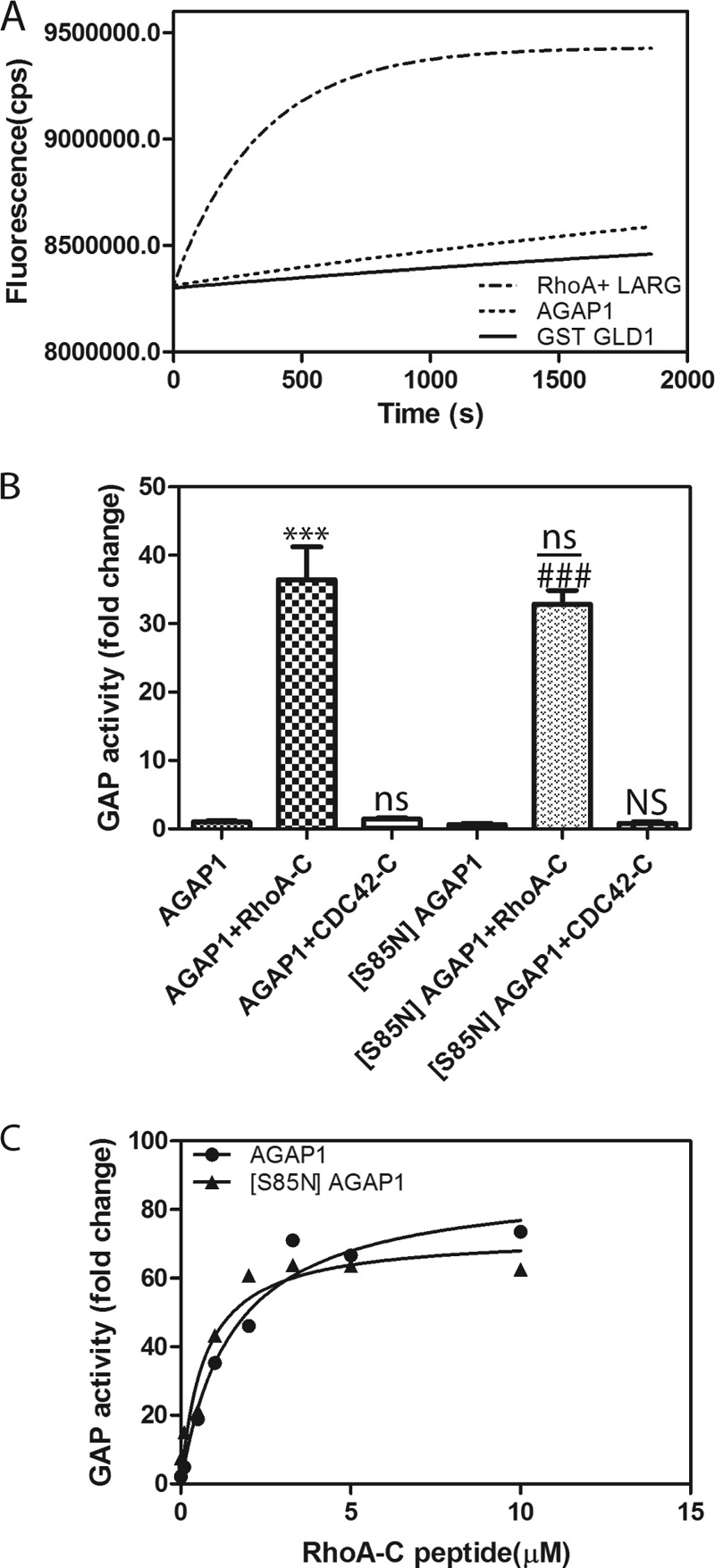
Tests for role for nucleotide binding to AGAP1. A, no association with BODIPY-GTPγS detected. Nucleotide binding was measured by incubating 0.5 μm his10AGAP1 or GST-GLD with 17.5 nm BODIPY-GTPγS in a buffer containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 2 mm DTT, 1 mm EDTA, 0.5 mm MgCl2. Binding to RhoA catalyzed by LARG DH/PH is shown as a positive control. The reaction contained 2.5 μm RhoA, 200 nm leukemia-associated Rho guanine nucleotide exchange factor (LARG), and 17.5 nm BODIPY-GTPγS in a reaction mixture that was the same as for the AGAPs except 3 mm MgCl2 was present. B, P-loop mutation had no effect on activity. GAP activity of His10AGAP1 and His10 [S85N]AGAP1 was compared in reactions containing 1.1 nm concentrations of the indicated recombinant AGAP1 and, where indicated, 10 μm RhoA C-terminal peptide or Cdc42 C-terminal peptide. The data were analyzed by one way analysis of variance followed by the Bonferroni multiple comparison test. ***, p < 0.001 compared with AGAP1 alone; ns, not significantly different than AGAP1 alone; ###, p < 0.001 compared with [S85N]AGAP1 alone; NS, not significant compared with [S85N]AGAP1 alone; ns, not significant compared with AGAP1 + RhoA-C. C, titration of RhoA-C peptide into reaction with His-AGAP1 and His-[S85N]AGAP1 is shown. GAP activity was determined using a fixed time point assay, and reaction mixtures contained 1.1 nm AGAP1 or 1.1 nm [S85N]AGAP1 and 0.2 μm [α32P]GTP·Arf1 as the substrate and the indicated concentration of peptides from either the C terminus of RhoA (RhoA-C) or Cdc42 (Cdc42-C).
DISCUSSION
AGAP1 is the best documented case to date as far as we are aware of an ArfGAP being allosterically regulated by a protein interaction. The activity of other ArfGAPs, such as ArfGAP2 and ArfGAP3, are dependent on a second protein, such as coatomer (55–58). In these cases, the means of regulation are not clear. Coatomer may directly interact with the ArfGAP domain, and it is plausible that coatomer forms part of the substrate binding site. Cargo peptides have also been found to increase ArfGAP activity, but the effect required the presence of coat protein and, therefore, may have been mediated by an effect on the coat protein (56). Other mechanisms mediating the regulation of Arf GAPs that have been described involve lipids. ArfGAP1 is thought to preferentially bind to curved lipid surfaces, thereby being recruited to a surface containing the substrate Arf (59). Phosphoinositides are thought to also act as allosteric modifiers for the ArfGAP ASAP1 (60, 61). For AGAP1, given that there was specific activation for Arf1 over Arf6, the effect of the C-terminal peptide of RhoA is likely due to a conformational change affecting the active site rather than a change in recruitment to the membrane containing the substrate. The precise molecular mechanisms are now being examined.
