

Background: The influence of protein conformation of autoantigens to induce disease has not been assessed in detail.
Results: Protein conformation of myelin oligodendrocyte glycoprotein determined encephalitogenicity and the degree of T- and B-cell responses.
Conclusion: Protein conformation has a strong impact on the potential of an autoantigen to induce autoimmune disease.
Significance: Autoantigen conformation needs to be taken into consideration in studies regarding disease-inducing capacity.
Keywords: Antigen, Antigen Processing, Lymphocyte, Myelin, Protein Folding, Autoantigen, Encephalitogenicity, Experimental Autoimmune Encephalomyelitis, Myelin Oligodendrocyte Glycoprotein
Abstract
It has become increasingly clear that only antibodies recognizing conformation-dependent epitopes of myelin oligodendrocyte glycoprotein (MOG) have a demyelinating potential in the animal model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE). Nevertheless, for the induction of EAE, most studies to date have used MOG peptides or bacterially expressed MOG, neither of which contain the tertiary structure of the native antigen. Non-refolded recombinant human MOG does not induce EAE in DA rats. Therefore, we refolded this protein in order to assess the influence of MOG conformation on its pathogenicity in DA rats. DA rats immunized with refolded human MOG developed severe acute EAE. As expected, rats immunized with the refolded protein had a higher amount of conformational MOG antibodies present in serum. But in addition, a striking effect of MOG refolding on the generation of T-cell responses was found. Indeed, T-cell responses against the encephalitogenic MOG 91–108 epitope were greatly enhanced after refolding. Therefore, we conclude that refolding of MOG increases its pathogenicity both by generating conformation-dependent MOG antibodies and by enhancing its processing or/and presentation on MHC molecules. These data are important in regard to investigations of the pathogenic potential of many (auto)antigens.
Introduction
Multiple scleroris (MS)2 is a chronic demyelinating disease of the CNS. It is caused supposedly by an autoaggressive attack of T- and B-cells against components of the CNS (1). Identifying the targets of this autoimmune attack has been the topic of numerous studies. Recently, we have eluted naturally presented peptides from the CNS of patients with MS (2). Whereas T-cell responses against several different myelin antigens have been implicated in disease pathogenesis, research on pathogenic humoral responses has focused mainly on myelin oligodendrocyte glycoprotein (MOG) ever since it was shown that antibodies against MOG can induce demyelination (3–5). MOG is a minor CNS-specific myelin protein, but due to its localization at the outermost surface of the myelin sheath, it is accessible for antibodies (6). It is becoming increasingly clear though, that not all MOG antibodies are pathogenic: only antibodies recognizing conformational MOG epitopes are capable of inducing demyelination (7, 8). This might partially explain the controversy that exists around the contribution of MOG-antibodies to MS pathology since several studies used methods for measuring anti-MOG responses failing to discriminate between antibodies recognizing conformational and linear epitopes (9, 10). Unfortunately, more recent reports using conformation-sensitive methods for determination of MOG antibodies in MS patients still do not offer a consensus on the matter as cell-based assays detected significant amounts of MOG antibodies in certain subsets of MS patients (11–13), whereas liquid phase assays failed to do so (14–16). Taken together, the pathogenic role of MOG-antibodies in humans is still controversial and awaits further clarification. In addition, antibodies against different MOG variants have not been investigated as yet in a systematic way (17).
MOG-induced EAE mimics several aspects of MS such as demyelination and axonal loss (18–20). Disease is induced routinely in rats or mice by immunization with MOG peptides or with the bacterially expressed extracellular part of MOG (MOG 1–125). Studies with B-cell deficient mice have clearly demonstrated that B-cells are not critical for the development of MOG-peptide induced disease (21). Still, even the pathological significance of MOG antibodies in models of experimental autoimmune encephalomyelitis (EAE) induced with MOG protein is not clarified completely (22, 23). Thus, it was shown that induction of EAE in C57BL/6 mice with recombinant MOG 1–125 from human origin but not from rat origin is B-cell-dependent although both immunization protocols lead to the generation of comparable titers of anti-MOG antibodies (24, 25).
It is important to keep in mind though that bacterial overexpression of MOG 1–125 usually leads to the expression of the protein under the form of inclusion bodies. Provided an efficient protein refolding protocol is applied, the resulting protein does not have its native conformation and lacks the correct disulfide bond which is present in the extracellular part of the molecule. Currently, the most common way of preparing extracellular MOG, is to purify a His-tagged form of the protein from Escherichia coli by metal chromatography under denaturing conditions. Subsequent dialysis of purified MOG against phosphate-buffered saline (PBS) yields a partially precipitated preparation, whereas dialysis against an acidic acetate buffer yields a soluble form of the protein. Both preparations, lacking the native conformation, have been shown to induce EAE in susceptible strains of rats and mice, although the soluble form of the protein is generally considered more pathogenic (18, 26, 27).
We used correctly refolded recombinant human MOG (rHuMOG) to induce disease in DA rats and compared its effect on pathogenicity, B-cell, and T-cell responses relative to precipitated and soluble rHuMOG. Based on the current concepts about MOG conformation, we expected refolded rHuMOG to be more pathogenic than its non-refolded counterparts due to the presence of conformational MOG antibodies. As expected, refolded rHuMOG was extremely pathogenic in DA rats, and this correlated with the presence of conformational MOG antibodies in the serum of DA rats. Still, the strong pathogenicity of refolded rHuMOG could not be attributed solely to the presence of conformational MOG antibodies. Indeed, we equally found a strong contribution of MOG conformation on MOG-directed T-cell responses.
