

Abstract
The contractile response of the heart can be altered by disease-related protein modifications to numerous contractile proteins. By utilizing an IAANS labeled fluorescent troponin C, 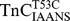 , we examined the effects of ten disease-related troponin modifications on the Ca2+ binding properties of the troponin complex and the reconstituted thin filament. The selected modifications are associated with a broad range of cardiac diseases: three subtypes of familial cardiomyopathies (dilated, hypertrophic and restrictive) and ischemia-reperfusion injury. Consistent with previous studies, the majority of the protein modifications had no effect on the Ca2+ binding properties of the isolated troponin complex. However, when incorporated into the thin filament, dilated cardiomyopathy mutations desensitized (up to 3.3-fold), while hypertrophic and restrictive cardiomyopathy mutations, and ischemia-induced truncation of troponin I, sensitized the thin filament to Ca2+ (up to 6.3-fold). Kinetically, the dilated cardiomyopathy mutations increased the rate of Ca2+ dissociation from the thin filament (up to 2.5-fold), while the hypertrophic and restrictive cardiomyopathy mutations, and the ischemia-induced truncation of troponin I decreased the rate (up to 2-fold). The protein modifications also increased (up to 5.4-fold) or decreased (up to 2.5-fold) the apparent rate of Ca2+ association to the thin filament. Thus, the disease-related protein modifications alter Ca2+ binding by influencing both the association and dissociation rates of thin filament Ca2+ exchange. These alterations in Ca2+ exchange kinetics influenced the response of the thin filament to artificial Ca2+ transients generated in a stopped-flow apparatus. Troponin C may act as a hub, sensing physiological and pathological stimuli to modulate the Ca2+-binding properties of the thin filament and influence the contractile performance of the heart.
, we examined the effects of ten disease-related troponin modifications on the Ca2+ binding properties of the troponin complex and the reconstituted thin filament. The selected modifications are associated with a broad range of cardiac diseases: three subtypes of familial cardiomyopathies (dilated, hypertrophic and restrictive) and ischemia-reperfusion injury. Consistent with previous studies, the majority of the protein modifications had no effect on the Ca2+ binding properties of the isolated troponin complex. However, when incorporated into the thin filament, dilated cardiomyopathy mutations desensitized (up to 3.3-fold), while hypertrophic and restrictive cardiomyopathy mutations, and ischemia-induced truncation of troponin I, sensitized the thin filament to Ca2+ (up to 6.3-fold). Kinetically, the dilated cardiomyopathy mutations increased the rate of Ca2+ dissociation from the thin filament (up to 2.5-fold), while the hypertrophic and restrictive cardiomyopathy mutations, and the ischemia-induced truncation of troponin I decreased the rate (up to 2-fold). The protein modifications also increased (up to 5.4-fold) or decreased (up to 2.5-fold) the apparent rate of Ca2+ association to the thin filament. Thus, the disease-related protein modifications alter Ca2+ binding by influencing both the association and dissociation rates of thin filament Ca2+ exchange. These alterations in Ca2+ exchange kinetics influenced the response of the thin filament to artificial Ca2+ transients generated in a stopped-flow apparatus. Troponin C may act as a hub, sensing physiological and pathological stimuli to modulate the Ca2+-binding properties of the thin filament and influence the contractile performance of the heart.
Introduction
The heart is a highly dynamic organ that can regulate both its contractile strength and speed to accommodate the demands of the body. Numerous cardiac diseases adversely alter the heart’s ability to properly maintain its performance [1]. It is well established that aberrant intracellular Ca2+ signaling is associated with systolic and diastolic cardiac dysfunctions [2], [3]. Cardiac diseases may also alter how the heart responds to the Ca2+ signal [4]. For instance, numerous reports have demonstrated that myofilament Ca2+ sensitivity is affected by cardiomyopathy associated protein mutations, truncations and post-translational modifications [5], [6], [7], [8]. Thus, contractile dysfunctions may be caused by both altered intracellular Ca2+ signaling and abnormal myofilament responsiveness to the Ca2+ signal [4], [9].
Troponin C (TnC) is the myofilament Ca2+ sensor in cardiac muscle responsible for translating the intracellular Ca2+ signal into mechanical force [10]. The Ca2+ sensitivity of TnC can be modulated by multiple factors, including its interactions with other myofilament proteins, post-translational modifications of the myofilament, as well as cardiac disease-related protein modifications [11], [12], [13], [14]. In this regard, TnC is not just a passive element that transmits the Ca2+ signal. Instead, it may act as a central hub that integrates information from the myofilament (beneficial or maligned) and adjusts its Ca2+ binding properties to regulate cardiac muscle mechanics [15].
While it is clear that myofilament disease-related protein modifications can alter the steady-state Ca2+ sensitivity of TnC, much less is known regarding their effects on TnC’s Ca2+ exchange kinetics [16], [17], [18]. The kinetics of Ca2+ exchange with TnC may be even more significant to how the heart performs since the heart is dynamic and does not function in a static steady-state. Furthermore, it is the kinetics of Ca2+ exchange with TnC, including the rate of Ca2+ association to and dissociation from TnC that determine its steady state Ca2+ sensitivity. The rate of Ca2+ association is 3 to 4 orders of magnitude faster than the rate of Ca2+ dissociation, and considered diffusion controlled (for review see [15]). Thus, it is generally assumed that changes in the steady-state Ca2+ sensitivity of TnC are caused exclusively by modulating the rate of Ca2+ dissociation. However, our previous studies suggest the rate of Ca2+ association to TnC can also be altered [19], [20].
In this work, disease-related protein modifications of troponin I (TnI) and troponin T (TnT) were selected to systematically study their effects on Ca2+ binding and exchange with the troponin complex (Tn) and the thin filament using a fluorescently labeled TnC. The protein modifications include five dilated cardiomyopathy (DCM) mutations (TnI K36Q, TnT R141W, TnT R131W, TnT R205L, and TnT ΔK210), two hypertrophic cardiomyopathy (HCM) mutations (TnI S166F and TnT R92Q), two restrictive cardiomyopathy (RCM) mutations (TnI D190H and TnI R192H), and an ischemia-induced truncation of TnI (residues 1-192). These protein modifications represent a broad spectrum of diseases that change the steady-state Ca2+ sensitivity of the actomyosin ATPase activity and/or force generation [12], [13], [21], [22]. More importantly, some of these protein modifications also alter the kinetics and amplitude of cardiac muscle contraction and/or relaxation [23], [24]. We demonstrate that these disease associated protein modifications adversely altered steady-state Ca2+ binding to TnC by influencing both its Ca2+ association and dissociation rates on the thin filament (with almost no effect when measured in the isolated Tn complex). These results suggest that TnC acts as a central hub on the thin filament by sensing pathological stimuli to alter cardiac contractile properties.
Materials and Methods
Materials
Phenyl-Sepharose CL-4B, Tween-20, and EGTA were purchased from Sigma Chemical Co. (St. Louis, MO). IAANS and phalloidin were purchased from Invitrogen (Carlsbad, CA). Affi-Gel 15 affinity media was purchased from Bio-Rad (Hercules, CA).
Protein Mutagenesis
The pET3a plasmid encoding human cardiac TnC was a generous gift from Dr. Lawrence Smillie (University of Alberta, Canada). The pET3a plasmids encoding human cardiac TnI and TnT were graciously provided by Dr. James Potter (University of Miami, FL). TnC, TnI and TnT mutants were constructed from their respective pET3a plasmids using the primer-based QuikChange Site-Directed Mutagenesis Kit (Stratagene, Santa Clara, CA) as previously described [11]. The mutations were confirmed by DNA sequence analysis at an on-site molecular genetics core facility.