We consider the increase in specificity for Arf1 on activation of AGAP1 to be significant for two reasons. First, the result supports the idea that the activation is due to a conformational change resulting in better substrate recognition with exclusion of irrelevant substrates. Our speculation is that at physiological concentrations of Arf and AGAP, the basal state AGAP does not regulate any of the Arfs because the proteins are present in insufficient concentrations. When stimulated, AGAP1 will specifically use Arf1 as a substrate. Second, these results highlight the fact that our current understanding of substrate specificity for the ArfGAPs may be flawed. If relatively inactive proteins are examined, real substrate specificity may be obscured. This phenomenon has been observed for another ArfGAP. Full-length ASAP1 is autoinhibited by an N-terminal extension of the BAR domain. The protein does not have strong preference for Arf1 over Arf6; however, with the autoinhibitory motif removed, ASAP1 uses Arf1 50–100-fold more efficiently than Arf6 (48, 62, 63). When trying to assess substrate specificity in cells, activity of an ArfGAP may be observed because of the large amount of protein expressed, but the results could be misleading because the allosteric activator is limiting.
Regulation of AGAP is unique among proteins with G-protein-like domains. Three modes of nucleotide-dependent dimerization have been described (16): (i) GTP-dependent dimerization of two proteins from the same family, e.g. signal recognition particle and signal recognition particle receptor; (ii) GTP-dependent formation of a dimer of identical G proteins (e.g. guanylate binding protein 1 or dynamin); (iii) dimerization through a domain adjacent to the G-protein domain (e.g. LRRK1/2) with subsequent GTP-dependent association of the G-protein domain. Among these proteins, LRRKs appeared to be analogous to AGAPs in that they contain multiple domains including a catalytic domain (17, 21, 50, 64). Although LRRK dimerizes through a COR domain, the kinase is inactive until the G-protein domains associate on GTP binding. Our results support a different model for AGAPs. RhoA·GDP appears to make the active complex, and the effects of the GLD are independent of nucleotide binding. The AGAP dependence of activity is sigmoidal in the absence of Rho but hyperbolic in the presence of RhoA, consistent with the possibility that RhoA affects dimerization of AGAP1. We are currently testing the idea of RhoA regulating dimerization of AGAP1 and determining if other proteins may have a similar regulatory interaction with AGAPs.
