

Background: The p53 and c-Jun N-terminal kinase (JNK) pathways both act to initiate an apoptotic response to genotoxic stress.
Results: The DNA binding domain of p53 binds to diphosphorylated JNK and prevents its dephosphorylation.
Conclusion: p53 potentiates the level of JNK activity.
Significance: Changes in p53 levels may coordinate the timing of an apoptotic response through regulation of JNK activity.
Keywords: Apoptosis, Dual Specificity Phosphoprotein Phosphatase, c-Jun N-terminal Kinase (JNK), p53, Stress
Abstract
c-Jun N-terminal kinase (JNK) is a serine/threonine phosphotransferase whose sustained activation in response to genotoxic stress promotes apoptosis. In Drosophila, the normally rapid JNK-dependent apoptotic response to genotoxic stress is significantly delayed in Dmp53 (Drosophila p53) mutants. Likewise, the extent of JNK activity after UV irradiation is dependent on p53 in murine embryonic fibroblasts with loss of p53 resulting in diminished JNK activity. Together, these results suggest that p53 potentiates the JNK-dependent response to genotoxic stress; however, the mechanism whereby p53 stimulates JNK activity remains undefined. Here, we demonstrate that both Drosophila and human p53 can directly stimulate JNK activity independently of p53-dependent gene transcription. Furthermore, we demonstrate that both the Drosophila and human p53 orthologs form a physical complex with diphosphorylated JNK (DPJNK) both in vivo and in vitro, suggesting that the interaction is evolutionarily conserved. Focusing on human p53, we demonstrate that the interaction maps to the DNA binding domain (hp53DBD). Intriguingly, binding of p53DBD alone to DPJNK prevented its inactivation by MAPK phosphatase (MKP)-5; however, JNK was still able to phosphorylate c-Jun while in a complex with the p53DBD. Apparent dissociation constants for the p53DBD·DPJNK (274 ± 14 nm) and MKP-5·DPJNK (55 ± 8 nm) complexes were established; however, binding of MKP-5 and p53 to JNK was not mutually exclusive. Together, these results suggest that stress-dependent increases in p53 levels potentiate JNK activation by preventing its rapid dephosphorylation by MKPs and that the simultaneous activation of p53 and JNK may constitute a “fail-safe” switch for the JNK-dependent apoptotic response.
Introduction
Programmed cell death (or apoptosis) plays a fundamental role during the development of multicellular organisms, serving to sculpt tissues by removing excess cells, establish appropriate immune cell complements through the elimination of self-reactive cells, and maintain tissue homeostasis through regulation of normal cell turnover (1–4). Importantly, dysregulation of the normal apoptotic response is an essential step during oncogenic transformation (3, 4), implying that the fidelity of the cell death process is of paramount importance.
Initiation of the apoptotic process depends on activation of either the extrinsic, intrinsic, or both cell death pathways, which differ in the mechanisms whereby the downstream caspase machinery in activated. The c-Jun N-terminal kinase (JNK) signaling cascade has unique roles in both pathways (5–9); thus, a detailed understanding of the role(s) of JNK and its regulation during both normal development and the apoptotic response to oncogenic and genotoxic stress is essential.
JNK was originally identified as a result of its ability to phosphorylate the transcription factor c-Jun at Ser-63 and Ser-73 (10). Additional studies have shown that JNK-dependent phosphorylation also regulates the activity of a series of other transcription factors, such as ATF2, Elk-1, p53, Pax2, c-Myc, FOXO4, STAT1/3, and PPAR-γ1 (for a review, see Ref. 11). Together, these studies suggest that JNK signaling affects cellular phenotypes, including an apoptotic response to genotoxic stress through the regulation of gene transcription. Consistent with this hypothesis, mutation of Ser-63 and Ser-73 to alanine blocks the apoptotic response of several different cell types to JNK stimulation (12–14). Alternatively, JNK can phosphorylate many cytoplasmic proteins, including members of the Bcl-2 and Bax families (15–17), which play a direct role in regulating the apoptotic response through modulation of mitochondrial outer membrane permeability and cytochrome c release. Thus, JNK signaling can stimulate apoptosis through mitochondrion-based mechanisms as well as traditional transcriptional (nucleus-based) mechanisms.
The MAPK phosphatases (MKPs)2 render extracellular signal-regulated kinase (ERK), JNK, and p38 kinases inactive through dephosphorylation of phosphothreonine and phosphotyrosine residues found in the TXY motif in the kinase activation loop (for a review, see Ref. 6). Individual MKPs show preferential activity toward specific MAPKs; however, the functional redundancy between MKPs has hindered assignment of their relevant functions during development (18). Nevertheless, elevated expression of MKPs, like MKP-1, have been shown to support oncogenic transformation by antagonizing the apoptotic response to oncogenic and/or genotoxic stress (19–21). Thus, modulation of phosphatase activity is a critical determinant of cell viability and transformation through its regulation of MAPKs.
p53, commonly referred to as “guardian of the genome,” is known to regulate cell cycle progression and apoptosis in mammals and is mutated in approximately half of all the known human tumors (22, 23). Like its ortholog, Drosophila p53 (Dmp53) is needed to induce apoptosis in cells that are subjected to genotoxic stress (24–29). In contrast, Dmp53 does not induce cell cycle arrest at the G1-to-S checkpoint when overexpressed (28), and it is not required for radiation-induced cell cycle arrest at the G2-to-M checkpoint (24). Therefore, p53-dependent induction of apoptosis is an evolutionarily conserved process, whereas p53-dependent regulation of the cell cycle evolved later.
In mammalian systems, it has been suggested that the basal level p53 activity is influenced in part by JNK as JNK interacts with and promotes ubiquitylation and degradation of p53 (30). However, under genotoxic stress, p53 is thought to both dissociate from and be phosphorylated by JNK at Ser-6 and Thr-81, which allow it to accumulate in cells (31–33). In Drosophila, we have previously demonstrated that p53 is required for activation of the JNK-dependent apoptosis in response to genotoxic stress (34). However, the mechanism whereby Dmp53 influenced Drosophila JNK (dJNK) activity is not known. Here, we establish an evolutionarily conserved mechanism whereby p53 can influence JNK activity. In Drosophila, we demonstrate that both Dmp53 and human p53 selectively bind phosphorylated dJNK, whereas phosphorylation does not influence binding of rat α-stress-activated protein kinase (SAPK; henceforth referred to as rJNK). Furthermore, we demonstrate that the DNA binding domain of p53 is capable of preventing MKP-5-mediated dephosphorylation of activated JNK. Although we demonstrate that MKP-5 binds with a 5-fold tighter affinity to JNK than does p53, binding of MKP-5 is not mutually exclusive with c-Jun or JNK. Based of these results, we propose a model whereby p53 binding to JNK regulates the level of kinase activity with stress-dependent stimulation of p53 levels resulting in sustained activation of JNK and the induction of apoptosis.
EXPERIMENTAL PROCEDURES
Drosophila Genetics
The following Drosophila strains were used in these studies: yw, Dmp53[5A-1–4], pucA251.1/TM6b, Tb, and arm-Gal411. All of the alleles are available from the Bloomington stock center and detailed descriptions can be found at FlyBase. Briefly, the Dmp53[5A-1–4] allele contains a deletion that starts 37 bp upstream of the transcription start site and continues to the last intron, leaving the last exon, which contains only seven codons prior to the stop codon, intact. The pucA251.1 allele is a recessive lethal insertional mutation in which the P{ArB} P-element has intron 2 of the puc locus inserted (35). The P{ArB} element carries a lacZ reporter gene and functions as an enhancer trap for the puc locus (35–37). The arm-Gal411 line carries a P-element that ubiquitously expresses the Gal4 transcription factor under the control of the armadillo promoter (38).
The pucA251.1 p53[5A-1–4] recombinant chromosome was generated through meiotic recombination. To generate flies carrying the UASp-FLAGDmp53 transgene, the Dmp53 open reading frame was amplified and cloned into the pENTR/TOPO vector (Invitrogen). The resulting pENTR/Dmp53 plasmid was sequenced and then recombined into the pPFW destination vector. The resulting transformation vector was then used to generate transformants through P-element-mediated transformation (39). Multiple independent lines were isolated and mapped, and expression was confirmed through Western blotting. To generate the flies harboring the UASp-HAhp53 transgene, the human p53 cDNA was isolated from the pUAS-p53 transformation vector (Ref. 40; a gift from M. Yamaguchi) and cloned into pBluescriptII. Site-directed mutagenesis was used to insert a BglII site just 5′ to the stop codon. The HA tag was inserted into the BglII site as a pair of oligonucleotides. The junction encodes for the following amino acids: … PDSDLYPYDVPDYA* (where * denotes the stop codon). The HA-tagged p53 open reading frame was then transferred to pUASp, and transformants were generated by P-element-mediated transformation. Again, multiple independent lines were isolated and mapped, and expression was tested. Additional details will be provided upon request.
Immunofluorescence/TUNEL Labeling
Wing discs from wandering third instar larvae were dissected and fixed in 3.7% formaldehyde, PBT×5 (phosphate-buffered saline (PBS) + 0.5% Triton X-100) for 20 min on ice. Tissues were then rinsed 3 × 20 min in PBT×5, blocked for at least 3 h at room temperature in PBT×5 + 10% BSA, and incubated with primary antibody overnight at 4 °C. The samples were then washed 3 × 20 min in PBT×5, incubated for 1 h at room temperature with the appropriate fluorescent secondary antibodies (1:1000; Molecular Probes), and washed 3 × 20 min in PBT×5 prior to equilibration in Aqua Poly/Mount (PolySciences, Inc.). TUNEL labeling was done essentially as described (34). Wing discs were then dissected away from the remainder of the larval tissues and mounted for confocal microscopy.
Images of wing discs were obtained with an Olympus FV1000 confocal microscope system equipped with argon (488 nm) and helium-neon (543 nm) lasers for the excitation and detection of FITC and Alexa Fluor 568, respectively. A 40× oil immersion objective (numerical aperture, 1.25) was used for obtaining images. Brightness and contrast levels were adjusted using Slidebook 4.0 (Intelligent Imaging Innovations) image analysis software.