EXPERIMENTAL PROCEDURES
Animals
Female DA rats were obtained from Harlan Winkelmann (Borchen, Germany). Rats were kept under specific pathogen-free conditions and obtained food and water ad libitum.
rHuMOG Preparations
The cDNA sequence encoding the extracellular domain (MOG 1–125) of the mature HuMOG protein was subcloned into the pQE60 vector (Qiagen, Hilden, Germany). The protein was overexpressed in inclusion bodies in Escherichia coli. The resulting C-terminal His-tagged fusion protein was purified by metal chelate affinity chromatography on Ni-NTA-agarose (Qiagen). Non-refolded rHuMOG was obtained by performing Ni-NTA chromatography under denaturing conditions in the presence of 8 m urea. Subsequently, the purified protein was dialyzed extensively against PBS, which leads to the formation of a precipitated rHuMOG preparation (rHuMOGPBS) or against 10 mm sodium acetate pH3, which lead to the formation of a soluble, non-refolded form of the protein (rHuMOGAC). Refolded rHuMOG (rHuMOGREF) was obtained by including a refolding step, whereas the protein was bound to the Ni-NTA column as described elsewhere (28) and was completely soluble in PBS. Purity of the obtained rHuMOG preparations was verified by SDS-PAGE. The extracellular part of rRMOG was purified as described previously (18).
Peptides
Rat MOG 73–90 (KESIGEGKVALRIQNVRF), rat MOG 91–108 (SDEGGYTCFFRDHSYQEE), and its human counterpart huMOG 93–108 (EGGFTCFFRDHSYQEE) were prepared by Karl-Heinz Wiesmüller (EMC Microcollections, Tübingen, Germany) by solid phase synthesis using F-moc/tBu chemistry. Peptides were purified by preparative high-performance liquid chromatography (HPLC) (Abimed, Langenfeld, Germany). The identity of the purified peptides was confirmed by electrospray mass spectrometry. The purity of the peptides was >95% as determined by analytical HPLC.
Induction and Scoring of EAE
Active EAE was induced by injection of 200 μl inoculum intradermally at the base of the tail. The inoculum consisted of 50 μg of rHuMOG in 100 μl emulsified with 100 μl of complete Freund's adjuvant (Sigma) containing 200 μg heat-inactivated Mycobacterium tuberculosis (strain H37RA; Difco).
For adoptive transfer experiments, rats were immunized with the different rHuMOG preparations as described above. Spleens were removed at day 11 post immunization (p.i.), and single-cell suspensions were cultured for 48 h at a concentration of 107 cells/ml in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 2 mm glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin (all from Invitrogen) and 5% FCS (PAA Laboratories, Linz, Austria) (complete medium; CM) in the presence of the antigen of immunization (20 μg/ml). 107 freshly restimulated cells were injected intraperitoneally in naïve rats.
Clinical signs were scored as follows: grade 1, tail weakness or tail paralysis; grade 2, hind leg paraparesis or hemiparesis; grade 3, hind leg paralysis or hemiparalysis; grade 4, complete paralysis, tetraplegia, moribund state, or death. Rats were scored up to 40 days.
Histopathology
Histopathological evaluation was performed on paraformaldehyde-fixed, paraffin-embedded sections of brains and spinal cords collected at day 13 p.i. Under deep anesthesia, the rats were perfused transcardially with 4% paraformaldehyde. Fixed brain tissues were embedded in paraffin. 3–5-μm-thick sections stained with hematoxylin and eosin (H&E), Luxol fast blue/periodic acid Schiff agent to assess the degree of inflammation and demyelination. In adjacent serial sections, immunohistochemistry was performed using antibodies for CD3+ T-cells (clone W3/13, CD43, Serotec, Oxford, UK), B-cells (clone HIS24, CD45R, BD Pharmingen), macrophages/activated microglia (clone ED1; Serotec, Oxford, UK). Deposition/infiltration of IgG was detected with biotinilated anti-rat-IgG (Sigma). For immunohistochemistry, sections were deparaffinized and pretreated with microwaving (3 × 5 min at 800 W) in citric acid buffer (10 mm, pH 6.0), and unspecific reactions were blocked with 10% FCS/PBS. Primary antibodies were applied and incubated over night at 4 °C. After application of the biotinylated secondary antibody, avidin peroxydase (Dako, Glostrup, Denmark) was added and developed with, 3,3′-diaminobenzidine hydrochloride (DAB, Sigma).
Negative controls were performed by omitting the primary antibody or applying non-immune sera or isotype control antibodies. Inflammatory lesions were quantified by counting inflammatory infiltrates in HE-stained spinal cord sections on overview photographs (×40). The inflammatory index was evaluated as follows: the mean number of perivascular inflammatory infiltrates from an average of 15 complete cross-sections of the spinal cord of an animal.
Isolation of Mononuclear Cells (MNCs) from Lymph Nodes (LNs) and Spleens from Rats
Draining inguinal LNs and spleens were dissected out under deep anesthesia. LNs were disrupted, and MNCs were washed twice in DMEM, resuspended in CM containing 50 μm 2-mercaptoethanol, and flushed through a 70-μm plastic strainer (Falcon; BD Biosciences). MNCs from spleen were prepared in the same way as from LNs with the difference that RBCs were lysed with lysis buffer containing 0.15 m NH4Cl, 10 mm KHCO3, and 0.1 mm EDTA adjusted to pH 7.4.
Enzyme-linked Immunosorbent Spot (ELISPOT)
Nitrocellulose-bottomed 96-well plates (MAHA; Millipore, Molsheim, France) were coated with an anti-interferon-γ (anti-IFN-γ) mouse monoclonal antibody (mAb) DB1 (a generous gift from Peter van der Meide, Netherlands Organization for Applied Scientific Research (TNO), Primate Center, Rijswijk, The Netherlands). Following washing with PBS, plates were blocked with CM (Invitrogen). MNCs (4.105 per well) in 200 μl CM containing 50 μm 2-mercaptoethanol and antigen were added to the plates and incubated for 48 h at 37 °C in a humidified atmosphere containing 5% CO2. For each antigen, duplicate determinations were performed. rHuMOG preparations and rRMOG were used at a concentration of 5 μg/ml, peptides at a concentration of 10 μg/ml and concanavalin A (Sigma) at a concentration of 1 μg/ml. Cells were discarded, and plates were washed four times with PBS. Secreted IFN-γ was visualized with biotinylated DB12, a mAb against rat IFN-γ (Peter van der Meide), avidin-biotin peroxidase (Vector Laboratories, Burlingame, CA), and subsequently by staining with carbazole (Sigma).