The GLD of AGAP1 is different than other G protein domains in a second respect; we did not detect nucleotide binding. Although we cannot exclude nucleotide binding on the basis of our inability to detect it, the result may reflect another aspect of G protein domains. Although the focus is often on nucleotide binding, the G proteins are primarily protein binding motifs, not GTPases. Recent reexamination of LRRK1 has provided evidence that nucleotide binding is not necessary for the function (21). Given the conflicting reports, the role of GTP binding for dimerization in LRRK remains to be determined. These recent results together with ours raise the possibility that among G proteins and G protein domains, which evolved to bind to other proteins, some have retained protein binding while losing the ability to bind nucleotide.
The association of AGAP1 and RhoA is intriguing, with two possible connections to the biological function. The first connection is to lysosomal maturation. RhoA has been implicated in lysosomal distribution (39–42). AGAP1 binds to AP-3, which affects lysosome function (6, 65). Reduced expression of AP-3 has been examined in Natural Killer cells. The cytolytic granules, which are lysosome-related organelles, formed, but they were not able to distribute appropriately to immunologic synapse, the site of release of the contents of the cytolytic granules (66). AGAP2, which binds to AP-1, could also interact with a Rho family member to affect early endocytic trafficking. For instance, RhoB affects trafficking steps that might be AP-1-dependent. These links to membrane traffic are being examined.
The second connection between AGAPs and Rho family proteins is related to the actin cytoskeleton and focal adhesions. Epitope-tagged AGAP2 (67) associates with fatty acids, and overexpression of epitope-tagged AGAP1 causes disruption of stress fibers (23). AGAP2, which is reported to bind to focal adhesion kinase, also affects the morphology of focal adhesions and cell migration (67). In addition, Rock1 was identified as a potential binding partner of AGAP2 in two-hybrid screens using the PH domain as bait.3 A model that we will investigate is that the ternary complex of RhoA-AGAP2-Rock coordinates Arf and Rho signaling to control fatty acid and stress fiber dynamics as has been described for ARAP subtype ArfGAPs (47, 68, 69).
In summary, we have identified the GLD of AGAP1 as a protein binding motif that allosterically regulates catalytic activity. This mechanism for regulating an ArfGAP has not been previously described and could generalize to the other members of the AGAP subfamily, the largest ArfGAP subfamily, as well as other multidomain proteins containing G-protein domains.
Acknowledgment
We thank Richard A. Kahn for discussions.
This work was supported, in whole or in part, by the National Institutes of Health Grant BC 007365 (Intramural Program of the NCI).
Z. Nie and P. A. Randazzo, unpublished information.
- GAP
- GTPase-activating protein
- GLD
- G-protein-like domain
- LRRK
- leucine-rich repeat kinase
- PH
- pleckstrin homology
- IP
- immunoprecipitation
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- C50
- concentration of GAP that induces hydrolysis of 50% of the GTP bound to Arf in a fixed time.