Cell Culture
Murine embryonic fibroblasts were maintained in DMEM supplemented with 10% fetal calf serum, penicillin, and streptomycin. Cultures were grown in 5% CO2 under 3% oxygen tension to mimic physiological conditions (41). Prior to treatment with UV light, cells were washed with 1× Hanks' balanced salt solution, medium was removed, and the culture was exposed to 200 J/m2 UV light using a Spectrolinker XL-1500 UV cross-linker. Fresh medium was added back to the cells, and cultures were returned to 3% oxygen tension until lysates were prepared at the indicated times. S2 cells were maintained in Schneider's medium supplemented with 10% fetal calf serum, penicillin, and streptomycin and transfected as described (42). HCT116 (p53−/−) and HEK 293 cells were maintained at 5% CO2 in DMEM supplemented with 10% fetal calf serum and penicillin. HCT116 (p53−/−) and HEK 293 cells were transfected using the TransIT-LT1 transfection reagent (Mirus) according to the manufacturer's recommendations. SP600125 was resuspended in DMSO, and cells were treated either with drug or vehicle alone (DMSO) for 24 h prior to harvesting cell lysates.
Lysates (in triplicate) were collected from transfected cells ∼48 h after transfection. Luciferase and β-galactosidase activities were determined as described (42). Luciferase activity was normalized to β-galactosidase activity, and data are presented as the average luc/β-gal ratio ±S.D. except in Fig. 3B where luc activity ±S.D. is plotted directly as the activity of the control pCS2+cytoβ-gal reporter was down-regulated by SP600125 itself.
FIGURE 3.

p53 expression stimulates JNK activity independently of p53-dependent changes in gene transcription. A and B, FLAGp53 (A) or FLAGDmp53 (B) was co-transfected with pAP1-luc into HCT116 (p53−/−) cells or S2 cells, respectively. AP1-dependent changes in luciferase expression were monitored, and the dependence on JNK activity was established by addition of SP600125. Transfection of the pAP1-luc alone with carrier DNA served to establish the basal level of JNK activity in the respective cell lines. Note that the p53-dependent increase in luciferase activity depended on JNK signaling as addition of SP600125 suppressed luciferase expression. C and D, FLAGp53MTS (C) or FLAGDmp53BaxTM (D) was co-transfected with pG13-luc, a p53-dependent luciferase reporter, into HEK 293 or S2 cells, respectively. Luciferase expression was compared with their otherwise wild-type p53 counterpart. Note that relocalization of p53 to the mitochondrial membrane failed to stimulate p53-dependent transcription. E and F, FLAGp53MTS (E) or FLAGDmp53BaxTM (F) was co-transfected with pAP1-luc, a JNK-dependent luciferase reporter into HEK 293 or S2 cells, respectively. Luciferase expression was compared with their otherwise wild-type p53 counterpart. Note that relocalization of p53 to the mitochondrial membrane robustly stimulated AP1-dependent luciferase expression. RLU, relative light units. BSSK-II, pBluescript II SK. Data plotted as average, with error bars denoting standard deviation.
Antibodies
The following antibodies were used: mouse α-β-galactosidase (Promega; 1:1000), mouse α-Myc (9E10; 1:5), mouse α-FLAG (Sigma-Aldrich; 1:3000), rabbit α-JNK (Santa Cruz Biotechnology; 1:500), rabbit α-DPJNK (Cell Signaling Technology; 1:500), mouse α-glutathione S-transferase (GST) (Santa Cruz Biotechnology; 1:1000), rabbit α-c-Jun and rabbit α-phospho-c-Jun (Cell Signaling Technology; 1:1000), mouse α-p38 and rabbit α-phospho-p38 (Santa Cruz Biotechnology; 1:1000), and mouse α-p53 (Cell Signaling Technology; 1:2000). Alexa Fluor 568-goat α-mouse (Molecular Probes) and Hoechst dye (Sigma-Aldrich) were used at 1:1000 to visualize primary antibodies and DNA, respectively.
Cloning, Expression, and Purification of Recombinant Proteins
Expression plasmids for producing diphosphorylated rat JNK and diphosphorylated murine p38 were provided by M. Cobb and have been described (43). Briefly, the appropriate plasmid combinations were co-transformed into Escherichia coli BL21(DE3) cells, selected with carbenicillin and kanamycin, and induced for 4 h at 37 °C with 0.8 mm isopropyl 1-thio-β-d-galactopyranoside after reaching an A600 of 0.5. Hexahistidine (His)-tagged rat JNK and murine p38 proteins were purified via nickel affinity chromatography under native conditions followed by ion exchange chromatography using a HiTrap Q HP column (GE Healthcare) and a linear salt gradient. Recombinant proteins were then dialyzed into the appropriate reaction buffer. The DNA binding domain (amino acids 94–312) of human p53 and the full-length coding sequence of human MKP-5 were cloned in pET-15b vector, yielding His-tagged fusion proteins that were purified as described above except that a HiTrap S HP column was used for ion exchange chromatography. Site-directed mutagenesis was used to generate the C408SMKP-5 mutant. Dmp53 (amino acids 1–344) and human p53 (amino acids 1–306) lacking their C-terminal tetramerization domains (ΔC) were cloned in pGEX-KG vector, transformed in E. coli BL21(DE3) cells, selected with carbenicillin, and induced with 0.8 mm isopropyl 1-thio-β-d-galactopyranoside. Glutathione-agarose beads were used in purifying GST-fused proteins after solubilization in NETN buffer (20 mm Tris, 1 mm EDTA, 100 mm NaCl, 0.5% Nonidet P-40 supplemented with protease inhibitors). Proteins were either used immediately or stored at −80 °C in pellets after freezing in liquid nitrogen.
DPrJNK Phosphorylation Analysis
To confirm dual phosphorylation, recombinant JNK protein was resolved by 10% SDS-PAGE and visualized by Coomassie Blue staining, and the protein was excised from the gel and subjected to in-gel tryptic digestion. The resulting peptides were subjected to HPLC-electrospray ionization-MS/MS, and the identity of the protein was confirmed through comparison of experimental data with predicted peptide masses. Phosphorylation sites were mapped through analysis of the tandem MS data, which yielded distinct fragmentation patterns associated with each of the phosphoresidues. Mass spectrometry, data collection, and analysis were conducted by the University of Texas Health Science Center at San Antonio Institutional Mass Spectrometry Core in collaboration with Dr. Susan Weintraub.
Immunoprecipitation and GST Pulldown Studies
The Gal4/UAS system was used to overexpress both the FLAGDmp53 and HA-tagged human p53 (HAhp53) protein in developing Drosophila embryos (38). Embryos from overnight collections were dechorionated in 50% bleach, washed with 0.1% Triton X-100, and lysed in NET buffer (50 mm Tris, pH 7.5, 400 mm NaCl, 5 mm EDTA, 1% Nonidet P-40). Nuclei were removed with a low speed spin, and then mouse α-FLAG (Sigma-Aldrich; M2) or rabbit α-HA antibody was added to the lysates and incubated for 1 h at 4 °C. Protein A-Sepharose was then added to the lysate/antibody suspension and incubated for an additional 1.5 h at 4 °C. Protein complexes were isolated by centrifugation, and the pellets were washed three to six times with NET buffer. The final wash was then removed, the pelleted was resuspended in 2× SDS-PAGE loading buffer, and samples were denatured by heating at 95 °C for 5 min, resolved by SDS-PAGE, and transferred to nitrocellulose for Western blotting.
Similarly, GST pulldown assays involving glutathione-agarose beads, GST-tagged Dmp53ΔC, human p53ΔC, and/or rJNK were incubated overnight at 4 °C in NET buffer. Beads were isolated through centrifugation and washed thoroughly with 1× PBS containing 1% Triton X-100, and complexes were resolved by SDS-PAGE and transferred to nitrocellulose prior to Western blotting.
Kinase Assay
Recombinant rat JNK was incubated with GST-c-Jun(1–89) in kinase buffer (25 mm Tris, pH 7.5, 5 mm β-glycerol phosphate, 2 mm DTT, 0.1 mm Na3VO4, 10 mm MgCl2, 200 μm ATP) for 30 min at 30 °C. Reactions were terminated by adding 4× SDS sample buffer, resolved by SDS-PAGE, and transferred to nitrocellulose. The extent of kinase activity was measured via immunoblotting the membranes with mouse α-c-Jun and rabbit α-phospho-c-Jun antibodies. Pretreatment of diphosphorylated rat JNK with either MKP-5 or calf intestinal alkaline phosphatase (CIAP) was done prior to dilution in kinase buffer and addition of GST-c-Jun(1–89). Preincubation of JNK with or without hp53DBD was carried out in 10 mm HEPES, 150 mm NaCl, 3 mm EDTA for 1 h at 30 °C. MKP-5 was then added, and the reaction was supplemented with 25 mm MgCl2 and 1 mm DTT. Mixtures were then incubated for 2 h at 30 °C prior to dilution with kinase buffer and addition of GST-c-Jun(1–89).
Phosphatase Assay
The phosphatase activity of recombinant MKP-5 was initially characterized using p-nitrophenyl phosphate as a substrate. MKP-5 was incubated at room temperature in a reaction mixture containing p-nitrophenyl phosphate (Thermo Scientific), 10 mm DTT, and 10 mm MgCl2. Phosphatase activity was determined by measuring the absorbance at 405 nm. MKP-5 was also used to dephosphorylate dually phosphorylated rat JNK. A reaction mixture containing human MKP-5 with diphosphorylated rat JNK or diphosphorylated p38 was supplemented with 10 mm DTT and 10 mm MgCl2 prior to incubation at 25 °C for 4 h. The reaction was terminated by adding 4× SDS-PAGE dye and boiling the samples at 95 °C for 5 min. Samples were resolved by SDS-PAGE and transferred to nitrocellulose, and phosphatase activity was assessed by Western blotting with phosphospecific antibodies.