Protein Purification
The plasmid encoding human cardiac TnC was transformed into E. coli BL21(DE3)pLysS cells (Novagen, San Diego, CA), while the TnI and TnT plasmids were transformed into Rosetta™(DE3)pLysS cells (Novagen, San Diego, CA). The proteins were expressed and purified as previously described [25].
Rabbit skeletal actin and bovine ventricular tropomyosin (Tm) were purified from acetone powders as previously described [26], [27]. Fresh bovine cardiac muscle was purchased from The Herman Falter Packing Company (Columbus, OH). All the animal protocols and procedures were performed in accordance with the National Institutes of Health Guidelines and approved by the Institutional Laboratory Animal Care and Use Committee at The Ohio State University.
Fluorescent Labeling
TnCC35S,T53C,C84S (herein denoted as TnCT53C) was labeled with the environmentally sensitive thiol-reactive fluorescent probe IAANS as previously described [25].
Reconstitution of Troponin Complexes and Regulated Thin Filaments
The reconstituted Tn complexes and regulated thin filaments were prepared as previously described [25]. All the Tn complexes contain full length Tn subunits of recombinant human 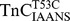 , TnI (except for the truncated TnI (1-192)) and TnT.
, TnI (except for the truncated TnI (1-192)) and TnT.
Steady-State Fluorescence Measurements
All steady-state fluorescence measurements were performed using a Perkin-Elmer LS55 spectrofluorimeter at 15°C. IAANS fluorescence was excited at 330 nm and monitored at 450 nm as microliter amounts of CaCl2 were added to 2 ml of each labeled Tn complex (0.15 µM) in a titration buffer (200 mM MOPS (to prevent pH changes upon addition of Ca2+), 150 mM KCl, 2 mM EGTA, 1 mM DTT, 3 mM MgCl2, 0.02% Tween-20, pH 7.0) with constant stirring. Reconstituted thin filaments were titrated in an identical buffer composition (excluding Tween-20). The [Ca2+]free was calculated using the computer program EGCA02 developed by Robertson and Potter [28]. The Ca2+ sensitivities were reported as a dissociation constant Kd, representing a mean of three to four separate titrations ± S.E.M. The data were fit with a logistic sigmoid function (mathematically equivalent to the Hill equation), as previously described [29].
Stopped-Flow Fluorescent Measurements
Ca2+ exchange rates were characterized using an Applied Photophysics model SX.20 stopped-flow instrument with a dead time of 1.4 ms at 15°C. IAANS fluorescence was excited at 330 nm. The IAANS emission was monitored through either a 420-470 nm band-pass interference filter for 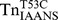 , or a 510 nm broad band-pass interference filter for the thin filament. The filters were purchased from Oriel (Stratford, CT). Data traces (an average of 3 to 5 individual traces) were fit with a single exponential equation to calculate the kinetic rates. The working buffer used for the kinetic measurements was 10 mM MOPS, 150 mM KCl, 1 mM DTT, 3 mM MgCl2, 0.02% Tween-20 (excluded for thin filament kinetic measurements), at pH 7.0. 10 mM EGTA was utilized to remove 200 µM Ca2+ from the Tn complexes or thin filaments.
, or a 510 nm broad band-pass interference filter for the thin filament. The filters were purchased from Oriel (Stratford, CT). Data traces (an average of 3 to 5 individual traces) were fit with a single exponential equation to calculate the kinetic rates. The working buffer used for the kinetic measurements was 10 mM MOPS, 150 mM KCl, 1 mM DTT, 3 mM MgCl2, 0.02% Tween-20 (excluded for thin filament kinetic measurements), at pH 7.0. 10 mM EGTA was utilized to remove 200 µM Ca2+ from the Tn complexes or thin filaments.
In order to measure the rate of Ca2+ association to the thin filament, a minimal concentration of EGTA was added to the thin filament mixture to remove contaminating Ca2+. The amount of EGTA added was determined by steady state titrations. For the control, TnI (1-192), TnT ΔK210, TnT R131W thin filaments, 5 µM EGTA was sufficient. For TnI R192H and TnI D190H thin filaments, 10 µM EGTA was required. The observed rates at various Ca2+ concentrations were plotted against the Ca2+ concentration and fit with a linear regression. The rate of Ca2+ association to the thin filament was obtained by calculating the slope of the fit [30]. The buffer used for the Ca2+ association rate measurements was the same as that used for the Ca2+ dissociation rate measurements.
Exposure of the Thin Filament to Artificial Ca2+ Transients (ACTs)
In order to determine the potential effects of altering the Ca2+ association rate to the thin filament, the IAANS labeled TnCs on the thin filament were exposed to artificial Ca2+ transients (ACTs) of increasing amplitude as previously described [19], [31]. ACTs were generated in the stopped-flow apparatus by rapidly mixing the thin filament (at a concentration and buffer described above for the rates of Ca2+ dissociation) in the presence of 500 µM EGTA (after mixing) against increasing [Ca2+]. This technique exposes the thin filament to rapid transient fluxes of Ca2+ (∼ 1 ms half life). A relatively high concentration of EGTA (500 µM after mixing) was utilized to ensure that the thin filament is not pre-bound by Ca2+ and to provide a large enough Ca2+ sink to effectively remove the free [Ca2+] after mixing, effectively generating a Ca2+ transient. The amplitude of the Ca2+ transient can be adjusted by rapidly mixing the thin filament with different amounts of Ca2+. The ability of a Ca2+-binding protein (in this case, TnC on the thin filament) to respond to these rapid Ca2+ transients, or become transiently occupied by Ca2+, is a function of the Ca2+ association rate of the Ca2+-binding protein [31]. In order to determine the maximal response of the thin filaments to Ca2+, high [Ca2+] (1 mM after mixing) was used to exceed the [EGTA] and saturate the thin filament. Analysis of the response of the thin filament to the ACTs is described in the legend of Figure 7.
Figure 7. Response of disease-related protein modifications to ACTs with increasing amplitude.

Panels A, B and C show responses of thin filament bound control 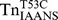 , TnI (1-192)
, TnI (1-192) 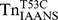 and TnT ▵K210
and TnT ▵K210 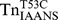 to ACTs, respectively. The peak transient occupancy was determined at ∼3 ms for each sub-saturating [Ca2+]. 100% occupancy was determined at the plateau of the trace in which saturating Ca2+ (1000 µM after mixing) was rapidly mixed with the thin filament. 0% occupancy was determined by mixing the thin filament without Ca2+ (data not shown). Each transient occupancy calculation was an average of three separate experiments repeated twice, with each trace being an average of at least 5 separate traces. The 25 µM Ca2+ data for TnT ▵K210 is not shown for clarity. All
to ACTs, respectively. The peak transient occupancy was determined at ∼3 ms for each sub-saturating [Ca2+]. 100% occupancy was determined at the plateau of the trace in which saturating Ca2+ (1000 µM after mixing) was rapidly mixed with the thin filament. 0% occupancy was determined by mixing the thin filament without Ca2+ (data not shown). Each transient occupancy calculation was an average of three separate experiments repeated twice, with each trace being an average of at least 5 separate traces. The 25 µM Ca2+ data for TnT ▵K210 is not shown for clarity. All 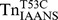 complexes consist of the full length Tn subunits of
complexes consist of the full length Tn subunits of 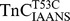 , TnI and TnT, except for ischemic related truncated TnI (1-192). The disease related modification is either in TnI or TnT, in either case, the other protein (TnT or TnI) was wild type.