REFERENCES
- 1. Kahn R. A., Bruford E., Inoue H., Logsdon J. M., Jr., Nie Z., Premont R. T., Randazzo P. A., Satake M., Theibert A. B., Zapp M. L., Cassel D. (2008) Consensus nomenclature for the human ArfGAP domain-containing proteins. J. Cell Biol. 182, 1039–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kahn R. A., Cherfils J., Elias M., Lovering R. C., Munro S., Schurmann A. (2006) Nomenclature for the human Arf family of GTP-binding proteins. ARF, ARL, and SAR proteins. J. Cell Biol. 172, 645–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Randazzo P. A., Hirsch D. S. (2004) Arf GAPs. Multifunctional proteins that regulate membrane traffic and actin remodeling. Cell. Signal. 16, 401–413 [DOI] [PubMed] [Google Scholar]
- 4. Randazzo P. A., Inoue H., Bharti S. (2007) Arf GAPs as regulators of the actin cytoskeleton. Biol. Cell 99, 583–600 [DOI] [PubMed] [Google Scholar]
- 5. Inoue H., Randazzo P. A. (2007) Arf GAPs and their interacting proteins. Traffic 8, 1465–1475 [DOI] [PubMed] [Google Scholar]
- 6. Nie Z., Boehm M., Boja E. S., Vass W. C., Bonifacino J. S., Fales H. M., Randazzo PA. (2003) Specific regulation of the adaptor protein complex AP-3 by the Arf GAP AGAP1. Dev. Cell 5, 513–521 [DOI] [PubMed] [Google Scholar]
- 7. Robinson M. S., Bonifacino J. S. (2001) Adaptor-related proteins. Curr. Opin. Cell Biol. 13, 444–453 [DOI] [PubMed] [Google Scholar]
- 8. Boehm M., Bonifacino J. S. (2001) Adaptins. The final recount. Mol. Biol. Cell 12, 2907–2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahn J. Y., Rong R., Kroll T. G., Van Meir E. G., Snyder S. H., Ye K. (2004) PIKE (phosphatidylinositol 3-kinase enhancer)-A GTPase stimulates Akt activity and mediates cellular invasion. J. Biol. Chem. 279, 16441–16451 [DOI] [PubMed] [Google Scholar]
- 10. Nie Z., Fei J., Premont R. T., Randazzo P. A. (2005) The Arf GAPs AGAP1 and AGAP2 distinguish between the adaptor protein complexes AP-1 and AP-3. J. Cell Sci. 118, 3555–3566 [DOI] [PubMed] [Google Scholar]
- 11. Shiba Y., Römer W., Mardones G. A., Burgos P. V., Lamaze C., Johannes L. (2010) AGAP2 regulates retrograde transport between early endosomes and the TGN. J. Cell Sci. 123, 2381–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reis K., Fransson A., Aspenström P. (2009) The Miro GTPases. At the heart of the mitochondrial transport machinery. FEBS Lett. 583, 1391–1398 [DOI] [PubMed] [Google Scholar]
- 13. Fransson S., Ruusala A., Aspenström P. (2006) The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 344, 500–510 [DOI] [PubMed] [Google Scholar]
- 14. Koshiba T., Holman H. A., Kubara K., Yasukawa K., Kawabata S., Okamoto K., MacFarlane J., Shaw J. M. (2011) Structure-function analysis of the yeast mitochondrial Rho GTPase, Gem1p. Implications for mitochondrial inheritance. J. Biol. Chem. 286, 354–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frederick R. L., Okamoto K., Shaw J. M. (2008) Multiple pathways influence mitochondrial inheritance in budding yeast. Genetics 178, 825–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gasper R., Meyer S., Gotthardt K., Sirajuddin M., Wittinghofer A. (2009) It takes two to tango. Regulation of G proteins by dimerization. Nat. Rev. Mol. Cell Biol. 10, 423–429 [DOI] [PubMed] [Google Scholar]
- 17. Guo L., Gandhi P. N., Wang W., Petersen R. B., Wilson-Delfosse A. L., Chen S. G. (2007) The Parkinson disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp. Cell Res. 313, 3658–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greggio E., Zambrano I., Kaganovich A., Beilina A., Taymans J. M., Daniëls V., Lewis P., Jain S., Ding J., Syed A., Thomas K. J., Baekelandt V., Cookson M. R. (2008) The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J. Biol. Chem. 283, 16906–16914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Berger Z., Smith K. A., Lavoie M. J. (2010) Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 49, 5511–5523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ito G., Okai T., Fujino G., Takeda K., Ichijo H., Katada T., Iwatsubo T. (2007) GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson disease. Biochemistry 46, 1380–1388 [DOI] [PubMed] [Google Scholar]