Coumarin Labeling and Fluorescence Quenching Analysis
To label DPJNK with the thiol-reactive reagent 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin (CPM) (Molecular Probes), 19.8 nmol of DPJNK was resuspended in HBS (10 mm HEPES, 150 mm NaCl, pH 7.4) and mixed with a ∼10-fold excess of CPM. The reaction was incubated on ice until the fluorescence intensity reached a maximum. Excess CPM was removed with a Sephadex G-25 column, and the concentration of labeled protein was estimated using the Bradford assay. The extent of thiol labeling was determined to be four of nine potential cysteine residues using an extinction coefficient of 33,000 m−1 cm−1 for CPM.
Fluorescence titration was used to determine the dissociation constants (Kd) for the DPrJNK·hp53DBD and the DPrJNK·MKP-5 complexes. CPM-labeled rat DPJNK (0.1 μm in HBS) was titrated with increasing concentrations of either recombinant hp53DBD or recombinant MKP-5 (0.001–0.6 μm in HBS), and fluorescence emission spectra were obtained at 25 °C after excitation at 340 nm. Corrections for inner filter effects were made as described (44), and the Kd values were calculated for p53·DPJNK and MKP-5·DPJNK complexes using non-linear regression to fit the data to the following equation: F°/Fcorr = (Bmax × [X])/(Kd + [X]) + 1 where X is the concentration of either p53DBD or MKP-5 in μm.
RESULTS
p53 Influences JNK Activity in Vivo
Previously, we used genetics to assess how JNK signaling and p53 function are integrated in the hierarchy of signaling pathways that regulate radiation-induced apoptosis (34). In Drosophila, the apoptotic response (assessed with TUNEL labeling (45)) throughout the development of imaginal wing discs began ∼4 h after exposure to 40 grays of x-irradiation, whereas unirradiated wild-type controls showed little or no TUNEL labeling (Fig. 1, compare B with A). Likewise, JNK activity monitored by a JNK-dependent lacZ enhancer trap inserted into the puc locus (JNKREP; Refs. 35–37) paralleled TUNEL labeling (Fig. 1, compare B with A) and was required to initiate the apoptotic response 4 h after exposure to 40 grays of x-irradiation (34). This initial apoptotic response to irradiation as well as the rapid increase in JNK activity required Dmp53 as little, if any, TUNEL labeling or JNKREP activity were observed 4 h after x-irradiation of Dmp53[5A-1–4]/Dmp53[5A-1–4] mutant wing discs (Fig. 1, compare C with B; Ref. 34). However, both JNK activity and TUNEL labeling were restored to wild-type levels in Dmp53[5A-1–4]/Dmp53[5A-1–4] mutant wing discs some 20–24 h after exposure to 40 grays of x-irradiation (Fig. 1, compare E with D). These results are consistent with those of Wichmann et al. (46) and suggest that Dmp53 is required for the initial JNK-dependent induction of apoptosis that occurs within hours of exposure to genotoxic stress; however, these data demonstrate that Dmp53 is not essential for either the JNK or apoptotic responses observed at later times and instead may only affect the timing of the apoptotic response.
FIGURE 1.

JNK signaling promotes apoptosis in absence of p53 after exposure to x-ray radiation. A–E, third instar wing discs labeled for JNKREP (red), TUNEL (green), and DNA (blue). A, B, and D, wild-type wing discs harboring the JNKREP (pucA251.1/+) at 0, 4, and 20 h, respectively, after exposure to 4000 rads of x-ray radiation. C and E, p53[5A-1–4]/p53[5A-1–4] wing discs harboring the JNKREP (pucA251.1/+) at 4 and 24 h, respectively, after 4000 rads of x-irradiation.
We also examined JNK activity in both wild-type and p53−/− mutant murine embryonic fibroblasts (MEFs) to assess whether p53 functions to regulate JNK activity in vertebrates. First, the basal level of diphosphorylated JNK (DPJNK) was higher in wild-type MEFs as compared with p53−/− MEFs (Fig. 2A, compare lanes 1 and 4). Exposure of wild-type MEFs to UV irradiation resulted in both a modest increase in p53 levels and a substantial increase in DPJNK levels after only 30 min; however, only a slight increase in DPJNK levels was observed in p53−/− MEFs at 30 min (Fig. 2A, compare lane 2 with lane 5). Finally, DPJNK levels remained elevated in wild-type MEFs as p53 continued to accumulate, whereas all DPJNK was lost in p53−/− MEFs by 6 h after irradiation (Fig. 2A, compare lane 3 with lane 6). Importantly, total JNK levels were not influenced by UV irradiation (Fig. 2A, bottom panel), demonstrating that the increases in DPJNK levels in wild-type MEFs were not the result of an increase in JNK protein expression. Together, these results suggest that the ability of p53 to regulate JNK activity is evolutionarily conserved with p53 functioning to increase the initial rate at which DPJNK accumulates and/or to stabilize DPJNK in response to genotoxic stress.
FIGURE 2.

DPJNK activity is potentiated by p53 in MEFs. Both wild-type (WT) and p53-null (p53−/−) MEFs were irradiated with a dose of 200 J/m2 UV light. The cells were allowed to grow for an additional 30 min or 6 h. Lysates were then harvested and subjected to SDS-PAGE and Western blot analysis using α-p53, α-DPrJNK, and α-JNK antibodies. Note that the level of JNK activity is consistently higher in WT MEFs as compared with p53-null MEFs, whereas total JNK levels remain unaffected.
p53 Expression Stimulates JNK Transcriptional Activity in Vivo
To assess whether p53 expression can influence JNK-dependent transcriptional activity in vivo, we transiently transfected both mammalian and Drosophila cells with the appropriate p53 expression construct and monitored changes in AP1-dependent luciferase expression using the pAP1-luc reporter plasmid. pAP1-luc contains a seven-copy multimeric array of the core c-Jun binding site and has been used extensively to monitor activation of the JNK pathway. Restoration of p53 expression in HCT116 (p53−/−) cells, a human colorectal cancer cell line that lacks p53 (47), resulted in an increase in AP1-luc reporter activity when compared with the basal activity observed in control transfected cells (Fig. 3A). The p53-dependent increase in luciferase activity was dependent on JNK activity as treatment with SP600125, a selective inhibitor of JNK (48), prevented the increase in JNK-dependent luciferase activity (Fig. 3A).
In addition to HCT116 (p53−/−) cells, we tested the effects of FLAGDmp53 expression on pAP1-luc activity is Drosophila S2 cells. As with the HCT116 (p53−/−) cells, elevated expression of FLAGDmp53 stimulated AP1-dependent luciferase activity in S2 cells, whereas treatment with SP600125 prevented the p53-dependent expression of luciferase (Fig. 3B).
These results demonstrate that p53 expression can stimulate JNK activity. Importantly, these results do not distinguish between transcriptional and non-transcriptional roles for the p53 orthologs. To address this issue, we fused either the mitochondrial targeting sequence of Bcl-2 (MTS; Ref. 49) or the Bax transmembrane domain (BaxTM; Ref. 50) to the C-terminal end of FLAG-tagged p53 (FLAGp53) or FLAGDmp53, respectively, and tested the effects of nuclear exclusion of p53 on both p53- and AP1-dependent transcriptional activity.
As expected, expression of either wild-type FLAGp53 or FLAGDmp53 stimulated a robust increase in p53-dependent luciferase expression from the pG13-luc reporter plasmid (Fig. 3, C and D), which harbors 13 copies of the established p53 binding half-site (51). Localization of either FLAGp53MTS or FLAGDmp53BaxTM to the mitochondrial membrane resulted in little to no induction of p53-dependent luciferase expression from the pG13-luc reporter plasmid (Fig. 3, C and D, respectively), demonstrating that p53 when localized to the outer mitochondrial membrane is unable to promote transcriptional activity through the p53 binding half-site. Nevertheless, both human and Drosophila p53 when sequestered at the outer mitochondrial membrane retained the ability to stimulate AP1-dependent transcriptional activity independently of the p53 binding half-site (Fig. 3, E and F).
These data suggest that p53-dependent changes in gene transcription are not responsible for the increased JNK activity as otherwise wild-type p53, which is sequestered outside of the nucleus and unable to stimulate p53-dependent changes in gene transcription, was capable of stimulating an AP1-dependent increase in luciferase expression. Thus, we explored non-transcriptional mechanisms for p53-mediated regulation of JNK activity following genotoxic stress.
p53 and JNK Orthologs Form Physical Complex in Vivo
Previously, Fuchs et al. (30) have shown that non-phosphorylated mammalian JNK and p53 form a complex in vivo, but their results suggested that JNK functions as part of an E3 ubiquitin ligase complex to promote ubiquitylation and the subsequent degradation of p53 in non-stressed cells. However, JNK-dependent phosphorylation of p53 has also been shown to inhibit both Mdm2 binding and ubiquitylation (31), suggesting that JNK-dependent phosphorylation contributes to the apoptotic response by stabilizing p53. Thus, we utilized immunoprecipitation to assess whether reciprocal regulation of JNK activity by p53 results from the formation of a physical complex between p53 and JNK in vivo.
To examine this potential interaction in mammalian cells, HA-tagged JNK (HAJNK) was co-expressed with either FLAGp53 alone or FLAGp53 together with constitutively activated MEK1 in HEK 293 cells. As expected, little DPJNK was found in the lysates without MEK1, whereas a substantial increase in DPJNK was noted in the MEK1-expressing lysates (Fig. 4A, upper panel, lanes 1 and 5, respectively). FLAGp53 levels were also elevated in MEK1 lysates, consistent with JNK-dependent stabilization of p53 protein (Fig. 4A, lower panel, lanes 1 and 5, respectively). However, overall levels of JNK protein were similar in both lysates (Fig. 4A, middle panel, compare lanes 1 and 5).
FIGURE 4.