, TnI and TnT, except for ischemic related truncated TnI (1-192). The disease related modification is either in TnI or TnT, in either case, the other protein (TnT or TnI) was wild type.
Data Analysis and Statistics
Statistical significance was determined by ANOVA followed by a Dunnett’s post-hoc t-test using the statistical analysis software Minitab (State College, PA). Two means were considered to be significantly different when the P value was < 0.05. The data is shown as a mean value ± S.E.M.
Results
Effects of the Protein Modifications on the Ca2+ Sensitivity of the Tn Complex
The data were divided into four subgroups according to the disease subtypes to facilitate data presentation. The effects of the disease-related protein modifications on the Ca2+ binding properties of the Tn complex were examined since Tn is the simplest biochemical system to test the effects of TnI and TnT modifications on TnC. Our laboratory has developed a fluorescent TnC, 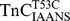 (which is specifically labeled with the fluorescent probe IAANS on Cys 53) that minimally affects TnC function, and reports the structural changes that occur within the regulatory domain of TnC upon Ca2+ binding and dissociation [11].
(which is specifically labeled with the fluorescent probe IAANS on Cys 53) that minimally affects TnC function, and reports the structural changes that occur within the regulatory domain of TnC upon Ca2+ binding and dissociation [11]. 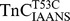 was incorporated into all the Tn complexes (denoted as
was incorporated into all the Tn complexes (denoted as 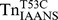 ), which enabled us to follow Ca2+ binding and exchange with isolated or thin filament bound Tn complexes.
), which enabled us to follow Ca2+ binding and exchange with isolated or thin filament bound Tn complexes.
The Ca2+ sensitivity of TnC within the various Tn complexes was measured by following the Ca2+ dependent decrease in IAANS fluorescence. Control 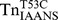 exhibited a Ca2+ induced half-maximal fluorescence decrease at 0.66±0.03 µM (Figure 1 and Table 1). As shown in figure 1, only HCM TnI S166F significantly increased the Ca2+ sensitivity of the Tn complex (∼ 1.5-fold; Figure 1 and Table 1).
exhibited a Ca2+ induced half-maximal fluorescence decrease at 0.66±0.03 µM (Figure 1 and Table 1). As shown in figure 1, only HCM TnI S166F significantly increased the Ca2+ sensitivity of the Tn complex (∼ 1.5-fold; Figure 1 and Table 1).
Figure 1. Effect of disease-related protein modifications on the Ca2+ sensitivity of the Tn complex.

Panel A shows the Ca2+ dependent decreases in IAANS fluorescence for control 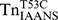 (□), and the DCM mutants, TnI K36Q
(□), and the DCM mutants, TnI K36Q 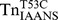 (?), TnT R131W
(?), TnT R131W 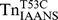 (○), TnT R141W
(○), TnT R141W 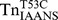 (•), TnT R205L
(•), TnT R205L 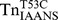 (▪) and TnT ΔK210
(▪) and TnT ΔK210 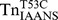 (⋆) as a function of pCa. Panel B shows the Ca2+ dependent decreases in IAANS fluorescence for control
(⋆) as a function of pCa. Panel B shows the Ca2+ dependent decreases in IAANS fluorescence for control 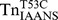 (□), and the HCM mutants, TnT R92Q
(□), and the HCM mutants, TnT R92Q 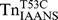 (▾) and TnI S166F
(▾) and TnI S166F 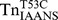 (▴) as a function of pCa. Panel C shows the Ca2+ dependent decreases in IAANS fluorescence for control
(▴) as a function of pCa. Panel C shows the Ca2+ dependent decreases in IAANS fluorescence for control 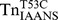 (□), and the RCM mutants, TnI D190H
(□), and the RCM mutants, TnI D190H 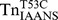 (▽) and TnI R192H
(▽) and TnI R192H 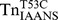 (▵) as a function of pCa. Panel D shows the Ca2+ dependent decreases in IAANS fluorescence for control
(▵) as a function of pCa. Panel D shows the Ca2+ dependent decreases in IAANS fluorescence for control 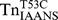 (□) and ischemic related truncated TnI (1-192)
(□) and ischemic related truncated TnI (1-192) 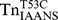 (
(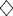 ) as a function of pCa. The data sets were normalized individually for each mutant. All
) as a function of pCa. The data sets were normalized individually for each mutant. All 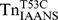 complexes consist of the full length Tn subunits of
complexes consist of the full length Tn subunits of 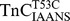 , TnI and TnT, except for ischemic related truncated TnI (1-192). The disease related modification is either in TnI or TnT, in either case, the other protein (TnT or TnI) was wild type. The Ca2+ sensitivities were reported as a dissociation constant Kd, representing a mean of three to four separate titrations ± S.E.M.
, TnI and TnT, except for ischemic related truncated TnI (1-192). The disease related modification is either in TnI or TnT, in either case, the other protein (TnT or TnI) was wild type. The Ca2+ sensitivities were reported as a dissociation constant Kd, representing a mean of three to four separate titrations ± S.E.M.
Table 1. Effect of disease-related protein modifications on the Ca2+ binding properties of the Tn complex.
| Protein | Disease Subtype | Tn complex Ca2+ Kd (µM) | Tn complex nH | Tn complex Ca2+ koff (/s) |
| Control | – | 0.66±0.03 | 0.90±0.02 | 40.8±0.4 |
| TnI K36Q | DCM | 0.68±0.07 | 0.95±0.05 | 48.5±0.3* |
| TnT R141W | DCM | 0.71±0.09 | 0.87±0.04 | 39.3±0.3 |
| TnT R131W | DCM | 0.9±0.2 | 0.80±0.04 | 42.0±0.6 |
| TnT R205L | DCM | 0.80±0.06 | 0.86±0.06 | 41.6±0.5 |
| TnT ΔK210 | DCM | 0.61±0.01 | 0.86±0.01 | 40.7±0.5 |
| TnT R92Q | HCM | 0.73±0.06 | 0.85±0.05 | 40±2 |
| TnI S166F | HCM | 0.43±0.04* | 0.96±0.04 | 22.5±0.1* |
| TnI D190H | RCM | 0.62±0.05 | 0.95±0.04 | 40.3±0.4 |
| TnI R192H | RCM | 0.674±0.007 | 0.89±0.03 | 41.2±0.6 |
| TnI (1-192) | Ischemia injury | 0.59±0.03 | 1.00±0.07 | 42.0±0.8 |
Values marked with * are significantly different from their respective control values (p<0.05).
Effects of the Protein Modifications on the Rate of Ca2+ Dissociation from the Tn Complex
Previously, we demonstrated that the fluorescence of 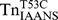 reported the actual rate of Ca2+ dissociation from unlabeled wild type Tn and a series of rationally engineered mutant Tn complexes with high fidelity [11], [25]. In this study, fluorescence stopped-flow measurements were performed to determine the rate of Ca2+ dissociation from the disease-related
reported the actual rate of Ca2+ dissociation from unlabeled wild type Tn and a series of rationally engineered mutant Tn complexes with high fidelity [11], [25]. In this study, fluorescence stopped-flow measurements were performed to determine the rate of Ca2+ dissociation from the disease-related 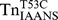 complexes. Figure 2 shows that the rate of Ca2+ dissociation from control
complexes. Figure 2 shows that the rate of Ca2+ dissociation from control 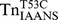 was 40.8±0.4/s (Table 1). Consistent with its effect on the Ca2+ sensitivity of Tn, HCM TnI S166F slowed the rate of Ca2+ dissociation ∼2-fold (Figure 2 and Table 1). Besides TnI S166F, only TnI K36Q slightly, but significantly altered the rate of Ca2+ dissociation from the Tn complex (∼ 1.2-fold faster; Figure 2A and Table 1).
was 40.8±0.4/s (Table 1). Consistent with its effect on the Ca2+ sensitivity of Tn, HCM TnI S166F slowed the rate of Ca2+ dissociation ∼2-fold (Figure 2 and Table 1). Besides TnI S166F, only TnI K36Q slightly, but significantly altered the rate of Ca2+ dissociation from the Tn complex (∼ 1.2-fold faster; Figure 2A and Table 1).