- 21. Liu M., Dobson B., Glicksman M. A., Yue Z., Stein R. L. (2010) Kinetic mechanistic studies of wild-type leucine-rich repeat kinase 2. Characterization of the kinase and GTPase activities. Biochemistry 49, 2008–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ye K., Hurt K. J., Wu F. Y., Fang M., Luo H. R., Hong J. J., Blackshaw S., Ferris C. D., Snyder S. H. (2000) Pike. A nuclear gtpase that enhances PI 3-kinase activity and is regulated by protein 4.1N. Cell 103, 919–930 [DOI] [PubMed] [Google Scholar]
- 23. Nie Z., Stanley K. T., Stauffer S., Jacques K. M., Hirsch D. S., Takei J., Randazzo P. A. (2002) AGAP1, an endosome-associated, phosphoinositide-dependent ADP-ribosylation factor GTPase-activating protein that affects actin cytoskeleton. J. Biol. Chem. 277, 48965–48975 [DOI] [PubMed] [Google Scholar]
- 24. Soundararajan M., Yang X., Elkins J. M., Sobott F., Doyle D. A. (2007) The centaurin γ-1 GTPase-like domain functions as an NTPase. Biochem. J. 401, 679–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ridley A. J. (2001) Rho GTPases and cell migration. J. Cell Sci. 114, 2713–2722 [DOI] [PubMed] [Google Scholar]
- 26. Jaffe A. B., Hall A. (2005) Rho GTPases. Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 27. Etienne-Manneville S., Hall A. (2002) Rho GTPases in cell biology. Nature 420, 629–635 [DOI] [PubMed] [Google Scholar]
- 28. Gampel A., Parker P. J., Mellor H. (1999) Regulation of epidermal growth factor receptor traffic by the small GTPase rhoB. Curr. Biol. 9, 955–958 [DOI] [PubMed] [Google Scholar]
- 29. Qualmann B., Mellor H. (2003) Regulation of endocytic traffic by Rho GTPases. Biochem. J. 371, 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Symons M., Rusk N. (2003) Control of vesicular trafficking by Rho GTPases. Curr. Biol. 13, R409–R418 [DOI] [PubMed] [Google Scholar]
- 31. Kaneko T., Maeda A., Takefuji M., Aoyama H., Nakayama M., Kawabata S., Kawano Y., Iwamatsu A., Amano M., Kaibuchi K. (2005) Rho mediates endocytosis of epidermal growth factor receptor through phosphorylation of endophilin A1 by Rho kinase. Genes Cells 10, 973–987 [DOI] [PubMed] [Google Scholar]
- 32. Khelfaoui M., Pavlowsky A., Powell A. D., Valnegri P., Cheong K. W., Blandin Y., Passafaro M., Jefferys J. G., Chelly J., Billuart P. (2009) Inhibition of RhoA pathway rescues the endocytosis defects in Oligophrenin1 mouse model of mental retardation. Hum. Mol. Gen. 18, 2575–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wherlock M., Gampel A., Futter C., Mellor H. (2004) Farnesyltransferase inhibitors disrupt EGF receptor traffic through modulation of the RhoB GTPase. J. Cell Sci. 117, 3221–3231 [DOI] [PubMed] [Google Scholar]
- 34. Mellor H., Flynn P., Nobes C. D., Hall A., Parker P. J. (1998) PRK1 is targeted to endosomes by the small GTPase, RhoB. J. Biol. Chem. 273, 4811–4814 [DOI] [PubMed] [Google Scholar]
- 35. Rondanino C., Rojas R., Ruiz W. G., Wang E., Hughey R. P., Dunn K. W., Apodaca G. (2007) RhoB-dependent modulation of postendocytic traffic in polarized Madin-Darby canine kidney cells. Traffic 8, 932–949 [DOI] [PubMed] [Google Scholar]
- 36. Leung S. M., Rojas R., Maples C., Flynn C., Ruiz W. G., Jou T. S., Apodaca G. (1999) Modulation of endocytic traffic in polarized Madin-Darby canine kidney cells by the small GTPase RhoA. Mol. Biol. Cell 10, 4369–4384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Apodaca G. (2001) Endocytic traffic in polarized epithelial cells. Role of the actin and microtubule cytoskeleton. Traffic 2, 149–159 [DOI] [PubMed] [Google Scholar]