Both human and Drosophila p53 interact with DPJNK in vivo. A, FLAGp53 was co-expressed either with HAJNK or HAJNK and constitutively active MEK1 in HEK 293 cells. FLAGp53-containing complexes were then isolated from the two lysates by immunoprecipitation with the mouse α-FLAG (M2) antibody, and proteins were resolved by SDS-PAGE prior to Western blotting with rabbit α-DPJNK, rabbit α-JNK, and rabbit α-DDDYK (FLAG) antibodies. B, FLAGDmp53 was expressed in Drosophila embryos using the Gal4/UAS system. The mouse α-FLAG (M2) antibody was used to immunoprecipitate FLAGDmp53 from embryo lysates. Immunoprecipitated proteins were resolved by SDS-PAGE prior to Western blotting with a rabbit α-JNK antibody. Note the apparent shift in molecular weight associated with immunoprecipitated dJNK (filled triangles) compared with the non-phosphorylated isoform (open triangles). C, FLAGDmp53 was immunoprecipitated from Drosophila embryo lysates and resolved as in A, but the Western blot was probed with a rabbit α-DPJNK-specific polyclonal antibody. D, HAhp53 was expressed in Drosophila embryos using the Gal4/UAS system, immunoprecipitated from Drosophila embryo lysates, resolved by SDS-PAGE, and subjected to Western blotting with either rabbit α-JNK (upper panel) or rabbit α-DPJNK (lower panel) antibody. Note that only the phosphorylated form of dJNK associated with Drosophila and human p53. WCL, whole cell lysate; FT, flow-through; IP, immunoprecipitate.
DPJNK selectively interacted with FLAGp53 as evidenced by its immunoprecipitation with α-FLAG antibodies in the MEK1-containing lysates (Fig. 4A, upper panel, lane 8). However, an interaction between non-phosphorylated JNK and FLAGp53 was not observed in either lysate even though significant levels of JNK and FLAGp53 were present in the lysate (Fig. 4A, middle panel, lane 1) and FLAGp53 was enriched in the immunoprecipitate (Fig. 4A, lower panel, lane 4). These results demonstrate that DPJNK and p53 are capable of forming a stable complex in vivo.
To characterize the interaction in Drosophila, FLAGDmp53 was expressed in Drosophila embryos. dJNK immunoreactivity with a retarded migration suggestive of phosphorylation was found exclusively in the FLAGDmp53 immunoprecipitate (Fig. 4B, closed triangles). To address whether this band represents DPdJNK, we repeated the FLAGDmp53 immunoprecipitation and probed the Western blot with an α-DPJNK-specific antibody (Fig. 4C). DPdJNK-specific immunoreactivity was only observed in the FLAGDmp53 immunoprecipitate, confirming that Dmp53 preferentially binds to DPdJNK. Importantly, we attempted to immunoprecipitate FLAGDmp53 using the α-DPJNK-specific antibody; however, we were unable to detect FLAGDmp53 in the immunoprecipitate. This suggests that Dmp53 when bound may regulate access to the diphosphorylated TPY motif found in DPdJNK. Furthermore, these results suggest that Dmp53 and DPdJNK bind with a relatively strong affinity as little or no detectable DPdJNK was observed in the whole cell lysates of non-stressed cells (Fig. 4, B and C); however, DPdJNK was highly enriched in the FLAGDmp53 immunoprecipitate.
Finally, we examined whether HAhp53 interacts with DPdJNK in Drosophila embryos to assess whether the functional motif(s) required for this interaction is evolutionarily conserved. Again, DPdJNK was selectively immunoprecipitated with HAhp53 in vivo (Fig. 4D, closed triangles), suggesting that the relevant binding motif for DPdJNK is functionally conserved between Drosophila and human p53 orthologs even though these proteins exhibit limited sequence identity (∼20%) and similarity (∼35%) overall (24).
Recombinant Rat JNK Binds to Both Drosophila and Human p53 in Vitro
To obtain sufficient quantities of recombinant JNK protein to further study these interactions in vitro, we utilized an established bacterium-based expression system (43) to produce fully functional recombinant rJNK (∼75% identical/∼87% similar to dJNK) in both the diphosphorylated (active; DPrJNK) and non-phosphorylated (inactive; rJNK) forms. The phosphorylation status of DPrJNK was analyzed through electrospray ionization-tandem mass spectrometry, and both the threonine and tyrosine residues in the TPY motif of the activation loop were phosphorylated when rJNK was co-expressed with constitutively active MEKK-C and wild-type MEK4 (supplemental Fig. 1A). Both rJNK and DPrJNK were then incubated with GST-tagged c-Jun (residues 1–89; GST-c-Jun(1–89)), and complexes were isolated with glutathione-agarose to confirm both binding ability and kinase activity. As expected, both rJNK and DPrJNK bound to GST-c-Jun(1–89) (supplemental Fig. 1B, α-JNK Western blot), whereas only DPrJNK was able to phosphorylate GST-c-Jun(1–89) (supplemental Fig. 1B, α-Pc-Jun Western blot). To confirm the relative importance of threonine and tyrosine residues in the activation loop, we tested the effect of a series of rJNK site-directed mutants (T192A, Y194A, and T192A/Y194A) on kinase activity using GST-c-Jun(1–89) as a substrate. Mutation of the tyrosine, threonine, or both residues to alanine reduced kinase activity to background levels seen in inactive rJNK samples (supplemental Fig. 1C) even though intact tyrosine and threonine residues were phosphorylated in the T192A and Y194A mutant rJNK isoforms (data not shown).
To determine whether DPrJNK binds to Dmp53 or hp53, we utilized GST-tagged forms of Dmp53 and hp53 lacking the C-terminal tetramerization domains, which increased their solubility by decreasing multimer/aggregate formation. GST alone and GST-Dmp53ΔC (residues 1–344) and GST-hp53ΔC (residues 1–306) fusion proteins were incubated with DPrJNK, and then glutathione-agarose was used to isolate potential p53·DPJNK complexes. Samples, including the relevant protein inputs (Fig. 5A, lanes 1–4), were then resolved by SDS-PAGE, and the constitution of the complex was assessed by Western blotting. GST-Dmp53ΔC and GST-hp53ΔC both were able to pull down DPrJNK (Fig. 5A, lanes 6 and 7, respectively); however, DPrJNK failed to interact with GST alone (Fig. 5A, lane 5).
FIGURE 5.

Recombinant DPJNK interacts with GST-tagged Drosophila and human p53 proteins. A, DPrJNK was incubated with either GST alone, GST-hp53ΔC, or GST-Dmp53ΔC, and then complexes were isolated with glutathione-agarose. Note that DPrJNK interacted with both Drosophila and human p53 but failed to interact with GST alone. Lanes were rearranged for clarity. B, non-phosphorylated rJNK was incubated with GST alone, GST-hp53ΔC, and GST-Dmp53ΔC, and then complexes were isolated with glutathione-agarose. Note that JNK bound to both human and Drosophila p53 but failed to interact with GST alone. * denotes nonspecific cleavage products. C, DPJNK was incubated with either GST alone or GST-hp53DBD. GST-containing complexes were isolated with glutathione-agarose. Note that DPJNK associated with the DNA binding domain of human p53.
Human p53 has previously been shown to also bind to non-phosphorylated JNK; however, we failed to observe this in vivo in the HEK 293 and S2 cells (Fig. 4, A and C, respectively). To reassess this interaction in vitro, we tested whether GST-hp53ΔC and GST-Dmp53 formed a complex with non-phosphorylated rJNK, which we isolated with glutathione-agarose. Samples, including the relevant inputs, were then characterized by Western blotting. As expected, rJNK was isolated with both GST-hp53ΔC and GST-Dmp53ΔC in a manner similar to DPrJNK (Fig. 5B, lanes 6 and 7, respectively) but failed to interact with GST alone (Fig. 5B, lane 5).
The JNK/p53 interaction was previously mapped to amino acids 97–116 of hp53, which corresponds to part of the DNA binding domain of p53 (hp53DBD) (52). As this domain folds independently of the N- and C-terminal segments, we tested whether the p53DBD (residues 94–312) was able to bind to DPrJNK. Like GST-hp53ΔC, DPrJNK co-purified with GST-hp53DBD on glutathione-agarose (Fig. 5C, lane 5), whereas GST alone failed to pull down DPrJNK (Fig. 5C, lane 4). Relevant protein inputs are shown in Fig. 5C, lanes 1–3.
Together, these results suggest that DPJNK interacts with p53 in vivo, whereas both diphosphorylated and non-phosphorylated rJNK binds to both hp53 and Dmp53 in vitro. Furthermore, these results demonstrate that the hp53DBD is capable of binding to DPrJNK as well as non-phosphorylated JNK (30). As the vertebrate p53DBD and JNK orthologs are predicted to exhibit extensive structural homology to their Drosophila counterparts, we chose to focus the remaining studies on the vertebrate proteins as hp53DBD and DPrJNK are easily produced in E. coli.
p53 Binding to DPrJNK Prevents Phosphatase-mediated Inactivation of JNK Kinase
The MKPs are the primary antagonists of MAPKs; they promote kinase inactivation through dephosphorylation of the TXY motif. Many of the MKPs are immediate early genes whose expression is up-regulated in response to MAPK activation; however, their expression completes a negative feedback loop that limits the duration and/or peak amplitude of MAPK activity in response to stimuli (53). Our previous genetic data (34) suggest that Dmp53 is capable of overriding MKP-mediated inactivation of dJNK, whereas our data in MEFs clearly demonstrate that the level of JNK activation depends on p53 (Fig. 2). This led us to hypothesize that p53 when bound to DPJNK potentiates JNK activity by limiting access to the TPY in the kinase activation loop; this is consistent with our observation that α-DPJNK antibodies were unable to immunoprecipitate the p53·DPJNK complex.
To test this hypothesis directly, both His-tagged human MKP-5, which preferentially inactivates the JNK and p38 stress-activated MAPKs, and a catalytically inactive mutant form (C408SMKP-5; Ref. 54) were expressed in and purified from E. coli. Recombinant MKP-5 was active and hydrolyzed p-nitrophenyl phosphate (a substrate mimic) in a dose-dependent manner, whereas the C408S mutant isoform showed little to no phosphatase activity (supplemental Fig. 2).