Assays of Antigen-induced Proliferation
All proliferative experiments were performed in triplicates in 96-well round-bottomed microtiter plates. 2 × 105 MNC/well in 100 μl of CM were cultured with or without the relevant antigen for 60 h and, subsequently, pulsed with 0.5 mCi [3H]TdR (GE Healthcare, Freiburg, Germany) per well for 12 h. DNA was collected on glass fiber filters (Skatron, Sterling, USA) and [3H]TdR incorporation was measured in a β-counter (Beckman, Palo Alto, CA). Proliferation of T-cells was expressed as a stimulation index, indicating the [3H]TdR uptake after exposure with antigen/[3H]TdR uptake without exposure of antigen.
ELISA
For detection of the different rHuMOG preparations by 8-18C5, 96-well Maxisorb plates (Nunc, Roskilde, Denmark) were coated overnight at 4 °C with serial dilutions of the proteins in 0.1 m NaHCO3, pH 8.3. Plates were washed with PBS/0.05% Tween and blocked for 1 h at room temperature with 5% BSA in PBS. After washing, wells were incubated for 1 h with 8-18C5 (2 μg/ml) in PBS. Plates were washed once more, and bound 8–18C5 antibody was detected with a polyclonal sheep anti-mouse IgG (1/10,000 in PBS; Amersham Biosciences) and incubated on the plates for 1 h at room temperature. After a final washing step, the reaction was visualized with 3,3′,5,5′-tetramethyl benzidine (TMB) (Sigma). The enzymatic reaction was stopped after 10 min by addition of 1 m HCl, and the optical density at 450 nm was measured.
For detection of rHuMOGREF IgG antibodies in rat serum, 96-well plates were coated overnight at 4 °C with 3 μg/ml rHuMOGREF in 0.1 m NaHCO3, pH 8.3. Plates were washed with PBS/0.05% Tween and blocked for 1 h at room temperature with 5% milk powder in PBS. After washing, wells were incubated for 1 h with rat serum, serially diluted in PBS. Plates were washed and a rabbit anti-rat IgG (1/2000 in PBS, Nordic, Tilburg, The Netherlands) was added and incubated for 1 h at room temperature. Plates were washed prior to the addition of peroxidase-conjugated goat anti-rabbit antiserum (1/10,000 in PBS, Nordic). After 30 min of incubation, plates were washed, and bound antibodies were visualized by addition of TMB (Sigma). The enzymatic reaction was stopped after 10 min by addition of 1 m HCl, and the optical density at 450 nm was measured.
Flow Cytometric Cell Sorting
L cells containing thymidine kinase (LTK cells) transfected with complete human MOG cDNA were a kind gift of Professor C. Linington (University of Glasgow). LTK-MOG cells were maintained in complete DMEM with 10% FCS and 1 mg/ml G418 (Sigma). To detect antibody responses to native MOG, transfected and non-transfected LTK cells were washed with PBS/1% BSA/0.02% NaN3 and incubated with rat serum (1/500) on ice for 1 h. Subsequently, cells were stained with biotinylated anti-rat IgG (Sigma) and labeled with streptavidine-phycoerytrin (Pharmingen, San Diego, CA). Finally, cells were analyzed by FACS (BD Biosciences). Negative controls were done with secondary antibody alone.
Data Analysis
Statistical analysis was performed using Sigmastat software (version 2.0, Systat, San Jose, CA). The Mann-Whitney Rank Sum test or Student's t test were used for statistical comparisons between different study groups. Data are presented as mean values ± S.E. (n.s., not significant; *, p < 0.05; **, p < 0.01).
RESULTS
Characterization of rHuMOGREF
rHuMOG was expressed in E. coli and preparations of rHuMOGPBS, rHuMOGAC, and rHuMOGREF were generated as described under “Experimental Procedures.” The purified proteins were subjected to SDS-PAGE (Fig. 1). rHuMOGPBS and rHuMOGAC clearly contain multimeric forms of the proteins, which are probably generated by incorrect pairing of the cysteine residues present in the extracellular MOG domain. In contrast, only monomeric rHuMOG was detected by SDS-PAGE after refolding.
FIGURE 1.
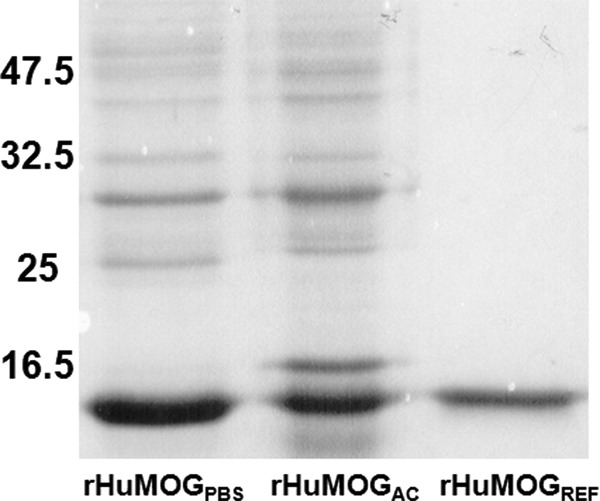
rHuMOGREF migrates as a monomeric form under reducing SDS-PAGE. Samples of the different rHuMOG preparations were electrophoresed on a reducing 15% SDS-polyacrylamide gel and visualized by Coomassie Blue staining. rHuMOGREF migrates as a monomer, whereas rHuMOGPBS and rHuMOGAC still contain multimeric forms of the protein.
Next, we measured the binding of the mouse monoclonal MOG antibody 8-18C5 to the different rHuMOG preparations by ELISA. Although 8-18C5 has often been used to detect MOG in Western blotting (i.e. under denaturing conditions), it originally was raised against rat cerebellar glycoproteins (29) and has been shown subsequently to recognize a conformational MOG epitope (30). Therefore, we reasoned that 8-18C5 would have a stronger affinity for rHuMOGREF than for rHuMOGPBS or for rHuMOGAC. We coated different concentrations of the rHuMOG preparations and detected binding of 8-18C5 to the proteins by ELISA. Fig. 2 clearly shows that 8-18C5 weakly reacts with rHuMOGPBS and rHuMOGAC but is much better at recognizing the refolded form of the protein.
FIGURE 2.