Figure 2. Effect of disease-related protein modifications on the rate of Ca2+ dissociation from the Tn complex.

Panel A shows the time courses of the increase in IAANS fluorescence as Ca2+ was removed by EGTA from control 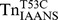 , and the DCM mutants, TnI K36Q
, and the DCM mutants, TnI K36Q 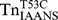 , TnT R131W
, TnT R131W 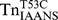 , TnT R141W
, TnT R141W 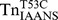 , TnT R205L
, TnT R205L 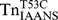 and TnT ΔK210
and TnT ΔK210 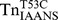 . Panel B shows the time courses of the increase in IAANS fluorescence as Ca2+ was removed by EGTA from control
. Panel B shows the time courses of the increase in IAANS fluorescence as Ca2+ was removed by EGTA from control 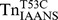 , and the HCM mutants, TnT R92Q
, and the HCM mutants, TnT R92Q 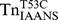 and TnI S166F
and TnI S166F 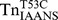 . Panel C shows the time courses of the increase in IAANS fluorescence as Ca2+ was removed by EGTA from control
. Panel C shows the time courses of the increase in IAANS fluorescence as Ca2+ was removed by EGTA from control 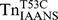 , and the RCM mutants, TnI D190H
, and the RCM mutants, TnI D190H 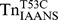 and TnI R192H
and TnI R192H 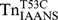 . Panel D shows the time courses of the increase in IAANS fluorescence as Ca2+ was removed by EGTA from control
. Panel D shows the time courses of the increase in IAANS fluorescence as Ca2+ was removed by EGTA from control 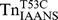 and ischemic related truncated TnI (1-192)
and ischemic related truncated TnI (1-192) 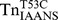 . All
. All 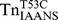 complexes consist of the full length Tn subunits of
complexes consist of the full length Tn subunits of 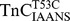 , TnI and TnT, except for ischemic related truncated TnI (1-192). The disease related modification is either in TnI or TnT, in either case, the other protein (TnT or TnI) was wild type. Data traces (an average of 3 to 5 individual traces collected at least 10 times) were fit with a single exponential equation to calculate the kinetic rates. The data traces have been staggered and normalized for clarity.
, TnI and TnT, except for ischemic related truncated TnI (1-192). The disease related modification is either in TnI or TnT, in either case, the other protein (TnT or TnI) was wild type. Data traces (an average of 3 to 5 individual traces collected at least 10 times) were fit with a single exponential equation to calculate the kinetic rates. The data traces have been staggered and normalized for clarity.
Effects of the Protein Modifications on the Ca2+ Sensitivity of the Thin Filament
Consistent with previous studies [6], [12], [13], our results indicate that the majority of the disease-related protein modifications have no effect on the Ca2+ binding properties of Tn. However, Tn is part of the thin filament system. Accordingly, we examined the effects of TnI and TnT modifications on the Ca2+ binding properties of the reconstituted thin filament.
Thin filament bound control 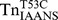 exhibited a Ca2+-dependent half-maximal fluorescence increase at 4.5±0.2 µM (Figure 3 and Table 2). Consistent with the general Ca2+ desensitizing effects of DCM mutations [5], [13], all five DCM mutations studied here desensitized Ca2+ binding to the thin filament. TnT R131W, R141W and R205L slightly desensitized Ca2+ binding to the thin filament by ∼ 1.5 fold; TnI K36Q had an intermediate Ca2+ desensitizing effect of ∼ 2-fold; while TnT ΔK210 had the largest Ca2+ desensitizing effect of ∼ 3.5-fold (Figure 3C and Table 2). Of the Ca2+ desensitizing modifications, only TnT ΔK210 significantly decreased the Ca2+ binding cooperativity of the thin filament by ∼ 1.3-fold (Figure 3C and Table 2).
exhibited a Ca2+-dependent half-maximal fluorescence increase at 4.5±0.2 µM (Figure 3 and Table 2). Consistent with the general Ca2+ desensitizing effects of DCM mutations [5], [13], all five DCM mutations studied here desensitized Ca2+ binding to the thin filament. TnT R131W, R141W and R205L slightly desensitized Ca2+ binding to the thin filament by ∼ 1.5 fold; TnI K36Q had an intermediate Ca2+ desensitizing effect of ∼ 2-fold; while TnT ΔK210 had the largest Ca2+ desensitizing effect of ∼ 3.5-fold (Figure 3C and Table 2). Of the Ca2+ desensitizing modifications, only TnT ΔK210 significantly decreased the Ca2+ binding cooperativity of the thin filament by ∼ 1.3-fold (Figure 3C and Table 2).
Figure 3. Effect of disease-related protein modifications on the Ca2+ binding sensitivity of the thin filament.

Panel A shows the Ca2+ dependent increases in IAANS fluorescence for thin filament bound control 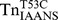 (□), and the DCM mutants, TnI K36Q
(□), and the DCM mutants, TnI K36Q 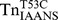 (
( ), TnT R131W (○)
), TnT R131W (○)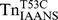 , TnT R141W
, TnT R141W 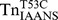 (•), TnT R205L
(•), TnT R205L 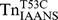 (▪) and TnT ΔK210
(▪) and TnT ΔK210 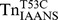 (⋆) as a function of pCa. Panel B shows the Ca2+ dependent increases in IAANS fluorescence for thin filament bound control
(⋆) as a function of pCa. Panel B shows the Ca2+ dependent increases in IAANS fluorescence for thin filament bound control 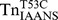 (□), and the HCM mutants, TnT R92Q
(□), and the HCM mutants, TnT R92Q 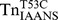 (▾) and TnI S166F
(▾) and TnI S166F 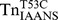 (▴) as a function of pCa. Panel C shows the Ca2+ dependent increases in IAANS fluorescence for thin filament bound control
(▴) as a function of pCa. Panel C shows the Ca2+ dependent increases in IAANS fluorescence for thin filament bound control 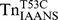 (□), and the RCM mutants, TnI D190H
(□), and the RCM mutants, TnI D190H 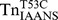 (▽) and TnI R192H
(▽) and TnI R192H 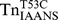 (▵) as a function of pCa. Panel D shows the Ca2+ dependent increases in IAANS fluorescence for thin filament bound control
(▵) as a function of pCa. Panel D shows the Ca2+ dependent increases in IAANS fluorescence for thin filament bound control 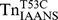 (□) and ischemic related truncated TnI (1-192)
(□) and ischemic related truncated TnI (1-192) 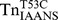 (
( ) as a function of pCa. The data sets were normalized individually for each mutant. All
) as a function of pCa. The data sets were normalized individually for each mutant. All 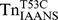 complexes consist of the full length Tn subunits of
complexes consist of the full length Tn subunits of 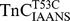 , TnI and TnT, except for ischemic related truncated TnI (1-192). The disease related modification is either in TnI or TnT, in either case, the other protein (TnT or TnI) was wild type. The Ca2+ sensitivities were reported as a dissociation constant Kd, representing a mean of three to four separate titrations ± S.E.M.
, TnI and TnT, except for ischemic related truncated TnI (1-192). The disease related modification is either in TnI or TnT, in either case, the other protein (TnT or TnI) was wild type. The Ca2+ sensitivities were reported as a dissociation constant Kd, representing a mean of three to four separate titrations ± S.E.M.