Recombinant DPrJNK was then incubated with GST-c-Jun(1–89), and the entire complex was purified with glutathione-agarose. Samples were then resolved by SDS-PAGE, and the constituents were identified via Western blotting. As expected, DPrJNK formed a stable complex with the N-terminal segment of c-Jun (Fig. 6A, lane 1, α-DPJNK), which it also phosphorylated (Fig. 6A, lane 1, α-P-c-Jun). Whereas preincubation of DPrJNK with C408SMKP-5 failed to affect either DPrJNK activity (Fig. 6A, lane 2, α-DPJNK) or its ability to bind and phosphorylate the N terminus of c-Jun (Fig. 6A, lane 2, α-P-c-Jun), incubation of DPrJNK with wild-type MKP-5 both inactivated (dephosphorylated) DPrJNK and prevented the phosphorylation of GST-c-Jun(1–89) (Fig. 6A, compare lanes 2 and 3). Importantly, neither MKP-5 nor C408SMKP-5 appreciably altered the formation of a JNK·c-Jun complex as addition of either to the binding reaction failed to affect the association of rJNK with GST-c-Jun(1–89) (Fig. 6A, lanes 2 and 3, α-JNK).
FIGURE 6.

hp53DBD prevents MKP-5 from dephosphorylating DPJNK. A, either MKP-5 or C408SMKP-5 was incubated with DPrJNK and GST-c-Jun(1–89) for 30 min at 30 °C. To determine whether p53 could prevent dephosphorylation of DPrJNK, replicate samples of DPrJNK were preincubated with a 1–3-fold molar equivalent of hp53DBD prior to addition of MKP-5 and GST-c-Jun(1–89). The resulting complexes were isolated with glutathione-agarose and resolved via SDS-PAGE, and the various samples were assessed with α-JNK, α-p53, and α-GST antibodies. The phosphorylation status of GST-c-Jun(1–89) and JNK was assessed with a phosphospecific α-c-Jun antibody and α-DPrJNK antibody. Note that C408SMKP-5 did not dephosphorylate JNK, whereas hp53DBD effectively protected DPrJNK from dephosphorylation mediated by wild-type MKP-5. B, CIAP was incubated for 30 min at 30 °C with DPrJNK and GST-c-Jun(1–89) or with a mixture of DPrJNK and GST-c-Jun(1–89) that had been preincubated with increasing molar ratios of hp53DBD. Complexes were then characterized by Western blotting as described in A. Note that CIAP readily dephosphorylated DPrJNK; however, hp53DBD prevented this dephosphorylation.
To assess whether hp53DBD had any affect on the ability of MKP-5 to regulate DPrJNK activity, we preincubated DPrJNK with various molar ratios of hp53DBD prior to the addition of both GST-c-Jun(1–89) and MKP-5 to the binding/kinase reaction (Fig. 6A, lanes 4–6). Importantly, preincubation of DPrJNK with hp53DBD prevented MKP-5-dependent dephosphorylation of activated JNK (Fig. 6A, lanes 4–6, α-DPJNK), suggesting that DPrJNK when sequestered in a complex with hp53DBD is resistant to dephosphorylation by MKP-5. Furthermore, the hp53DBD·DPrJNK complex both co-purified with and phosphorylated GST-c-Jun(1–89) (Fig. 6A, lanes 4–6, α-P-c-Jun), demonstrating that hp53DBD does not affect the ability of DPrJNK to associate with and phosphorylate c-Jun.
We also examined whether hp53DBD could sustain DPrJNK activity in the presence of CIAP, a nonspecific phosphatase capable of removing phosphates from various cellular substrates, including proteins. Like MKP-5, CIAP inactivated DPrJNK; however, it had little to no effect on the ability of rJNK to bind to GST-c-Jun(1–89) (Fig. 6B, lane 3, α-JNK). Preincubation with hp53DBD prevented CIAP-mediated dephosphorylation of DPrJNK (Fig. 6B, lanes 4–6, α-DPJNK); however, CIAP treatment did not affect the association of hp53DBD with DPrJNK and GST-c-Jun(1–89) (Fig. 6B, lanes 4–6, α-p53 and α-DPrJNK). As CIAP is not predicted to bind to DPrJNK, these results further suggest that hp53DBD inhibits dephosphorylation of DPrJNK by regulating access to the TPY motif.
As the members of the MAPK family share a conserved tertiary structure, we also tested whether hp53DBD was able to prevent the dephosphorylation of p38, another stress-activated MAPK that is greater than 50% identical and 65% similar to JNK. Again, C408SMKP-5 had no effect on the phosphorylation status of DPp38 (Fig. 7, lane 2); however, MKP-5 readily dephosphorylated DPp38 (Fig. 7, lane 3). Whereas addition of hp53DBD to DPrJNK prevented its dephosphorylation (Fig. 6A, lanes 4–6), dephosphorylation of DPp38 by MKP-5 was not prevented by preincubation with hp53DBD even at a 3-fold molar excess (Fig. 7, lanes 4–6). Together, these results suggest that hp53DBD is not a direct inhibitor of MKP-5 as MKP-5 was capable of dephosphorylating p38 in the presence of hp53DBD. Instead, the data suggest that hp53DBD selectively protects DPrJNK from MKP-5-dependent dephosphorylation.
FIGURE 7.
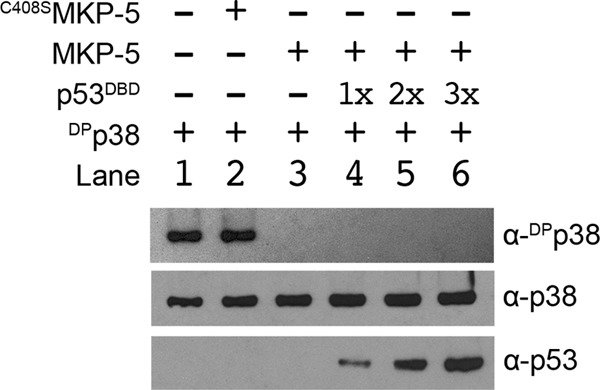
hp53DBD does not prevent MKP-5 from dephosphorylating DPp38. A, either MKP-5 or the catalytically inactive C408SMKP-5 mutant was incubated with DPp38 at 30 °C for 30 min. To test whether hp53DBD could prevent dephosphorylation, DPp38 was preincubated with various molar ratios of hp53DBD and then incubated with MKP-5 at 30 °C for 30 min. Reactions were then resolved by SDS-PAGE, and sample constituents were analyzed by Western blotting with α-p38 and α-p53 antibodies. The phosphorylation status of p38 was further assessed with a phosphospecific α-p38 antibody. Note that inclusion of hp53DBD in the reaction did not prevent the dephosphorylation of DPp38.
MKP-5 Has Higher Affinity for DPrJNK than hp53DBD
To assess whether competition between MKP-5 and hp53DBD for binding of DPrJNK could affect complex assembly, we first measured the apparent dissociation constants (Kd) for the MKP-5·DPrJNK and hp53DBD·DPrJNK complexes through the use of fluorescence quenching (55). Specifically, DPrJNK was fluorescently tagged with CPM at free thiol groups. CPM-labeled DPrJNK exhibited an emission maximum (λem) of 465 nm (Fig. 8, A and B).
FIGURE 8.

Determination of dissociation constants for hp53DBD·DPrJNK and MKP-5·DPrJNK complexes. A, increasing concentrations of hp53DBD (0.001–0.6 μm) were added to 0.1 μm CPM-labeled DPrJNK in 1× HBS (Imax = 3.1 × 105), and the fluorescence intensity was quantified at 465 nm. B, increasing concentrations of MKP-5 (0.001–0.6 μm) were added to 0.1 μm CPM-labeled DPrJNK in HBS (Imax = 3.5 × 105), and the fluorescence intensity was quantified at 465 nm. C, binding data from A plotted as percentage of CPM-DPJNK bound (F°/Fcorr) versus the concentration of hp53DBD. D, binding data from B plotted as percentage of CPM-DPJNK bound (F°/Fcorr) versus the concentration of MKP-5. Data from C and D were fit to a hyperbolic function using non-linear regression, and the apparent dissociation constants were determined. Data in C and D plotted as average ± standard deviation.
To measure the binding affinity of hp53DBD for DPrJNK, increasing concentrations of hp53DBD were then added to CPM-labeled DPrJNK, and the fluorescence intensity was measured at 465 nm. A distinct decrease in fluorescence intensity was observed as the hp53DBD titrant concentration increased from 0.001 to 0.6 μm (Fig. 8A) that allowed us to plot specific binding activity (F°/Fcorr) relative to the concentration of CPM-labeled DPrJNK after correcting for inner filter effects and sample dilution (Fig. 8C). Non-linear regression was then used to fit the data to a hyperbolic binding curve, and an apparent Kd of 274 ± 14 nm for the hp53DBD·CPM-DPrJNK complex was calculated.
Similar experiments were preformed using increasing concentrations of MKP-5 as the titrant to assess its ability to bind to DPrJNK. Again, a distinct decrease in fluorescence intensity was observed upon binding of MKP-5 to CPM-DPrJNK (Fig. 8B), and a range of MKP-5 concentrations from 0.001 to 0.6 μm was used to establish a binding curve. A non-linear regression-based fit of the data (F°/Fcorr versus [MKP-5]) yielded an apparent Kd of 55 ± 8 nm for the MKP-5/DPrJNK interaction (Fig. 9D). It should be noted that this experiment does not take into account cooperativity in binding that might be observed with wild-type p53 as hp53DBD is a monomer instead of the tetramer produced by full-length p53. However, these results still suggest that competition for a single binding site is not the mechanism whereby the p53DBD prevents MKP-5-dependent dephosphorylation of DPrJNK as the hp53DBD was capable of preventing any significant dephosphorylation of DPrJNK at a 1:1 molar ratio even though MKP-5 had approximately a 5-fold better affinity for DPrJNK.
FIGURE 9.