The conformational MOG antibody 8-18C5 preferentially recognizes rHuMOGREF by ELISA. Serial dilutions of the different rHuMOG preparations were coated on an ELISA plate. The mouse monoclonal antibody 8-18C5 was allowed to react with the protein in the ELISA plate. Detection of bound 8-18C5 occurred by using an HRP-coupled polyclonal sheep anti-mouse IgG. Data were consistent in three independent experiments.
Refolded rHuMOG Is More Pathogenic Than Non-refolded rHuMOG in DA Rats
Although on several occasions DA rats were immunized with rHuMOGPBS originating from different batches, they never developed overt signs of clinical EAE. In contrast, DA rats immunized with rHuMOGREF consistently developed severe clinical disease (Fig. 3A), which often was acutely lethal. Upon immunization with rHuMOGREF, disease onset varied between day 10 and day 18 p.i., and most rats attained an EAE score of 3 or more (Table 1). Immunization of DA rats with rHuMOGAC also led to EAE induction, although disease incidence and maximum disease score were reduced significantly as compared with the rHuMOGREF-immunized group (Table 1). Histopathological analysis revealed widespread inflammation and demyelination throughout the CNS of rHuMOGREF-immunized DA rats. In contrast in the rHuMOGPBS immunized rats inflammation was not found (inflammatory index, rHuMOGPBS (n = 4), 0 ± 0 versus rHuMOGREF (n = 5), 7.4 ± 0.7; p < 0.01). The inflammatory lesions were composed of lymphocytes and macrophages, and the proportion of B-cells (CD45) was high (data not shown). CNS tissue of rHuMOGREF immunized rats compared with rHuMOGPBS immunized rats was infiltrated widespread with IgG (Fig. 3B).
FIGURE 3.

rHuMOGREF induces acute EAE in DA rats. DA rats were immunized with 50 μg of rHuMOGPBS (n = 4) or rHuMOGREF (n = 5) in complete Freund's adjuvant. Clinical scores are shown (data are represented as mean values). p < 0.001 on sum scores of rHuMOGPBS compared with rHuMOGREF immunized rats (A). B, example of typical staining for IgG in CNS of rats immunized with either rHuMOGPBS or rHuMOGREF. In the CNS of rHuMOGREF but not rHuMOGPBS immunized rats, IgG was present widespread.
TABLE 1.
EAE induction with different rHuMOG preparations
Rats were immunized with 50 μg of the indicated rHuMOG preparation emulsified in complete Freund's adjuvant. Numbers are mean values (S.E.).
| Immunization | Disease incidence | Day of disease onset | Maximum disease score |
|---|---|---|---|
| rHuMOGPBS | 0/15 | 0 (0)a | |
| rHuMOGAC | 3/6 | 28.3 (0.9)b | 1 (0.5)c |
| rHuMOGREF | 15/16 | 14 (1.6) | 3.2 (0.3) |
a p < 0.001 (Mann-Whitney Rank Sum test, rHuMOGPBS versus rHuMOGREF).
b p = 0.024 (Mann-Whitney Rank Sum test, rHuMOGAC versus rHuMOGREF).
c p = 0.006 (Mann-Whitney Rank Sum test, rHuMOGAC versus rHuMOGREF).
Antibodies against Refolded rHuMOG Recognize Conformational MOG Epitopes
Sera of immunized rats taken at day 20 p.i. were analyzed by ELISA to measure the amount of anti-rHuMOGREF IgG present in the sera and by FACS to identify responses against eukaryotic native MOG protein expressed on the surface of MOG-transfected cells (7, 31).
All immunization protocols lead to the generation of high amounts of IgG antibodies against rHuMOGREF as measured by ELISA, irrespective of the protein preparation used for immunization. Surprisingly, anti-rHuMOGREF IgG antibodies were elevated slightly in the sera of rHuMOGAC immunized rats as compared with rHuMOGPBS and rHuMOGREF immunized rats on day 20 p.i., although these differences were not significant (Fig. 4A).
FIGURE 4.

The IgG responses against cell surface huMOG are highest in sera of DA rats immunized with rHuMOGREF. Antibody responses against rHuMOGREF and native human MOG in sera from rHuMOG immunized DA rats at day 20 p.i. A, IgG against rHuMOGREF was measured by ELISA. No significant differences could be detected in sera of rats immunized with rHuMOGPBS, rHuMOGAC, or rHuMOGREF. B, LTK cells transfected with human MOG were incubated with sera from immunized DA rats and binding of native MOG-specific antibodies was analyzed by flow cytometry. Data are representative of two independent experiments and are shown as mean values ± S.E. Asterisks indicate significance of difference with rHuMOGREF group (*, p < 0.05; **, p < 0.001). C, example of staining of sera by FACS of rHuMOGPBS and rHuMOGREF immunized rats to non-transfected LTK cells (left panel) or MOG-transfected LTK cells (right panel).
In addition, sera were analyzed by FACS to detect the presence of IgG antibodies against native MOG as expressed on the surface of MOG-transfected cells. Contrary to the results obtained by ELISA, rats immunized with rHuMOGREF raised a significantly higher IgG response against native MOG as rHuMOGPBS-immunized animals (p = 0.017) or rHuMOGAC-immunized animals (p < 0.001) (Fig. 4, B and C). It was shown previously that FACS-based assays using MOG transfectants primarily detect antibodies against conformation dependent epitopes, which are probably implicated in antibody-dependent demyelination (7). Thus, we conclude that immunization with rHuMOGREF leads to the generation of an antibody response, which contains a significantly higher proportion of conformational MOG antibodies than immunization with non-refolded preparations of rHuMOG.
T-cell Responses in rHuMOGPBS, rHuMOGAC, and rHuMOGREF-immunized DA Rats Are Qualitatively Different
DA rats immunized with rHuMOGREF develop much more disease than rats immunized either with rHuMOGPBS or rHuMOGAC. We questioned whether this effect could solely be attributed to the presence of higher amounts of conformational MOG antibodies in sera and CNS of rHuMOGREF immunized animals. Therefore, we also had to assess the influence of MOG refolding on MOG T-cell responses after immunization with different rHuMOG preparations. DA rats were immunized with rHuMOGPBS, rHuMOGAC, or rHuMOGREF in complete Freund's adjuvant and at day 12 p.i. T-cell responses in draining lymph nodes against MOG and MOG-derived peptides were analyzed by IFN-γ ELISPOT.