- 38. Khandelwal P., Ruiz W. G., Apodaca G. (2010) Compensatory endocytosis in bladder umbrella cells occurs through an integrin-regulated and RhoA- and dynamin-dependent pathway. EMBO J. 29, 1961–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nishimura Y., Itoh K., Yoshioka K., Ikeda K., Himeno M. (2002) A role for small GTPase RhoA in regulating intracellular membrane traffic of lysosomes in invasive rat hepatoma cells. Histochem. J. 34, 189–213 [DOI] [PubMed] [Google Scholar]
- 40. Nishimura Y., Itoh K., Yoshioka K., Tokuda K., Himeno M. (2003) Overexpression of ROCK in human breast cancer cells. Evidence that ROCK activity mediates intracellular membrane traffic of lysosomes. Pathol. Oncol. Res. 9, 83–95 [DOI] [PubMed] [Google Scholar]
- 41. Steffan J. J., Snider J. L., Skalli O., Welbourne T., Cardelli J. A. (2009) Na+/H+ exchangers and RhoA regulate acidic extracellular pH-induced lysosome trafficking in prostate cancer cells. Traffic 10, 737–753 [DOI] [PubMed] [Google Scholar]
- 42. Steffan J. J., Williams B. C., Welbourne T., Cardelli J. A. (2010) HGF-induced invasion by prostate tumor cells requires anterograde lysosome trafficking and activity of Na+/H+ exchangers. J. Cell Sci. 123, 1151–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reuther G. W., Lambert Q. T., Booden M. A., Wennerberg K., Becknell B., Marcucci G., Sondek J., Caligiuri M. A., Der C. J. (2001) Leukemia-associated Rho guanine nucleotide exchange factor, a Dbl family protein found mutated in leukemia, causes transformation by activation of RhoA. J. Biol. Chem. 276, 27145–27151 [DOI] [PubMed] [Google Scholar]
- 44. Luo R., Ahvazi B., Amariei D., Shroder D., Burrola B., Losert W., Randazzo P. A. (2007) Kinetic analysis of GTP hydrolysis catalysed by the Arf1-GTP-ASAP1 complex. Biochem. J. 402, 439–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Randazzo P. A., Fales H. M. (2002) in GTPase Protocols: The Ras Superfamily (Manser E., Leung T., eds.) Vol. 189, pp. 169–180, Humana Press Inc., Totowa, NJ [Google Scholar]
- 46. Ha V. L., Thomas G. M.., Stauffer S., Randazzo P. A. (2005) Preparation of myristoylated Arf1 and Arf6. Methods Enzymol. 404, 164–174 [DOI] [PubMed] [Google Scholar]
- 47. Miura K., Jacques K. M., Stauffer S., Kubosaki A., Zhu K., Hirsch D. S., Resau J., Zheng Y., Randazzo P. A. (2002) ARAP1. A point of convergence for Arf and Rho signaling. Mol. Cell 9, 109–119 [DOI] [PubMed] [Google Scholar]
- 48. Luo R., Jacques K., Ahvazi B., Stauffer S., Premont R. T., Randazzo P. A. (2005) Mutational analysis of the Arf1*GTP/Arf GAP interface reveals an Arf1 mutant that selectively affects the Arf GAP ASAP1. Curr. Biol. 15, 2164–2169 [DOI] [PubMed] [Google Scholar]
- 49. Nie Z., Hirsch D. S., Luo R., Jian X., Stauffer S., Cremesti A., Andrade J., Lebowitz J., Marino M., Ahvazi B., Hinshaw J. E., Randazzo P. A. (2006) A BAR domain in the N terminus of the Arf GAP ASAP1 affects membrane structure and trafficking of epidermal growth factor receptor. Curr. Biol. 16, 130–139 [DOI] [PubMed] [Google Scholar]
- 50. Korr D., Toschi L., Donner P., Pohlenz H. D., Kreft B., Weiss B. (2006) LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell. Signal. 18, 910–920 [DOI] [PubMed] [Google Scholar]
- 51. Vincent S., Settleman J. (1997) The PRK2 kinase is a potential effector target of both Rho and Rac GTPases and regulates actin cytoskeletal organization. Mol. Cell. Biol. 17, 2247–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tolias K. F., Cantley L. C., Carpenter C. L. (1995) Rho family GTPases bind to phosphoinositide kinases. J. Biol. Chem. 270, 17656–17659 [DOI] [PubMed] [Google Scholar]