Table 2. Effect of disease-related protein modifications on the Ca2+ binding properties of the thin filament.
| Protein | Disease Subtype | TF Ca2+ Kd (µM) | Relative Changein Kd | TF nH | TF Ca2+ koff (/s) | Relative Change in koff | Calculated TF Ca2+ kon (×106 M-1s-1) | TF Ca2+ kon (×106 M-1s-1) |
| Control | – | 4.5±0.2 | – | 1.4±0.1 | 109.7±0.7 | – | 24±1 | 9±1 |
| TnI K36Q | DCM | 8.4±0.3* | ↓1.9±0.1 | 1.1±0.1 | 279±5* | ↑2.54±0.05 | 33±1 | ND |
| TnT R141W | DCM | 6.2±0.1* | ↓1.38±0.07 | 1.44±0.04 | 115.0±0.8 | 1.05±0.01 | 18.5±0.3 | ND |
| TnT R131W | DCM | 6.2±0.4* | ↓1.4±0.1 | 1.16±0.04 | 138±1* | ↑1.26±0.01 | 22±1 | 12±1 |
| TnT R205L | DCM | 7.2±0.6* | ↓1.6±0.2 | 1.7±0.1 | 166±2* | ↑1.51±0.02 | 23±2 | ND |
| TnT ΔK210 | DCM | 15±1* | ↓3.3±0.3 | 1.05±0.06* | 252±6* | ↑2.30±0.06 | 17±1 | 3.6±0.6 |
| TnT R92Q | HCM | 3.7±0.1 | ↑1.22±0.06 | 1.05±0.04* | 107.5±0.8 | 1.02±0.01 | 29.1±0.8 | ND |
| TnI S166F | HCM | 3.1±0.1* | ↑1.45±0.08 | 1.24±0.04 | 65.2±0.4* | ↓1.68±0.01 | 21.0±0.7 | ND |
| TnI D190H | RCM | 1.62±0.03* | ↑2.8±0.1 | 1.2±0.1 | 87.9±0.6* | ↓1.24±0.01 | 54±1 | 36±3 |
| TnI R192H | RCM | 1.59±0.01* | ↑2.8±0.1 | 1.17±0.05 | 72±1* | ↓1.52±0.02 | 45.3±0.7 | 48±5 |
| TnI (1-192) | Ischemia injury | 0.71±0.05* | ↑6.3±0.5 | 1.21±0.06 | 55.3±0.4* | ↓1.98±0.02 | 78±6 | 49±11 |
Values marked with * are significantly different from their respective control values (p<0.05). ND: not determined.
Consistent with the general Ca2+ sensitizing effect of HCM, RCM and ischemia-induced truncation of TnI [5], [6], four of the five TnI and TnT modifications studied here sensitized Ca2+ binding to the thin filament. As shown in figure 3B, HCM TnI S166F slightly, but significantly sensitized Ca2+ binding to the thin filament by ∼ 1.4-fold. However, the other HCM mutation R92Q did not significantly alter thin filament Ca2+ sensitivity, but significantly decreased the Ca2+ binding cooperativity of the thin filament by ∼ 1.3-fold (Figure 3B and Table 2).
Figure 3C shows that both RCM mutations, TnI D190H and TnI R192H, increased the Ca2+ sensitivity of the thin filament by ∼ 2.8-fold (Figure 3C and Table 2). Compared to the HCM and RCM mutations, which had slight and intermediate Ca2+ sensitizing effects, the ischemia-induced truncation of TnI (1-192) hyper-sensitized Ca2+ binding to thin filament by ∼ 6.3-fold (Figure 3D and Table 2).
Effect of the Protein Modifications on the Rate of Ca2+ Dissociation from the Thin Filament
It is generally assumed that a change in the Ca2+ sensitivity of TnC is due to a change in the rate of Ca2+ dissociation. Figure 4A shows that the rate of Ca2+ dissociation from thin filament bound control 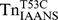 was 109.7±0.7/s (Table 2). Figure 4A also shows the effects of the five DCM mutations on the rate of Ca2+ dissociation from the thin filament. TnT R141W negligibly altered the rate of Ca2+ dissociation, while TnT R131W and R205L caused a slightly faster rate of Ca2+ dissociation from the thin filament (less than ∼ 1.5-fold; Figure 4A and Table 2). TnI K36Q and TnT ΔK210 had larger effects on accelerating the rate of Ca2+ dissociation from the thin filament (∼ 2.5-fold and ∼ 2.3-fold, respectively; Table 2).
was 109.7±0.7/s (Table 2). Figure 4A also shows the effects of the five DCM mutations on the rate of Ca2+ dissociation from the thin filament. TnT R141W negligibly altered the rate of Ca2+ dissociation, while TnT R131W and R205L caused a slightly faster rate of Ca2+ dissociation from the thin filament (less than ∼ 1.5-fold; Figure 4A and Table 2). TnI K36Q and TnT ΔK210 had larger effects on accelerating the rate of Ca2+ dissociation from the thin filament (∼ 2.5-fold and ∼ 2.3-fold, respectively; Table 2).
Figure 4. Effect of disease-related protein modifications on the rate of Ca2+ dissociation from the thin filament.

Panel A shows the time courses of the decrease in IAANS fluorescence as Ca2+ was removed by EGTA from thin filament bound control 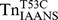 , and the DCM mutants, TnI K36Q
, and the DCM mutants, TnI K36Q 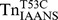 , TnT R131W
, TnT R131W 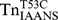 , TnT R141W
, TnT R141W 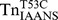 , TnT R205L
, TnT R205L 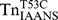 and TnT ΔK210
and TnT ΔK210 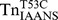 . Panel B shows the time courses of the decrease in IAANS fluorescence as Ca2+ was removed by EGTA from thin filament bound control
. Panel B shows the time courses of the decrease in IAANS fluorescence as Ca2+ was removed by EGTA from thin filament bound control 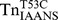 , and the HCM mutants, TnT R92Q
, and the HCM mutants, TnT R92Q 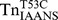 and TnI S166F
and TnI S166F 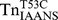 . Panel C shows the time courses of the decrease in IAANS fluorescence as Ca2+ was removed by EGTA from thin filament bound control
. Panel C shows the time courses of the decrease in IAANS fluorescence as Ca2+ was removed by EGTA from thin filament bound control 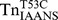 , and the RCM mutants, TnI D190H
, and the RCM mutants, TnI D190H 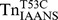 and TnI R192H
and TnI R192H 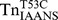 . Panel D shows the time courses of the decrease in IAANS fluorescence as Ca2+ was removed by EGTA from thin filament bound control
. Panel D shows the time courses of the decrease in IAANS fluorescence as Ca2+ was removed by EGTA from thin filament bound control 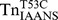 and ischemic related truncated TnI (1-192)
and ischemic related truncated TnI (1-192) 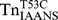 . All
. All 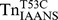 complexes consist of the full length Tn subunits of
complexes consist of the full length Tn subunits of 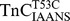 , TnI and TnT, except for ischemic related truncated TnI (1-192). The disease related modification is either in TnI or TnT, in either case, the other protein (TnT or TnI) was wild type. Data traces (an average of 3 to 5 individual traces collected at least 10 times) were fit with a single exponential equation to calculate the kinetic rates. The data traces have been staggered and normalized for clarity.
, TnI and TnT, except for ischemic related truncated TnI (1-192). The disease related modification is either in TnI or TnT, in either case, the other protein (TnT or TnI) was wild type. Data traces (an average of 3 to 5 individual traces collected at least 10 times) were fit with a single exponential equation to calculate the kinetic rates. The data traces have been staggered and normalized for clarity.