Irrespective of the protein used for immunization, IFN-γ secreting T-cells cross-reactive with recombinant rat MOG (rRMOG) could be detected in LNs from all animals. Although rHuMOGREF-immunized DA rats showed slightly higher responses to rRMOG, differences between groups were not significant. T-cell responses against rHuMOGPBS were significantly higher in the rHuMOGREF-immunized group as compared with the rHuMOGAC-immunized group (p = 0.006). Differences between the groups became even more pronounced when measuring recall responses against rHuMOGREF: IFN-γ responses were three to four times higher in rHuMOGREF-immunized animals (rHuMOGPBS versus rHuMOGREF, p < 0.001; rHuMOGAC versus rHuMOGREF, p < 0.001). Next, we studied IFN-γ responses against MOG 73–90 (rat sequence), MOG 91–108 (rat sequence), and MOG 93–108 (human sequence), which are dominant MOG epitopes in RT1av1 rats (32). No IFN-γ responses were detectable against MOG 73–90, which is known to be a non-encephalitogenic MOG determinant (33). In contrast, IFN-γ responses against the main encephalitogenic MOG determinant, MOG 91–108, varied significantly between groups and correlated quite well with the grade of disease obtained in each group (rHuMOGPBS versus rHuMOGREF, p = 0.004; rHuMOGPBS versus rHuMOGAC, p = 0.024; rHuMOGAC versus rHuMOGREF, p = 0.012). Similar results were obtained with the human variant of the peptide MOG 93–108 (rHuMOGPBS versus rHuMOGREF, p = 0.004; rHuMOGPBS versus rHuMOGAC, p = 0.001; and rHuMOGAC versus rHuMOGREF, p = 0.021) (Fig. 5). Similar results were obtained with proliferation assays measuring 3H-TdR uptake (data not shown). So obviously, responses against human MOG 93–108 are capable of cross-reacting with rat MOG 91–108.
FIGURE 5.

T-cell responses against the pathogenic MOG 91–108 epitope are lacking in DA rats immunized with rHuMOGPBS. MOG-specific T-cell responses were measured at day 12 p.i. by IFN-γ ELISPOT (data are represented as mean values ± S.E.). Asterisks indicate significance of difference with rHuMOGREF group for after restimulation with the antigen indicated on the x axis (rHuMOGPBS, n = 6; rHuMOGAC, n = 3; rHuMOGREF, n = 5; *, p < 0.05; **, p < 0.001;).
In view of these results, it seems most likely that the extreme encephalitogenic potential of rHuMOGREF in DA rats is not due exclusively to the presence of elevated levels of conformational MOG antibodies after rHuMOGREF immunization because refolding rHuMOG also leads to the generation of highly increased levels of T-cell responses specific for the main encephalitogenic MOG determinant MOG 91–108.
Adoptive Transfer of rHuMOG Lymphocytes
Based on our previous results, T-cells activated after immunization with rHuMOGREF should be more encephalitogenic than T-cells obtained after stimulation with rHuMOGPBS or rHuMOGAC. To test this hypothesis, DA rats were immunized with rHuMOGPBS, rHuMOGAC, or rHuMOGREF, lymphocytes were isolated out of the spleen of immunized animals at day 11 p.i. and then restimulated for 48 h with the antigen of immunization. Rats adoptively transferred with rHuMOGPBS lymphocytes remained completely healthy (n = 2). Both transfer of rHuMOGAC and rHuMOGREF lymphocytes induced a monophasic type of disease in DA rats (n = 2 each). Still, disease induced with rHuMOGAC lymphocytes was significantly milder (rHuMOGPBS compared with rHuMOGAC; n.s.) than disease obtained with rHuMOGREF lymphocytes (rHuMOGPBS compared with rHuMOGREF; p < 0.05) (Fig. 6). These results are in complete accordance with the IFN-γ responses measured against the dominant encephalitogenic MOG 91–108 peptide after immunization with the different rHuMOG preparations.
FIGURE 6.

Passive transfer of splenocytes from rHuMOGREF immunized DA rats leads to EAE induction. rHuMOG-specific splenocytes (107) were transferred to naïve recipients. DA rats receiving rHuMOGREF-specific splenocytes developed EAE (n = 2), whereas rats receiving rHuMOGPBS-specific splenocytes remained completely healthy (n = 2). Rats receiving rHuMOGAC developed an intermediate degree of disease (n = 2). Shown are rHuMOGPBS compared with rHuMOGREF, p < 0.05; rHuMOGPBS compared with rHuMOGAC was not significant. Data are representative of two independent experiments.
DISCUSSION
The data presented here demonstrate that refolding rHuMOG strongly enhances its encephalitogenicity. Surprisingly, this increase in pathogenic potential could not exclusively be attributed to the enhanced presence of antibodies recognizing the native form of the protein because we found an equally strong increase of the encephalitogenic potential of ensuing T-cell responses after immunization with rHuMOGREF.
Several recent studies in MS and EAE have emphasized the importance of MOG tertiary structure for detection of pathogenic conformational MOG antibodies (8, 11, 34). We wanted to apply this information in an EAE model and demonstrate an increased pathogenicity of bacterially produced extracellular MOG upon refolding because an increase in the presence of conformational MOG antibodies was to be expected. DA rats readily develop induced autoimmune diseases: immunization of these rats with rRMOG leads to a severe, chronic type of EAE, characterized by demyelination and axonal loss (18, 20). Despite a high sensitivity to rRMOG-induced EAE, we found that DA rats did not develop EAE after immunization with the equivalent human version of the protein. In this respect, it is interesting to mention that it was described previously that rat and human rMOG induce EAE in C57/BL6 mice by different mechanisms (22–24). More specifically, only rHuMOG induced EAE in these mice is B-cell-dependent. This B-cell dependence is strictly determined by the presence of a proline at position 42 of the human MOG protein (serine at position 42 of the rat protein). Moreover, it was shown that only immunization with rHuMOG and not rRMOG leads to the generation of a pathogenic antibody response (25). Still, the biochemical basis for a link between the presence of proline/serine at position 42 and the generation of a pathogenic antibody response remains to be elucidated. In conclusion, the combination of autoimmune prone DA rats, resistant to disease induction with rHuMOG seemed to us the ideal starting point for studying the potentially enhanced encephalitogenicity of a refolded version of rHuMOG.