MKP-5 and hp53DBD bind independently of each other to DPrJNK. To address whether MKP-5 and hp53DBD could bind to DPrJNK concurrently, GST-hp53ΔC was incubated together with MKP-5 alone, DPrJNK alone, or a mixture of MKP-5 and DPrJNK, whereas GST alone was used as a negative control. Complexes were allowed to assemble at 4 °C for 30 min and then isolated with glutathione-agarose. Pulldown samples as well as the appropriate input controls were then resolved via SDS-PAGE, and complex assembly was assessed with α-MKP-5, α-DPJNK, and α-GST antibodies. Note that MKP-5 failed to interact with either GST or GST-hp53ΔC and was only isolated in the presences of JNK. * denotes a nonspecific cleavage product of GST-hp53ΔC.
MKP-5 and hp53DBD Bind in Non-competitive Manner to Distinct Sites on JNK
Binding of hp53DBD to DPrJNK did not inhibit the ability of JNK to either bind or phosphorylate the c-Jun substrate (Fig. 6A), which is known to depend on a δ-domain binding motif. As MKP-5 is also suggested to utilize a δ-domain-like (D-box) binding motif to interact with JNK (56), it is possible that MKP-5 and hp53DBD bind concurrently to DPrJNK. To test this hypothesis, we utilized GST pulldown experiments to define direct interactions between MKP-5, DPrJNK, and hp53DBD (Fig. 9). As expected, DPrJNK interacted directly with GST-p53ΔC but was not associated with GST alone (Fig. 9, compare lanes 5 and 8). MKP-5 did not directly interact with either GST or GST-hp53ΔC (Fig. 9, lanes 6 and 7); however, MKP-5 was isolated with GST-hp53ΔC in the presence of DPrJNK (Fig. 9, lane 9). These results demonstrate that the recruitment of MKP-5 into the p53·JNK complex depends on JNK (i.e. JNK bridges hp53DBD and MKP-5) and that unique binding sites on DPrJNK are utilized by p53 and MKP-5. Furthermore, these results confirm that binding of hp53DBD and MKP-5 to DPrJNK is not mutually exclusive.
DISCUSSION
p53 Regulates JNK Activity in Vivo
The expression of puc, the sole JNK-specific MKP in Drosophila, is required to maintain cell viability as it prevents JNK-mediated apoptosis in response to the basal level of JNK activity (34). However, dJNK signaling promotes apoptosis in response to genotoxic stress even though puc expression is stimulated in response to dJNK activity (34). As genotoxic stress is known to induce apoptosis in a p53-dependent manner (24–29), we examined whether the initial dJNK-dependent induction of apoptosis after exposure to genotoxic stress requires Dmp53. Through these studies, we observed that the initial activation of dJNK ∼4 h after exposure to genotoxic stress requires Dmp53 (34). However, it is clear that both dJNK activation and apoptosis are delayed in the Dmp53 mutant background with JNK activity and apoptosis being initiated some 20–24 h after exposure to genotoxic stress (our results and Ref. 46). Together, these results suggest that Dmp53 accelerates both the accumulation of DPdJNK and induction of apoptosis in response to genotoxic stress. This rapid induction of apoptosis may serve to induce a significant amount of apoptosis prior to the accumulation of mutations associated with error-prone DNA repair mechanisms. Nevertheless, the genetic experiments did not define the relevant molecular mechanism(s).
Although Puc overexpression is capable of inhibiting apoptosis resulting from exposure to genotoxic stress, overexpression of Dmp53 stimulates apoptosis even when Puc is overexpressed in the same cells (34). This suggests that Dmp53 plays a role in overriding the repressive effects of Puc whose expression is induced by Dmp53 expression. To this end, we extended our studies and present a model describing the ability of p53 to reciprocally regulate JNK activity in Drosophila and vertebrates.
It is unlikely that the rapid elevation of DPJNK levels in MEFs 30 min after irradiation results from p53-dependent changes in gene transcription as UV-induced DNA damage blocks gene transcription (57, 58). Consistent with this hypothesis, we demonstrate that p53 plays a role in potentiating the JNK-dependent transcriptional response independently of p53-mediated changes in gene transcription, leading us to pursue non-transcriptional mechanisms for the p53-dependent regulation of JNK activity. We should acknowledge that our model for p53-dependent regulation of JNK activity is not mutually exclusive with other models postulating p53-dependent regulation of gene transcription.
p53 and DPrJNK Form Stable Complex Both in Vivo and in Vitro
The data presented here establish that p53 and JNK form a physical complex with p53 selectively binding to DPJNK in vivo. This interaction is direct as tested by in vitro binding studies. However, other cell type-specific factors may modify this interaction in vivo as we saw little to no binding of non-phosphorylated JNK to p53 under non-stressed conditions, which had been reported previously (30, 31, 52). Importantly, the in vitro data demonstrate that p53 is capable of maintaining JNK in its active state by sequestering it in a complex that is resistant to MKP-dependent dephosphorylation; otherwise, MKPs would rapidly render DPJNK inactive. Although a great deal of attention has been focused on the transcriptional role of p53 after exposure to genotoxic stress, these results present strong evidence for the reciprocal regulation of JNK activity by p53.
p53·JNK Complex Is Evolutionarily Conserved
Using either in vitro or in vivo experiments, we demonstrated that cross-species interactions between p53 and DPJNK orthologs are preserved, suggesting that the motif(s) in the p53 orthologs required for the association with JNK is evolutionarily conserved. We have noted that dJNK must be phosphorylated to form a complex with either Dmp53 or hp53 in vivo, whereas non-phosphorylated rat JNK bound to both human and Drosophila p53 in vitro. As previous results have demonstrated that p53 can bind non-phosphorylated JNK in vivo (30, 59), we speculate that phosphorylation-dependent binding of p53 to JNK in Drosophila may have been lost evolutionarily and may reflect the fact that the JNK-dependent binding to p53 has evolved in vertebrates to independently regulate basal p53 levels in the absence of JNK activation (30).
p53 is the archetypal member of a family of transcription factors that are involved in a series of different cellular processes; however, they share similar overall structure and binding specificity (60). As Dmp53 is most similar to vertebrate p63 (61), it will be interesting to see whether JNK associates with the p63 paralog and whether this interaction depends on the phosphorylation of JNK, especially as the DNA binding domain of p63 adopts a more rigid conformation than p53 (62). Furthermore, binding of p63 and p73 to DPJNK could influence JNK activity in response to different stimuli, leading to a mechanism for coordinating JNK activation with other parallel signaling pathways during development and cellular differentiation. Additional experiments will be required to address these possibilities.
Binding of p53DBD to DPJNK Directly Influences JNK Activity by Preventing Access to TPY Motif
As numerous kinases and other proteins can influence the function of p53 and JNK in vivo, we chose to define the p53/DPJNK interaction in vitro. Binding of the p53DBD to DPrJNK could prevent MKP-5-mediated inactivation of DPrJNK by masking the TPY motif or by competing for a common binding site that is independent of the TPY motif (63). MKPs utilize an N-terminal binding domain containing positively charged residues to associate with members of the MAPK family (64); however, our results demonstrate that MKP-5 and hp53DBD bind concurrently to DPrJNK. Thus, we conclude that MKP-5 and hp53DBD do not compete for the same binding site. Furthermore, hp53DBD prevented CIAP-mediated dephosphorylation of DPrJNK. As we do not expect CIAP to associate directly with DPrJNK, we conclude that the binding of hp53DBD to DPrJNK limits access to the phosphorylated TPY motif. This is consistent with the fact that we were unable to perform the reciprocal immunoprecipitation of the Dmp53·DPdJNK complex with the α-DPJNK antibody, which requires the TPY motif for binding.
In addition to characterizing complex assembly, we established the apparent Kd values for the hp53DBD·DPrJNK and MKP-5·DPrJNK complexes. Intriguingly, MKP-5 bound to DPrJNK approximately 5 times better than the hp53DBD; however, the hp53DBD was able to prevent the dephosphorylation of DPrJNK by MKP-5. As MKP-5 is thought to use a docking site separate from the active site to associate with MAPKs (56), we suspect that the affinity of the hp53DBD for the TPY motif in DPrJNK is greater that the affinity of the MKP-5 active site for the diphosphorylated TPY motif. Importantly, we speculate that simultaneous binding of both MKPs and p53 to DPJNK will still influence the rate of DPrJNK dephosphorylation as local concentrations of bound MKP will be high and will effectively dephosphorylate DPJNK as p53 dissociates from it. Thus, factors that affect p53 levels will dictate the duration of JNK activity in a concentration-dependent manner.
p53·DPJNK Complex Assembly May Regulate Substrate Specificity
JNK is a proline-directed serine/threonine kinase with consensus phosphorylation sites containing a PX(S/T)P motif (65, 66). However, the consensus phosphoacceptor motif is not sufficient for phosphorylation by JNK (64). Instead, additional motifs like the δ- or D-box in the substrate regulate kinase specificity toward various cellular substrates by serving as docking sites for JNK (10, 67–69). “Scaffolding” proteins may also regulate the specificity of substrate phosphorylation by localizing JNK and its substrates in physical proximity (68, 70, 71). Here, we have demonstrated that JNK kinase when bound by p53 is still able to bind to and phosphorylate the N-terminal segment of c-Jun. This suggests that p53 does not interfere with δ-domain-dependent substrate binding of c-Jun and may stimulate JNK-dependent changes in the gene transcriptional programs by sustaining JNK activation. In fact, p53 may act to recruit activated JNK to specific promoters where it might stimulate transcription through the selective activation of Fos-c-Jun heterodimers (AP1) bound at specific promoter sites. Alternatively, p53 is capable of binding to a number of different cellular substrates, formally raising the possibility that p53 acts as a scaffolding protein capable of recruiting a series of novel JNK substrates. For example, p53 accumulation at the mitochondrial outer membrane in response to genotoxic stress is known to promote apoptosis through an increase in mitochondrial outer membrane permeability (71); however, the mechanism whereby p53 regulates permeability remains to be established. Interestingly, DNA damage induces relocalization of JNK to the mitochondria where it can facilitate an apoptotic response by phosphorylating either Bcl-XL (72) at Thr-47 and Thr-115 or Bcl-2 at Thr-56 (16, 73). As p53 binds to both Bcl-2 and Bcl-XL at the mitochondrial membrane (74, 75), p53 may potentiate the apoptotic response by recruiting DPJNK to the mitochondria.