Figure 4B, C and D show the effects of the two HCM mutations, two RCM mutations and ischemia-induced truncation of TnI on the rate of Ca2+ dissociation from the thin filament. The HCM mutation TnI S166F slowed the rate of Ca2+ dissociation by ∼ 1.7-fold, while the HCM mutation TnT R92Q had no effect on the dissociation rate (Figure 4B and Table 2). Both RCM mutations slowed the rate of Ca2+ dissociation from the thin filament, ∼1.2-fold slower for TnI D190H and ∼1.5-fold slower for TnI R192H (Figure 4C and Table 2). The ischemia-induced truncation of TnI (1-192) slowed the rate of Ca2+ dissociation from the thin filament by ∼ 2-fold (Figure 4D and Table 2).
Effects of the Protein Modifications on the Rate of Ca2+ Association to the Thin Filament
The kinetic studies indicate that the changes in Ca2+ sensitivity of the thin filament caused by the protein modifications are in part due to changes in the rate of Ca2+ dissociation. However, as shown in table 2 and figure 5, there was only a weak correlation (r2 = 0.17, fit not shown) between the changes in the thin filament Ca2+ sensitivities and the changes in the Ca2+ dissociation rates for the different protein modifications. These results suggest that the rate of Ca2+ association to the thin filament can also be altered by the protein modifications. To test this hypothesis, stopped-flow experiments were performed to measure the rate of Ca2+ association to the thin filaments containing TnI R192H, D190H, TnI (1-192), TnT ΔK210 and TnT R131W. Based on the calculated rate of Ca2+ association, TnT R131W was predicted to have little effect on the rate of Ca2+ association while the rest of the protein modifications were expected to significantly change the rate of Ca2+ association (Table 2). Using a traditional approach to determine the apparent Ca2+ association rate to the thin filament, the measured rate of Ca2+ association to the control thin filament was 9±1×106 M−1s−1 (Figure 6A, 6C and Table 2). Consistent with the calculated Ca2+ association rates, TnI R192H, D190H and TnI (1-192) increased the apparent rate of Ca2+ association to the thin filament by ∼ 4- to 5-fold (Figure 6B, 6D and Table 2). In contrast, TnT ΔK210 slowed the apparent rate of Ca2+ association to the thin filament by ∼3-fold (Figure 6D and Table 2). On the other hand, the apparent rate of Ca2+ association to the thin filament containing TnT R131W was similar to that of the control thin filament, with the rate being 12±1×106 M−1s−1 (Figure 6D and Table 2). Thus, the experimental results indicate that the apparent rate of Ca2+ association to the thin filament can also be altered by the disease-related protein modifications.
Figure 5. The relationship between changes in the Ca2+ sensitivity and the rate of Ca2+ dissociation.

The changes in the thin filament Ca2+ sensitivity for the ten disease-related protein modifications are plotted against the changes in the rate of Ca2+ dissociation from the thin filament. The straight line in the figure represents a perfect correlation between the thin filament change in Ca2+ sensitivity and Ca2+ dissociation rate.
Figure 6. Effect of disease-related protein modifications on the rate of Ca2+ association to the thin filament.

Panel A shows the kinetic traces (an average of 3 to 5 individual traces collected at least 9 times) observed for Ca2+ association to thin filament bound control 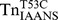 at 5 µM, 10 µM and 20 µM Ca2+ ([Ca2+] after mixing). Panel B shows the kinetic traces (an average of 3 to 5 individual traces collected at least 9 times) observed for Ca2+ association to thin filament bound TnI(1-192)
at 5 µM, 10 µM and 20 µM Ca2+ ([Ca2+] after mixing). Panel B shows the kinetic traces (an average of 3 to 5 individual traces collected at least 9 times) observed for Ca2+ association to thin filament bound TnI(1-192) 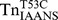 at 2.5 µM, 5 µM and 10 µM Ca2+. The rate of the fluorescence increase when Ca2+ associates with the thin filament was fit with a single exponential to calculate the observed rate (kobserved). All
at 2.5 µM, 5 µM and 10 µM Ca2+. The rate of the fluorescence increase when Ca2+ associates with the thin filament was fit with a single exponential to calculate the observed rate (kobserved). All 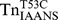 complexes consist of the full length Tn subunits of
complexes consist of the full length Tn subunits of 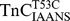 , TnI and TnT, except for ischemic related truncated TnI (1-192). The disease related modification is either in TnI or TnT, in either case, the other protein (TnT or TnI) was wild type. Panel C shows the plots of kobserved versus [Ca2+] for thin filament bound control
, TnI and TnT, except for ischemic related truncated TnI (1-192). The disease related modification is either in TnI or TnT, in either case, the other protein (TnT or TnI) was wild type. Panel C shows the plots of kobserved versus [Ca2+] for thin filament bound control 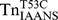 (□) and TnI (1-192)
(□) and TnI (1-192) 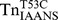 (
(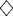 ). The rate of Ca2+ association to the thin filament was obtained by calculating the slope of a linear fit to the data. Panel D shows the plots of kobserved versus [Ca2+] for thin filament bound TnI R192H
). The rate of Ca2+ association to the thin filament was obtained by calculating the slope of a linear fit to the data. Panel D shows the plots of kobserved versus [Ca2+] for thin filament bound TnI R192H 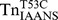 (▵), TnI D190H
(▵), TnI D190H 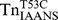 (▽), TnT ΔK210
(▽), TnT ΔK210 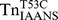 (⋆), and TnT R131W
(⋆), and TnT R131W 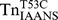 (○).
(○).
The Ca2+ association rate data suggest that the disease-related modifications alter the ability of the thin filament to respond to a Ca2+ transient. This idea was tested in a stopped-flow apparatus by exposing the thin filament to artificial Ca2+ transients (ACTs) of increasing amplitude (Figure 7). Due to the relatively slow Ca2+ association rate of EGTA, initially Ca2+ transiently binds to the thin filament (dependent on its Ca2+ association rate) until EGTA chelates the Ca2+ (∼ 1 ms half life). Although these are not physiological Ca2+ transients, it is expected that for a fixed Ca2+ transient amplitude, a faster rate of Ca2+ association should lead to a higher level of transient occupancy, while a slower rate of Ca2+ association should lead to a lower level of transient occupancy. Figure 7A shows the responses of thin filament bound control 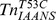 to ACTs. As the [Ca2+] was successively increased from 12.5 µM to 25 µM and then to 50 µM (after mixing), the percentage of transient occupancy increased from 17±1%, to 30±2% and then to 47±3%, respectively (Table 3). Compared to the control thin filament, the percentage of the transient occupancy was higher for TnI (1-192) and lower for TnT ΔK210 at all three Ca2+ concentrations (Figure 7B, 7C and Table 3). Thus, consistent with the Ca2+ association rate calculations and measurements, the ACT experiments support the idea that the TnI (1-192) containing thin filament has an apparent faster rate of Ca2+ association, while the TnT ΔK210 containing thin filament has an apparent slower rate of Ca2+ association than the control thin filament.
to ACTs. As the [Ca2+] was successively increased from 12.5 µM to 25 µM and then to 50 µM (after mixing), the percentage of transient occupancy increased from 17±1%, to 30±2% and then to 47±3%, respectively (Table 3). Compared to the control thin filament, the percentage of the transient occupancy was higher for TnI (1-192) and lower for TnT ΔK210 at all three Ca2+ concentrations (Figure 7B, 7C and Table 3). Thus, consistent with the Ca2+ association rate calculations and measurements, the ACT experiments support the idea that the TnI (1-192) containing thin filament has an apparent faster rate of Ca2+ association, while the TnT ΔK210 containing thin filament has an apparent slower rate of Ca2+ association than the control thin filament.