Refolding rHuMOG affected both solubility of the protein, i.e. it became completely soluble in PBS as well as the affinity of the conformational 8-18C5 antibody for the refolded protein. The MOG antibody 8-18C5 mediates in vitro and in vivo demyelination and recognizes a discontinuous epitope consisting of three different loops in the extracellular part of the protein (30). Although 8-18C5 recognizes a conformational MOG epitope, it has been widely used as a positive control in Western blotting assays (performed under denaturing conditions) for the detection of MOG antibodies. In line with these results, we show that although 8-18C5 has a residual affinity for non-refolded rHuMOGPBS and rHuMOGAC as measured by ELISA, the antibody reacts much stronger with rHuMOGREF.
It has been reported previously that MOG solubility influences EAE severity (19, 26, 35) in such a way that more soluble preparations tend to be more pathogenic. To distinguish effects due to protein solubility from effects due to protein conformation, we decided to include non-refolded rHuMOG solubilized by dialysis against a low pH acetate buffer in this study. Immunization of DA rats with rHuMOGREF lead to a rapid onset, acute type of EAE, which was frequently fatal and was histopathologically characterized by massive inflammation in the presence of sparse demyelination. These results are in line with previous observations showing a similar histopathological pattern in early stages of acutely progressing MOG-induced EAE (19, 35). The aggregated form of the protein could not induce disease in DA rats, whereas the non-refolded but soluble form of the protein led to a moderate type of EAE with late onset and with a lower incidence than rHuMOGREF. From these data, we conclude that both protein solubility and conformation influence disease outcome and severity.
We could not detect significant differences in anti-MOG IgG production between different groups as measured by ELISA, irrespective of the rHuMOG preparation that was used for coating the 96-well plates. Although it was previously suggested that even after extensive refolding, recombinant bacterial MOG denatures during ELISA protocols (34), we consistently measured strong reactions of polystyrene-coated rHuMOGREF with the conformational 8-18C5 antibody (36). An alternative explanation for these results might be offered by the fact that part of the antibody responses in the immunized animals could be raised against trace amounts of host-derived contaminants or against the His tag and thus are irrelevant for disease.
Alternatively, we measured anti-MOG IgG responses using FACS: this cell-based assay specifically measures antibodies against eukaryotic, conformationally folded, human MOG expressed in the cell-membrane of transfected LTK-cells (7, 31). Only DA rats immunized with rHuMOGREF had a markedly higher proportion of conformational MOG antibodies in their sera than rHuMOGPBS or rHuMOGAC immunized animals. Therefore, we conclude that rHuMOG conformation and not rHuMOG solubility has a crucial influence on the generation of conformational MOG antibodies.
Next, we explored whether MOG solubility/conformation also had an influence on MOG-specific T-cell responses. When measuring recall responses against different MOG preparations and peptides after immunization, we made the following observations: 1) lack of disease in DA rats immunized with the aggregated protein was not due to the lack of a MOG-specific T-cell response. T-cell responses were detectable both against rRMOG and rHuMOGPBS/REF, and these responses were comparable or even higher than responses in animals immunized with rHuMOGAC. 2) The human version of the main encephalitogenic peptide of rRMOG, MOG 91–108 cross-reacts with its rat equivalent. 3) T-cell responses against MOG 91-108 and MOG 93–108 are completely absent in rHuMOGPBS-immunized rats, whereas these responses are very high in rHuMOGREF-rats and intermediate in rHuMOGAC-immunized rats. These results suggest that in the case of MOG 91–108 peptide-specific T-cell responses, solubility as well as conformation contribute to immunogenicity of the MOG preparation used for immunization. The amount of T-cell responses generated against the main encephalitogenic MOG epitope as measured by IFN-γ ELISPOT correlates quite well with the degree of disease. Results obtained by adoptive transfer experiments with lymphocytes from rHuMOGPBS, rHuMOGAC, or rHuMOGREF-immunized animals are in complete agreement with these data as we found that rHuMOGREF lymphocytes are more encephalitogenic than rHuMOGAC lymphocytes, whereas rHuMOGPBS lymhocytes were non-encephalitogenic.
The question remains as to why such a marked influence of protein conformation and solubility is observed on the specificity of the ensuing T-cell responses. Mapping rHuMOGIgd T-cell epitope specifities after rHuMOGPBS and rHuMOGREF immunization using MOG peptides spanning the complete rHuMOGIgd region did not reveal the presence of qualitatively different responses between both groups. Indeed, the differences were restricted to a strongly enhanced T-cell response against the main encephalitogenic MOG 91–108 epitope present in rHuMOGREF-immunized rats. This quantitive effect of rHuMOG solubility/conformation on protein immunogenicity could be due to a combination of different factors related to antigen uptake and processing. First, one could envisage a differential uptake of the protein by antigen presenting cells depending on the aggregational state of the protein. Second, it is known that susceptibility of protein antigens to lysosomal proteolysis plays an important role in determining immunogenicity (37). In that respect, it is interesting to note that non-refolded aggregated rMOG as present in inclusion bodies consists of intermolecular associations of exposed hydrophobic surfaces on folding intermediates resistant to proteolytic degredation (38) and thus might be poorly available for antigen processing, which might explain the absent MOG 91–108 T-cell responses after immunization with the aggregated form of the protein.