p53 Binding to JNK Creates “Fail-safe” Mechanism for Regulating Induction of Apoptosis
Differences in the peak amplitude and duration of JNK activation are thought to underlie distinct cellular consequences with transient activation of JNK supporting a non-apoptotic response to various stimuli and sustained JNK activity promoting apoptosis. We have previously demonstrated that MKPs play a critical role in preventing JNK-dependent apoptosis in the absence of genotoxic stress by antagonizing the basal level of JNK activity in the cell, which we have shown depends on p53 activity even in the absence of exogenous stress (34). However, a mechanism(s) for overriding the antagonistic action of MKPs must exist to promote JNK-dependent apoptosis as the expression of many MKPs is up-regulated in response to JNK activation. The data presented here suggest that p53 when bound to DPJNK is capable of preventing the inactivation of DPJNK by MKPs. This interaction provides an attractive model for discriminating between transient and sustained activation of JNK. Specifically, our results would suggest that the commitment to the JNK-dependent apoptotic process might require simultaneous elevation of p53 levels and activation of JNK. p53 is then capable of sequestering DPJNK in a catalytically active complex that phosphorylates substrates but is resistant to MKP-mediated dephosphorylation as a result of masking of the TPY motif in the activation loop of JNK. By requiring the coordinated activation of both JNK and p53, the cell establishes a molecular fail-safe switch that restricts the induction of apoptosis to conditions where a sufficient stimulus exists to activate both signaling pathways simultaneously, whereas stimulation of the JNK pathway in the absence of p53 stabilization fails to induce apoptosis as induction of MKP activity supports only transient activation of JNK. Importantly, this fail-safe may contribute to oncogenic transformation as loss of p53 function would limit the ability of JNK to promote apoptosis in response to various chemotherapeutic modalities.
Supplementary Material
Acknowledgments
We thank M. Cobb and M. Yamaguchi for providing reagents used in this study as well as Dr. Mark Pandeshwar for help with the coumarin labeling and use of the spectrophotometer. We also thank M. Venkatachalam and B. Nicholson for critically reading the manuscript.
This work was supported in part by American Cancer Society Research Scholar Award RSG-10-049-01-DMC (to D. G. M.).

This article contains supplemental Figs. 1 and 2.
- MKP
- MAPK phosphatase
- dJNK
- Drosophila JNK
- DPJNK
- diphosphorylated JNK
- rJNK
- rat SAPK
- hp53
- human p53
- Dmp53
- Drosophila p53
- DBD
- DNA binding domain
- Puc
- puckered
- MEF
- murine embryonic fibroblast
- CIAP
- calf intestinal alkaline phosphatase
- CPM
- 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin
- luc
- luciferase
- UAS
- upstream activation sequence
- MTS
- mitochondrial targeting sequence of Bcl-2
- BaxTM
- Bax transmembrane domain.
REFERENCES
- 1. Jacobson M. D., Weil M., Raff M. C. (1997) Programmed cell death in animal development. Cell 88, 347–354 [DOI] [PubMed] [Google Scholar]
- 2. Kerr J. F., Wyllie A. H., Currie A. R. (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rinkenberger J. L., Korsmeyer S. J. (1997) Errors of homeostasis and deregulated apoptosis. Curr. Opin. Genet. Dev. 7, 589–596 [DOI] [PubMed] [Google Scholar]
- 4. Thompson C. B. (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267, 1456–1462 [DOI] [PubMed] [Google Scholar]
- 5. Chang L., Karin M. (2001) Mammalian MAP kinase signalling cascades. Nature 410, 37–40 [DOI] [PubMed] [Google Scholar]
- 6. Davis R. J. (2000) Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 [DOI] [PubMed] [Google Scholar]
- 7. Dhanasekaran D. N., Reddy E. P. (2008) JNK signaling in apoptosis. Oncogene 27, 6245–6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lin A. (2003) Activation of the JNK signaling pathway: breaking the brake on apoptosis. BioEssays 25, 17–24 [DOI] [PubMed] [Google Scholar]
- 9. Shaulian E., Karin M. (2002) AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–E136 [DOI] [PubMed] [Google Scholar]
- 10. Hibi M., Lin A., Smeal T., Minden A., Karin M. (1993) Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 7, 2135–2148 [DOI] [PubMed] [Google Scholar]
- 11. Bogoyevitch M. A., Kobe B. (2006) Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 70, 1061–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Behrens A., Sabapathy K., Graef I., Cleary M., Crabtree G. R., Wagner E. F. (2001) Jun N-terminal kinase 2 modulates thymocyte apoptosis and T cell activation through c-Jun and nuclear factor of activated T cell (NF-AT). Proc. Natl. Acad. Sci. U.S.A. 98, 1769–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bossy-Wetzel E., Bakiri L., Yaniv M. (1997) Induction of apoptosis by the transcription factor c-Jun. EMBO J. 16, 1695–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watson A., Eilers A., Lallemand D., Kyriakis J., Rubin L. L., Ham J. (1998) Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J. Neurosci. 18, 751–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maundrell K., Antonsson B., Magnenat E., Camps M., Muda M., Chabert C., Gillieron C., Boschert U., Vial-Knecht E., Martinou J. C., Arkinstall S. (1997) Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J. Biol. Chem. 272, 25238–25242 [DOI] [PubMed] [Google Scholar]
- 16. Yamamoto K., Ichijo H., Korsmeyer S. J. (1999) BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G2/M. Mol. Cell. Biol. 19, 8469–8478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu C., Minemoto Y., Zhang J., Liu J., Tang F., Bui T. N., Xiang J., Lin A. (2004) JNK suppresses apoptosis via phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol. Cell 13, 329–340 [DOI] [PubMed] [Google Scholar]
- 18. Franklin C. C., Kraft A. S. (1997) Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J. Biol. Chem. 272, 16917–16923 [DOI] [PubMed] [Google Scholar]
- 19. Wang Z., Xu J., Zhou J. Y., Liu Y., Wu G. S. (2006) Mitogen-activated protein kinase phosphatase-1 is required for cisplatin resistance. Cancer Res. 66, 8870–8877 [DOI] [PubMed] [Google Scholar]
- 20. Wang J., Zhou J. Y., Wu G. S. (2007) ERK-dependent MKP-1-mediated cisplatin resistance in human ovarian cancer cells. Cancer Res. 67, 11933–11941 [DOI] [PubMed] [Google Scholar]
- 21. Small G. W., Shi Y. Y., Higgins L. S., Orlowski R. Z. (2007) Mitogen-activated protein kinase phosphatase-1 is a mediator of breast cancer chemoresistance. Cancer Res. 67, 4459–4466 [DOI] [PubMed] [Google Scholar]
- 22. Vogelstein B. (1990) Cancer. A deadly inheritance. Nature 348, 681–682 [DOI] [PubMed] [Google Scholar]
- 23. Hollstein M., Sidransky D., Vogelstein B., Harris C. C. (1991) p53 mutations in human cancers. Science 253, 49–53 [DOI] [PubMed] [Google Scholar]
- 24. Brodsky M. H., Nordstrom W., Tsang G., Kwan E., Rubin G. M., Abrams J. M. (2000) Drosophila p53 binds a damage response element at the reaper locus. Cell 101, 103–113 [DOI] [PubMed] [Google Scholar]
- 25. Brodsky M. H., Weinert B. T., Tsang G., Rong Y. S., McGinnis N. M., Golic K. G., Rio D. C., Rubin G. M. (2004) Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol. Cell. Biol. 24, 1219–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jassim O. W., Fink J. L., Cagan R. L. (2003) Dmp53 protects the Drosophila retina during a developmentally regulated DNA damage response. EMBO J. 22, 5622–5632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee J. H., Lee E., Park J., Kim E., Kim J., Chung J. (2003) In vivo p53 function is indispensable for DNA damage-induced apoptotic signaling in Drosophila. FEBS Lett. 550, 5–10 [DOI] [PubMed] [Google Scholar]
- 28. Ollmann M., Young L. M., Di Como C. J., Karim F., Belvin M., Robertson S., Whittaker K., Demsky M., Fisher W. W., Buchman A., Duyk G., Friedman L., Prives C., Kopczynski C. (2000) Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 101, 91–101 [DOI] [PubMed] [Google Scholar]
- 29. Sogame N., Kim M., Abrams J. M. (2003) Drosophila p53 preserves genomic stability by regulating cell death. Proc. Natl. Acad. Sci. U.S.A. 100, 4696–4701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fuchs S. Y., Adler V., Buschmann T., Yin Z., Wu X., Jones S. N., Ronai Z. (1998) JNK targets p53 ubiquitination and degradation in nonstressed cells. Genes Dev. 12, 2658–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fuchs S. Y., Adler V., Pincus M. R., Ronai Z. (1998) MEKK1/JNK signaling stabilizes and activates p53. Proc. Natl. Acad. Sci. U.S.A. 95, 10541–10546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Buschmann T., Potapova O., Bar-Shira A., Ivanov V. N., Fuchs S. Y., Henderson S., Fried V. A., Minamoto T., Alarcon-Vargas D., Pincus M. R., Gaarde W. A., Holbrook N. J., Shiloh Y., Ronai Z. (2001) Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol. Cell. Biol. 21, 2743–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oleinik N. V., Krupenko N. I., Krupenko S. A. (2007) Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene 26, 7222–7230 [DOI] [PubMed] [Google Scholar]
- 34. McEwen D. G., Peifer M. (2005) Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development 132, 3935–3946 [DOI] [PubMed] [Google Scholar]