Table 3. Percent transient occupancy of the thin filament during artificial Ca2+ transients of increasing amplitude.
| Protein | % Transient Occupancywith 12.5 µM Ca2+ | % Transient Occupancywith 25 µM Ca2+ | % Transient Occupancywith 50 µM Ca2+ |
| Control | 17±1 | 30±2 | 47±3 |
| TnT ▵K210 | 5±3* | 20±2* | 28±6* |
| TnI (1–192) | 44±3* | 64±2* | 81±2* |
Values marked with * are significantly different from their respective control values (p<0.05).
Discussion
In this study, we examined the effects of cardiac disease-related TnI and TnT modifications on the Ca2+ binding properties of TnC in the Tn complex and on the thin filament. The selected ten protein modifications are associated with a broad range of cardiac diseases: three subtypes of familial cardiomyopathies (DCM, HCM and RCM), and ischemia-reperfusion injury. Most of the selected protein modifications have previously been shown to alter the Ca2+ sensitivity of skinned cardiac muscle force generation, and/or the actomyosin-ATPase activity [12], [13], [21], [22]. Transgenic mice models have been developed for TnI R192H, TnI (1-192), TnT R92Q, TnT ΔK210 and TnT R141W, all of which recapitulate the phenotypes of the human diseases [23], [24], [32], [33], [34]. In addition to altering the steady-state Ca2+ sensitivity of force generation, some of these protein modifications also alter the kinetics and magnitude of contraction and/or relaxation [23], [24]. Thus, it is important to understand not only the effects of these protein modifications on the Ca2+ sensitivity of TnC, but also the effects on the kinetics of Ca2+ exchange with TnC.
Previous studies utilized IAANS labeled fluorescent TnCs (at either Cys 35 or Cys 84) to measure the effects of disease-related protein modifications on the steady-state Ca2+ sensitivity of TnC [6], [12], [13], [22]. Our steady-state results were qualitatively consistent with these previous studies. For instance, we observed that the majority of the disease-related protein modifications did not alter the Ca2+ binding properties of the Tn complex [6], [12], [13], [22]. However, when reconstituted into the thin filament, the DCM mutations decreased the Ca2+ sensitivity of the thin filament, while most of the HCM and RCM mutations, as well as the ischemia-induced truncation of TnI, increased the Ca2+ sensitivity of the thin filament [6], [12], [13], [22]. Slight quantitative differences in the Ca2+ sensitivity observed for the same protein modifications in the current study, compared to previous studies [6], [12], [13], [22], might be due to the utilization of different fluorescent TnCs, protein isoforms from different species (human in this manuscript), buffer composition, temperature, or the integrity of the Tn complex and/or the reconstituted thin filament. Great care was taken in the current study to ensure that there was no contaminating free TnC within the Tn complex, or free Tn within the thin filament that would have compromised the results.
The steady-state Ca2+ sensitivity of TnC is determined by the kinetics of Ca2+ association and dissociation. It is generally assumed that alterations in the steady-state Ca2+ sensitivity of TnC operate exclusively through changes in the rate of Ca2+ dissociation, since Ca2+ association to TnC has been traditionally thought to be diffusion controlled (for review see [15]). Our data clearly indicate that the majority of the disease-related protein modifications altered the rate of Ca2+ dissociation from the thin filament, in a way that is consistent with their effect on Ca2+ sensitivity. However, the magnitude of the change in the Ca2+ dissociation rates does not always correlate with the magnitude of the Ca2+ sensitivity changes. This finding suggests that the Ca2+ association rate to TnC must also be affected by the disease-related protein modifications. Experimental measurements verified that the apparent rates of Ca2+ association were also altered by some of the disease-related protein modifications. These data are consistent with our previous results that demonstrated natural and engineered TnC mutations could also affect the rate of Ca association to TnC [19], [20]. Interestingly, the Ca2+ sensitizing protein modifications (TnI R192H, D190H, and ischemia-induced truncation of TnI) tend to alter the Ca2+ binding sensitivity by modulating both the association and dissociation rates; while the Ca2+ desensitizing mutations tend to predominantly affect the dissociation rates (with the exception of TnT ΔK210).
While the physiological significance of the Ca2+ dissociation rate from TnC remains controversial [15], it is striking that Tn modifications (disease or engineered) with slowed or accelerated Ca2+ dissociation rates prolonged or abbreviated relaxation [23], [24], [35]. Thus, an aberrant rate of Ca2+ dissociation from TnC could potentially contribute to the diastolic dysfunction typically observed with the various cardiomyopathies [36], [37]. On the other hand, the rate of Ca2+ association to the thin filament could affect the amount of maximal force generation or myocyte shortening. For instance, despite similar intracellular Ca2+ transients, myocytes transfected with TnI R192H had a larger shortening amplitude than control myocytes, especially under high stimulating frequencies [38]. Whereas, intact papillary muscles from TnT ΔK210 transgenic mice displayed the same amount of maximal force as control muscles, even though the amplitude of the intracellular Ca2+ transient was higher than that of the control [24]. These transgenic studies suggest that in addition to the intracellular Ca2+ transient, the response of the thin filament to Ca2+ may also regulate the extent and duration of force development. Consistent with this idea, the current study demonstrates that both TnI (1-192) and TnT ΔK210 altered the response of the thin filament to non-physiological, artificial Ca2+ transients in a way consistent with their contractile effects.
There are potentially multiple mechanisms that exist to alter the Ca2+ binding properties of TnC on the thin filament [15]. It is well established that the binding of TnI to TnC is critical in determining the Ca2+ sensitivity and kinetics of TnC [11], [15]. However, it appears that only TnI S166F altered the ability of TnI to directly change the Ca2+ binding properties of TnC, since it was the only protein modification that substantially altered the Ca2+ binding properties of the Tn complex. TnI S166F is located close to the switch region of TnI that binds to the hydrophobic pocket of the regulatory domain of TnC. Thus, TnI S166F might be influencing this critical TnC-TnI interaction to affect the Ca2+ binding properties of TnC.
The more distant C-terminal region of TnI (residues 188-210) is believed to contribute to the inhibition of actomyosin interactions during diastole by direct interactions with actin-Tm [39]. The ability of TnI to bind actin-Tm may reduce the ability of the switch region of TnI to interact with the hydrophobic pocket of TnC, in essence reducing the apparent Ca2+ sensitivity of TnC [11]. TnI D190H, R192H and the removed residues from TnI (1-192) are located within this region of TnI and may weaken the interactions between TnI and actin-Tm. In this regard, these protein modifications may facilitate the switching of TnI from actin-Tm to TnC, causing an enhanced apparent sensitivity for Ca2+ only when the Tn complex is incorporated in the thin filament. Similarly, the TnT mutations may also change the ability of TnI to bind to actin-Tm. This hypothesis would be consistent with the TnT modifications only affecting the apparent Ca2+ sensitivity of the thin filament and not the Tn complex.
In summary, the disease-related protein modifications studied in this work adversely altered the steady-state Ca2+ sensitivity of the thin filament by influencing both the association and dissociation rates of Ca2+ exchange. The protein modifications modified the Ca2+ exchange properties of TnC in a way consistent with their effect on cardiac muscle physiology. Therefore, TnC may act as a central hub that converges these pathological stimuli to affect cardiac contractile properties. The abnormal myofilament Ca2+ binding along with aberrant intra-cellular Ca2+ signaling could contribute to contractile dysfunctions leading to cardiac remodeling and disease phenotypes. Thus, pharmaceutically [40] or genetically targeting TnC to correct the abnormal myofilament Ca2+ binding properties may represent a beneficial therapeutic strategy.