In conclusion, our results demonstrate that in EAE, the conformation of the rHuMOG protein used for immunization strongly influences the presence of conformational antibodies. Importantly, rHuMOG conformation also has a very strong impact on T-cell immunogenicity. Whether this effect is directly due to protein conformation, which might influence APC uptake and antigen processing, or whether altered T-cell immunogenicity is mainly a consequence of protein solubility, remains to be determined. It was previously shown that, at least in the case of myelin basic protein, the native protein and the recombinant protein behave antigenically similar as far as B- and T-cell responses are concerned (39). This is in stark contrast to our findings with MOG. We believe that our results have a wide relevance since the data imply that it is important to use antigens in a form which is similar to the native antigen not only when looking at antibody responses, but even when considering T-cell responses to this (auto)antigen.
This work was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft).
- MS
- multiple sclerosis
- DA
- dark agouti
- CM
- complete medium
- EAE
- experimental autoimmune encephalomyelitis
- LN
- lymph node
- MOG
- myelin oligodendrocyte glycoprotein
- MNC
- mononuclear cell
- rHuMOG
- recombinant human MOG 1–125
- rRMOG
- recombinant rat MOG 1–125
- RT1av1
- MHC of the DA rat
- Ni-NTA
- nickel-nitrilotriacetic acid.
REFERENCES
- 1. Sospedra M., Martin R. (2005) Immunology of multiple sclerosis. Annu. Rev. Immunol. 23, 683–747 [DOI] [PubMed] [Google Scholar]
- 2. Fissolo N., Haag S., de Graaf K. L., Drews O., Stevanovic S., Rammensee H. G., Weissert R. (2009) Naturally presented peptides on major histocompatibility complex I and II molecules eluted from central nervous system of multiple sclerosis patients. Mol. Cell Proteomics 8, 2090–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schluesener H. J., Sobel R. A., Linington C., Weiner H. L. (1987) A monoclonal antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central nervous system autoimmune disease. J. Immunol. 139, 4016–4021 [PubMed] [Google Scholar]
- 4. Linington C., Bradl M., Lassmann H., Brunner C., Vass K. (1988) Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am. J. Pathol. 130, 443–454 [PMC free article] [PubMed] [Google Scholar]
- 5. Genain C. P., Nguyen M. H., Letvin N. L., Pearl R., Davis R. L., Adelman M., Lees M. B., Linington C., Hauser S. L. (1995) Antibody facilitation of multiple sclerosis-like lesions in a nonhuman primate. J. Clin. Invest. 96, 2966–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brunner C., Lassmann H., Waehneldt T. V., Matthieu J. M., Linington C. (1989) Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2′,3′-cyclic nucleotide 3′-phosphodiesterase in the CNS of adult rats. J. Neurochem. 52, 296–304 [DOI] [PubMed] [Google Scholar]
- 7. Brehm U., Piddlesden S. J., Gardinier M. V., Linington C. (1999) Epitope specificity of demyelinating monoclonal autoantibodies directed against the human myelin oligodendrocyte glycoprotein (MOG). J. Neuroimmunol. 97, 9–15 [DOI] [PubMed] [Google Scholar]
- 8. von Büdingen H. C., Hauser S. L., Ouallet J. C., Tanuma N., Menge T., Genain C. P. (2004) Frontline: Epitope recognition on the myelin/oligodendrocyte glycoprotein differentially influences disease phenotype and antibody effector functions in autoimmune demyelination. Eur. J. Immunol. 34, 2072–2083 [DOI] [PubMed] [Google Scholar]
- 9. Berger T., Rubner P., Schautzer F., Egg R., Ulmer H., Mayringer I., Dilitz E., Deisenhammer F., Reindl M. (2003) Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N. Engl. J. Med. 349, 139–145 [DOI] [PubMed] [Google Scholar]
- 10. Lindert R. B., Haase C. G., Brehm U., Linington C., Wekerle H., Hohlfeld R. (1999) Multiple sclerosis: B- and T-cell responses to the extracellular domain of the myelin oligodendrocyte glycoprotein. Brain 122, 2089–2100 [DOI] [PubMed] [Google Scholar]
- 11. Lalive P. H., Menge T., Delarasse C., Della Gaspera B., Pham-Dinh D., Villoslada P., von Büdingen H. C., Genain C. P. (2006) Antibodies to native myelin oligodendrocyte glycoprotein are serologic markers of early inflammation in multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 103, 2280–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou D., Srivastava R., Nessler S., Grummel V., Sommer N., Brück W., Hartung H. P., Stadelmann C., Hemmer B. (2006) Identification of a pathogenic antibody response to native myelin oligodendrocyte glycoprotein in multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 103, 19057–19062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McLaughlin K. A., Chitnis T., Newcombe J., Franz B., Kennedy J., McArdel S., Kuhle J., Kappos L., Rostasy K., Pohl D., Gagne D., Ness J. M., Tenembaum S., O'Connor K. C., Viglietta V., Wong S. J., Tavakoli N. P., de Seze J., Idrissova Z., Khoury S. J., Bar-Or A., Hafler D. A., Banwell B., Wucherpfennig K. W. (2009) Age-dependent B-cell autoimmunity to a myelin surface antigen in pediatric multiple sclerosis. J. Immunol. 183, 4067–4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lampasona V., Franciotta D., Furlan R., Zanaboni S., Fazio R., Bonifacio E., Comi G., Martino G. (2004) Similar low frequency of anti-MOG IgG and IgM in MS patients and healthy subjects. Neurology 62, 2092–2094 [DOI] [PubMed] [Google Scholar]
- 15. O'Connor K. C., McLaughlin K. A., De Jager P. L., Chitnis T., Bettelli E., Xu C., Robinson W. H., Cherry S. V., Bar-Or A., Banwell B., Fukaura H., Fukazawa T., Tenembaum S., Wong S. J., Tavakoli N. P., Idrissova Z., Viglietta V., Rostasy K., Pohl D., Dale R. C., Freedman M., Steinman L., Buckle G. J., Kuchroo V. K., Hafler D. A., Wucherpfennig K. W. (2007) Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat. Med. 13, 211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gaertner S., de Graaf K. L., Greve B., Weissert R. (2004) Antibodies against glycosylated native MOG are elevated in patients with multiple sclerosis. Neurology 63, 2381–2383 [DOI] [PubMed] [Google Scholar]