- 53. Tolias K. F., Couvillon A. D., Cantley L. C., Carpenter C. L. (1998) Characterization of a Rac1- and RhoGDI-associated lipid kinase signaling complex. Mol. Cell. Biol. 18, 762–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Saci A., Cantley L. C., Carpenter C. L. (2011) Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol. Cell 42, 50–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Luo R., Randazzo P. A. (2008) Kinetic analysis of Arf GAP1 indicates a regulatory role for coatomer. J. Biol. Chem. 283, 21965–21977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luo R., Ha V. L., Hayashi R., Randazzo P. A. (2009) Arf GAP2 is positively regulated by coatomer and cargo. Cell. Signal. 21, 1169–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kliouchnikov L., Bigay J., Mesmin B., Parnis A., Rawet M., Goldfeder N., Antonny B., Cassel D. (2009) Discrete determinants in ArfGAP2/3 conferring Golgi localization and regulation by the COPI coat. Mol. Biol. Cell 20, 859–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Weimer C., Beck R., Eckert P., Reckmann I., Moelleken J., Brügger B., Wieland F. (2008) Differential roles of ArfGAP1, ArfGAP2, and ArfGAP3 in COPI trafficking. J. Cell Biol. 183, 725–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bigay J., Gounon P., Robineau S., Antonny B. (2003) Lipid packing sensed by ArfGAP1 couples COPI coat disassembly to membrane bilayer curvature. Nature 426, 563–566 [DOI] [PubMed] [Google Scholar]
- 60. Che M. M., Boja E. S., Yoon H. Y., Gruschus J., Jaffe H., Stauffer S., Schuck P., Fales H. M., Randazzo P. A. (2005) Regulation of ASAP1 by phospholipids is dependent on the interface between the PH and Arf GAP domains. Cell. Signal. 17, 1276–1288 [DOI] [PubMed] [Google Scholar]
- 61. Luo R., Miller Jenkins L. M., Randazzo P. A., Gruschus J. (2008) Dynamic interaction between Arf GAP and PH domains of ASAP1 in the regulation of GAP activity. Cell. Signal. 20, 1968–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jian X., Cavenagh M., Gruschus J. M., Randazzo P. A., Kahn R. A. (2010) Modifications to the C terminus of Arf1 alter cell functions and protein interactions. Traffic 11, 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brown M. T., Andrade J., Radhakrishna H., Donaldson J. G., Cooper J. A., Randazzo P. A. (1998) ASAP1, a phospholipid-dependent arf GTPase-activating protein that associates with and is phosphorylated by Src. Mol. Cell. Biol. 18, 7038–7051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Deng J., Lewis P. A., Greggio E., Sluch E., Beilina A., Cookson M. R. (2008) Structure of the ROC domain from the Parkinson disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc. Natl. Acad. Sci. U.S.A. 105, 1499–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Janvier K., Bonifacino J. S. (2005) Role of the endocytic machinery in the sorting of lysosome-associated membrane proteins. Mol. Biol. Cell 16, 4231–4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bossi G., Griffiths G. M. (2005) CTL secretory lysosomes. biogenesis and secretion of a harmful organelle. Semin. Immunol. 17, 87–94 [DOI] [PubMed] [Google Scholar]
- 67. Zhu Y., Wu Y., Kim J. I., Wang Z., Daaka Y., Nie Z. (2009) Arf GTPase-activating protein AGAP2 regulates focal adhesion kinase activity and focal adhesion remodeling. J. Biol. Chem. 284, 13489–13496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yoon H. Y., Miura K., Cuthbert E. J., Davis K. K., Ahvazi B., Casanova J. E., Randazzo P. A. (2006) ARAP2 effects on the actin cytoskeleton are dependent on Arf6-specific GTPase-activating protein activity and binding to RhoA-GTP. J. Cell Sci. 119, 4650–4666 [DOI] [PubMed] [Google Scholar]
- 69. Yoon H. Y., Lee J. S., Randazzo P. A. (2008) ARAP1 regulates endocytosis of EGFR. Traffic 9, 2236–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]