- 35. Martín-Blanco E., Gampel A., Ring J., Virdee K., Kirov N., Tolkovsky A. M., Martinez-Arias A. (1998) puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 12, 557–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ring J. M., Martinez Arias A. (1993) puckered, a gene involved in position-specific cell differentiation in the dorsal epidermis of the Drosophila larva. Dev. Suppl. 251–259 [PubMed] [Google Scholar]
- 37. McEwen D. G., Cox R. T., Peifer M. (2000) The canonical Wg and JNK signaling cascades collaborate to promote both dorsal closure and ventral patterning. Development 127, 3607–3617 [DOI] [PubMed] [Google Scholar]
- 38. Brand A. H., Manoukian A. S., Perrimon N. (1994) Ectopic expression in Drosophila. Methods Cell Biol. 44, 635–654 [DOI] [PubMed] [Google Scholar]
- 39. Rubin G. M., Spradling A. C. (1982) Genetic transformation of Drosophila with transposable element vectors. Science 218, 348–353 [DOI] [PubMed] [Google Scholar]
- 40. Yamaguchi M., Hirose F., Inoue Y. H., Shiraki M., Hayashi Y., Nishi Y., Matsukage A. (1999) Ectopic expression of human p53 inhibits entry into S phase and induces apoptosis in the Drosophila eye imaginal disc. Oncogene 18, 6767–6775 [DOI] [PubMed] [Google Scholar]
- 41. Ravi D., Chen Y., Karia B., Brown A., Gu T. T., Li J., Carey M. S., Hennessy B. T., Bishop A. J. (2011) 14-3-3 σ expression effects G2/M response to oxygen and correlates with ovarian cancer metastasis. PLoS One 6, e15864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McEwen D. G., Ornitz D. M. (1998) Regulation of the fibroblast growth factor receptor 3 promoter and intron I enhancer by Sp1 family transcription factors. J. Biol. Chem. 273, 5349–5357 [DOI] [PubMed] [Google Scholar]
- 43. Khokhlatchev A., Xu S., English J., Wu P., Schaefer E., Cobb M. H. (1997) Reconstitution of mitogen-activated protein kinase phosphorylation cascades in bacteria. Efficient synthesis of active protein kinases. J. Biol. Chem. 272, 11057–11062 [DOI] [PubMed] [Google Scholar]
- 44. Yin H., Zhou Q., Panda M., Yeh L. C., Zavala M. C., Lee J. C. (2007) A fluorescence study of type I and type II receptors of bone morphogenetic proteins with bis-ANS (4,4′-dianilino-1,1′-bisnaphthyl-5,5′-disulfonic acid). Biochim. Biophys. Acta 1774, 493–501 [DOI] [PubMed] [Google Scholar]
- 45. Gavrieli Y., Sherman Y., Ben-Sasson S. A. (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 119, 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wichmann A., Jaklevic B., Su T. T. (2006) Ionizing radiation induces caspase-dependent but Chk2- and p53-independent cell death in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 103, 9952–9957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J. P., Sedivy J. M., Kinzler K. W., Vogelstein B. (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501 [DOI] [PubMed] [Google Scholar]
- 48. Bennett B. L., Sasaki D. T., Murray B. W., O'Leary E. C., Sakata S. T., Xu W., Leisten J. C., Motiwala A., Pierce S., Satoh Y., Bhagwat S. S., Manning A. M., Anderson D. W. (2001) SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U.S.A. 98, 13681–13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nguyen M., Millar D. G., Yong V. W., Korsmeyer S. J., Shore G. C. (1993) Targeting of Bcl-2 to the mitochondrial outer membrane by a COOH-terminal signal anchor sequence. J. Biol. Chem. 268, 25265–25268 [PubMed] [Google Scholar]
- 50. Schinzel A., Kaufmann T., Schuler M., Martinalbo J., Grubb D., Borner C. (2004) Conformational control of Bax localization and apoptotic activity by Pro168. J. Cell Biol. 164, 1021–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kern S. E., Pietenpol J. A., Thiagalingam S., Seymour A., Kinzler K. W., Vogelstein B. (1992) Oncogenic forms of p53 inhibit p53-regulated gene expression. Science 256, 827–830 [DOI] [PubMed] [Google Scholar]
- 52. Adler V., Pincus M. R., Minamoto T., Fuchs S. Y., Bluth M. J., Brandt-Rauf P. W., Friedman F. K., Robinson R. C., Chen J. M., Wang X. W., Harris C. C., Ronai Z. (1997) Conformation-dependent phosphorylation of p53. Proc. Natl. Acad. Sci. U.S.A. 94, 1686–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Owens D. M., Keyse S. M. (2007) Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 26, 3203–3213 [DOI] [PubMed] [Google Scholar]
- 54. Tanoue T., Moriguchi T., Nishida E. (1999) Molecular cloning and characterization of a novel dual specificity phosphatase, MKP-5. J. Biol. Chem. 274, 19949–19956 [DOI] [PubMed] [Google Scholar]
- 55. Kojima K., Kitada S., Shimokata K., Ogishima T., Ito A. (1998) Cooperative formation of a substrate binding pocket by α- and β-subunits of mitochondrial processing peptidase. J. Biol. Chem. 273, 32542–32546 [DOI] [PubMed] [Google Scholar]
- 56. Theodosiou A., Smith A., Gillieron C., Arkinstall S., Ashworth A. (1999) MKP5, a new member of the MAP kinase phosphatase family, which selectively dephosphorylates stress-activated kinases. Oncogene 18, 6981–6988 [DOI] [PubMed] [Google Scholar]
- 57. Ljungman M., Zhang F. (1996) Blockage of RNA polymerase as a possible trigger for u.v. light-induced apoptosis. Oncogene 13, 823–831 [PubMed] [Google Scholar]
- 58. Ljungman M., Zhang F., Chen F., Rainbow A. J., McKay B. C. (1999) Inhibition of RNA polymerase II as a trigger for the p53 response. Oncogene 18, 583–592 [DOI] [PubMed] [Google Scholar]
- 59. Buschmann T., Minamoto T., Wagle N., Fuchs S. Y., Adler V., Mai M., Ronai Z. (2000) Analysis of JNK, Mdm2 and p14ARF contribution to the regulation of mutant p53 stability. J. Mol. Biol. 295, 1009–1021 [DOI] [PubMed] [Google Scholar]
- 60. Levrero M., De Laurenzi V., Costanzo A., Gong J., Wang J. Y., Melino G. (2000) The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J. Cell Sci. 113, 1661–1670 [DOI] [PubMed] [Google Scholar]
- 61. Yang A., McKeon F. (2000) p63 and p73: p53 mimics, menaces and more. Nat. Rev. Mol. Cell Biol. 1, 199–207 [DOI] [PubMed] [Google Scholar]
- 62. Klein C., Georges G., Künkele K. P., Huber R., Engh R. A., Hansen S. (2001) High thermostability and lack of cooperative DNA binding distinguish the p63 core domain from the homologous tumor suppressor p53. J. Biol. Chem. 276, 37390–37401 [DOI] [PubMed] [Google Scholar]
- 63. Tanoue T., Nishida E. (2002) Docking interactions in the mitogen-activated protein kinase cascades. Pharmacol. Ther. 93, 193–202 [DOI] [PubMed] [Google Scholar]
- 64. Tanoue T., Adachi M., Moriguchi T., Nishida E. (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2, 110–116 [DOI] [PubMed] [Google Scholar]
- 65. Clark-Lewis I., Sanghera J. S., Pelech S. L. (1991) Definition of a consensus sequence for peptide substrate recognition by p44mpk, the meiosis-activated myelin basic protein kinase. J. Biol. Chem. 266, 15180–15184 [PubMed] [Google Scholar]
- 66. Gonzalez F. A., Raden D. L., Davis R. J. (1991) Identification of substrate recognition determinants for human ERK1 and ERK2 protein kinases. J. Biol. Chem. 266, 22159–22163 [PubMed] [Google Scholar]
- 67. Jacobs D., Glossip D., Xing H., Muslin A. J., Kornfeld K. (1999) Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 13, 163–175 [PMC free article] [PubMed] [Google Scholar]
- 68. Kallunki T., Deng T., Hibi M., Karin M. (1996) c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell 87, 929–939 [DOI] [PubMed] [Google Scholar]
- 69. Adler V., Franklin C. C., Kraft A. S. (1992) Phorbol esters stimulate the phosphorylation of c-Jun but not v-Jun: regulation by the N-terminal δ domain. Proc. Natl. Acad. Sci. U.S.A. 89, 5341–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee C. M., Onésime D., Reddy C. D., Dhanasekaran N., Reddy E. P. (2002) JLP: a scaffolding protein that tethers JNK/p38MAPK signaling modules and transcription factors. Proc. Natl. Acad. Sci. U.S.A. 99, 14189–14194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schuler M., Bossy-Wetzel E., Goldstein J. C., Fitzgerald P., Green D. R. (2000) p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J. Biol. Chem. 275, 7337–7342 [DOI] [PubMed] [Google Scholar]
- 72. Kharbanda S., Saxena S., Yoshida K., Pandey P., Kaneki M., Wang Q., Cheng K., Chen Y. N., Campbell A., Sudha T., Yuan Z. M., Narula J., Weichselbaum R., Nalin C., Kufe D. (2000) Translocation of SAPK/JNK to mitochondria and interaction with Bcl-xL in response to DNA damage. J. Biol. Chem. 275, 322–327 [DOI] [PubMed] [Google Scholar]
- 73. Srivastava R. K., Sollott S. J., Khan L., Hansford R., Lakatta E. G., Longo D. L. (1999) Bcl-2 and Bcl-XL block thapsigargin-induced nitric oxide generation, c-Jun NH2-terminal kinase activity, and apoptosis. Mol. Cell. Biol. 19, 5659–5674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mihara M., Erster S., Zaika A., Petrenko O., Chittenden T., Pancoska P., Moll U. M. (2003) p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 11, 577–590 [DOI] [PubMed] [Google Scholar]
- 75. Bonafé M., Salvioli S., Barbi C., Trapassi C., Tocco F., Storci G., Invidia L., Vannini I., Rossi M., Marzi E., Mishto M., Capri M., Olivieri F., Antonicelli R., Memo M., Uberti D., Nacmias B., Sorbi S., Monti D., Franceschi C. (2004) The different apoptotic potential of the p53 codon 72 alleles increases with age and modulates in vivo ischaemia-induced cell death. Cell Death Differ. 11, 962–973 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.