- 17. Allamargot C., Gardinier M. V. (2007) Alternative isoforms of myelin/oligodendrocyte glycoprotein with variable cytoplasmic domains are expressed in human brain. J. Neurochem. 101, 298–312 [DOI] [PubMed] [Google Scholar]
- 18. Weissert R., Wallström E., Storch M. K., Stefferl A., Lorentzen J., Lassmann H., Linington C., Olsson T. (1998) MHC haplotype-dependent regulation of MOG-induced EAE in rats. J. Clin. Invest. 102, 1265–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Storch M. K., Stefferl A., Brehm U., Weissert R., Wallström E., Kerschensteiner M., Olsson T., Linington C., Lassmann H. (1998) Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol. 8, 681–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kornek B., Storch M. K., Weissert R., Wallstroem E., Stefferl A., Olsson T., Linington C., Schmidbauer M., Lassmann H. (2000) Multiple sclerosis and chronic autoimmune encephalomyelitis: A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 157, 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hjelmström P., Juedes A. E., Fjell J., Ruddle N. H. (1998) B-cell-deficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J. Immunol. 161, 4480–4483 [PubMed] [Google Scholar]
- 22. Lyons J. A., San M., Happ M. P., Cross A. H. (1999) B-cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur. J. Immunol. 29, 3432–3439 [DOI] [PubMed] [Google Scholar]
- 23. Lyons J. A., Ramsbottom M. J., Cross A. H. (2002) Critical role of antigen-specific antibody in experimental autoimmune encephalomyelitis induced by recombinant myelin oligodendrocyte glycoprotein. Eur. J. Immunol. 32, 1905–1913 [DOI] [PubMed] [Google Scholar]
- 24. Oliver A. R., Lyon G. M., Ruddle N. H. (2003) Rat and human myelin oligodendrocyte glycoproteins induce experimental autoimmune encephalomyelitis by different mechanisms in C57BL/6 mice. J. Immunol. 171, 462–468 [DOI] [PubMed] [Google Scholar]
- 25. Marta C. B., Oliver A. R., Sweet R. A., Pfeiffer S. E., Ruddle N. H. (2005) Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc. Natl. Acad. Sci. U.S.A. 102, 13992–13997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adelmann M., Wood J., Benzel I., Fiori P., Lassmann H., Matthieu J. M., Gardinier M. V., Dornmair K., Linington C. (1995) The N-terminal domain of the myelin oligodendrocyte glycoprotein (MOG) induces acute demyelinating experimental autoimmune encephalomyelitis in the Lewis rat. J. Neuroimmunol. 63, 17–27 [DOI] [PubMed] [Google Scholar]
- 27. Amor S., Groome N., Linington C., Morris M. M., Dornmair K., Gardinier M. V., Matthieu J. M., Baker D. (1994) Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J. Immunol. 153, 4349–4356 [PubMed] [Google Scholar]
- 28. Liñares D., Echevarria I., Mañá P. (2004) Single-step purification and refolding of recombinant mouse and human myelin oligodendrocyte glycoprotein and induction of EAE in mice. Protein Expr. Purif. 34, 249–256 [DOI] [PubMed] [Google Scholar]
- 29. Linnington C., Webb M., Woodhams P. L. (1984) A novel myelin-associated glycoprotein defined by a mouse monoclonal antibody. J. Neuroimmunol. 6, 387–396 [DOI] [PubMed] [Google Scholar]
- 30. Breithaupt C., Schubart A., Zander H., Skerra A., Huber R., Linington C., Jacob U. (2003) Structural insights into the antigenicity of myelin oligodendrocyte glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 100, 9446–9451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bourquin C., Schubart A., Tobollik S., Mather I., Ogg S., Liblau R., Linington C. (2003) Selective unresponsiveness to conformational B cell epitopes of the myelin oligodendrocyte glycoprotein in H-2b mice. J. Immunol. 171, 455–461 [DOI] [PubMed] [Google Scholar]
- 32. Weissert R., de Graaf K. L., Storch M. K., Barth S., Linington C., Lassmann H., Olsson T. (2001) MHC class II-regulated central nervous system autoaggression and T-cell responses in peripheral lymphoid tissues are dissociated in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J. Immunol. 166, 7588–7599 [DOI] [PubMed] [Google Scholar]
- 33. Herrmann M. M., Gaertner S., Stadelmann C., van den Brandt J., Böscke R., Budach W., Reichardt H. M., Weissert R. (2005) Tolerance induction by bone marrow transplantation in a multiple sclerosis model. Blood 106, 1875–1883 [DOI] [PubMed] [Google Scholar]
- 34. Mathey E., Breithaupt C., Schubart A. S., Linington C. (2004) Commentary: Sorting the wheat from the chaff: Identifying demyelinating components of the myelin oligodendrocyte glycoprotein (MOG)-specific autoantibody repertoire. Eur. J. Immunol. 34, 2065–2071 [DOI] [PubMed] [Google Scholar]
- 35. Sakuma H., Kohyama K., Park I. K., Miyakoshi A., Tanuma N., Matsumoto Y. (2004) Clinicopathological study of a myelin oligodendrocyte glycoprotein-induced demyelinating disease in LEW.1AV1 rats. Brain 127, 2201–2213 [DOI] [PubMed] [Google Scholar]
- 36. Breithaupt C., Schäfer B., Pellkofer H., Huber R., Linington C., Jacob U. (2008) Demyelinating myelin oligodendrocyte glycoprotein-specific autoantibody response is focused on one dominant conformational epitope region in rodents. J. Immunol. 181, 1255–1263 [DOI] [PubMed] [Google Scholar]
- 37. Delamarre L., Couture R., Mellman I., Trombetta E. S. (2006) Enhancing immunogenicity by limiting susceptibility to lysosomal proteolysis. J. Exp. Med. 203, 2049–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carrió M. M., Villaverde A. (2002) Construction and deconstruction of bacterial inclusion bodies. J. Biotechnol. 96, 3–12 [DOI] [PubMed] [Google Scholar]
- 39. Oettinger H. F., al-Sabbagh A., Jingwu Z., LaSalle J. M., Weiner H. L., Hafler D. A. (1993) Biological activity of recombinant human myelin basic protein. J. Neuroimmunol. 44, 157–162 [DOI] [PubMed] [Google Scholar]