Acknowledgments
We thank Dr. Jianchao Zhang, Sean Little and Vikram Shettigar for critical reading of the manuscript.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This study was supported by the American Heart Association (to BL and JPD), National Institues of Health grants HL091986 (to JPD) and HL087462 (to SBT). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Janssen PM. Myocardial contraction-relaxation coupling. Am J Physiol Heart Circ Physiol. 2010;299:H1741–1749. doi: 10.1152/ajpheart.00759.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, et al. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res. 1999;84:562–570. doi: 10.1161/01.res.84.5.562. [DOI] [PubMed] [Google Scholar]
- 3.Bers DM, Despa S, Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. 2006;1080:165–177. doi: 10.1196/annals.1380.015. [DOI] [PubMed] [Google Scholar]
- 4.van der Velden J. Diastolic myofilament dysfunction in the failing human heart. Pflugers Arch. 2011;462:155–163. doi: 10.1007/s00424-011-0960-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, et al. Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J Mol Cell Cardiol. 2010;48:882–892. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 6.Tachampa K, Kobayashi T, Wang H, Martin AF, Biesiadecki BJ, et al. Increased cross-bridge cycling kinetics after exchange of C-terminal truncated troponin I in skinned rat cardiac muscle. J Biol Chem. 2008;283:15114–15121. doi: 10.1074/jbc.M801636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mirza M, Marston S, Willott R, Ashley C, Mogensen J, et al. Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J Biol Chem. 2005;280:28498–28506. doi: 10.1074/jbc.M412281200. [DOI] [PubMed] [Google Scholar]
- 8.Gomes AV, Liang J, Potter JD. Mutations in human cardiac troponin I that are associated with restrictive cardiomyopathy affect basal ATPase activity and the calcium sensitivity of force development. J Biol Chem. 2005;280:30909–30915. doi: 10.1074/jbc.M500287200. [DOI] [PubMed] [Google Scholar]
- 9.Tardiff JC. Thin filament mutations: developing an integrative approach to a complex disorder. Circ Res. 2011;108:765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 11.Davis JP, Norman C, Kobayashi T, Solaro RJ, Swartz DR, et al. Effects of thin and thick filament proteins on calcium binding and exchange with cardiac troponin C. Biophys J. 2007;92:3195–3206. doi: 10.1529/biophysj.106.095406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi T, Solaro RJ. Increased Ca2+ affinity of cardiac thin filaments reconstituted with cardiomyopathy-related mutant cardiac troponin I. J Biol Chem. 2006;281:13471–13477. doi: 10.1074/jbc.M509561200. [DOI] [PubMed] [Google Scholar]
- 13.Robinson P, Griffiths PJ, Watkins H, Redwood CS. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res. 2007;101:1266–1273. doi: 10.1161/CIRCRESAHA.107.156380. [DOI] [PubMed] [Google Scholar]
- 14.Lu QW, Hinken AC, Patrick SE, Solaro RJ, Kobayashi T. Phosphorylation of cardiac troponin I at protein kinase C site threonine 144 depresses cooperative activation of thin filaments. J Biol Chem. 2010;285:11810–11817. doi: 10.1074/jbc.M109.055657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis JP, Tikunova SB. Ca(2+) exchange with troponin C and cardiac muscle dynamics. Cardiovasc Res. 2008;77:619–626. doi: 10.1093/cvr/cvm098. [DOI] [PubMed] [Google Scholar]
- 16.Dong WJ, Xing J, Ouyang Y, An J, Cheung HC. Structural kinetics of cardiac troponin C mutants linked to familial hypertrophic and dilated cardiomyopathy in troponin complexes. J Biol Chem. 2008;283:3424–3432. doi: 10.1074/jbc.M703822200. [DOI] [PubMed] [Google Scholar]
- 17.Iorga B, Blaudeck N, Solzin J, Neulen A, Stehle I, et al. Lys184 deletion in troponin I impairs relaxation kinetics and induces hypercontractility in murine cardiac myofibrils. Cardiovasc Res. 2008;77:676–686. doi: 10.1093/cvr/cvm113. [DOI] [PubMed] [Google Scholar]
- 18.Kruger M, Zittrich S, Redwood C, Blaudeck N, James J, et al. Effects of the mutation R145G in human cardiac troponin I on the kinetics of the contraction-relaxation cycle in isolated cardiac myofibrils. J Physiol. 2005;564:347–357. doi: 10.1113/jphysiol.2004.079095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tikunova SB, Davis JP. Designing calcium-sensitizing mutations in the regulatory domain of cardiac troponin C. J Biol Chem. 2004;279:35341–35352. doi: 10.1074/jbc.M405413200. [DOI] [PubMed] [Google Scholar]
- 20.Liang B, Chung F, Qu Y, Pavlov D, Gillis TE, et al. Familial hypertrophic cardiomyopathy-related cardiac troponin C mutation L29Q affects Ca2+ binding and myofilament contractility. Physiol Genomics. 2008;33:257–266. doi: 10.1152/physiolgenomics.00154.2007. [DOI] [PubMed] [Google Scholar]
- 21.Morimoto S, Lu QW, Harada K, Takahashi-Yanaga F, Minakami R, et al. Ca(2+)-desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proc Natl Acad Sci U S A. 2002;99:913–918. doi: 10.1073/pnas.022628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carballo S, Robinson P, Otway R, Fatkin D, Jongbloed JD, et al. Identification and functional characterization of cardiac troponin I as a novel disease gene in autosomal dominant dilated cardiomyopathy. Circ Res. 2009;105:375–382. doi: 10.1161/CIRCRESAHA.109.196055. [DOI] [PubMed] [Google Scholar]
- 23.Du J, Liu J, Feng HZ, Hossain MM, Gobara N, et al. Impaired relaxation is the main manifestation in transgenic mice expressing a restrictive cardiomyopathy mutation, R193H, in cardiac TnI. Am J Physiol Heart Circ Physiol. 2008;294:H2604–2613. doi: 10.1152/ajpheart.91506.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du CK, Morimoto S, Nishii K, Minakami R, Ohta M, et al. Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ Res. 2007;101:185–194. doi: 10.1161/CIRCRESAHA.106.146670. [DOI] [PubMed] [Google Scholar]
- 25.Tikunova SB, Liu B, Swindle N, Little SC, Gomes AV, et al. Effect of calcium-sensitizing mutations on calcium binding and exchange with troponin C in increasingly complex biochemical systems. Biochemistry. 2010;49:1975–1984. doi: 10.1021/bi901867s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smillie LB. Pt B. Vol. 85. Methods Enzymol; 1982. Preparation and identification of alpha- and beta-tropomyosins. pp. 234–241. [DOI] [PubMed] [Google Scholar]
- 27.Pardee JD, Spudich JA. Methods Enzymol 85 Pt B; 1982. Purification of muscle actin. pp. 164–181. [DOI] [PubMed] [Google Scholar]
- 28.Robertson S, Potter JD. The regulation of free Ca2+ ion concentration by metal chelators. Methods in Pharmacology. 1984;5:63–75. [Google Scholar]
- 29.Tikunova SB, Rall JA, Davis JP. Effect of hydrophobic residue substitutions with glutamine on Ca(2+) binding and exchange with the N-domain of troponin C. Biochemistry. 2002;41:6697–6705. doi: 10.1021/bi011763h. [DOI] [PubMed] [Google Scholar]
- 30.Kasturi R, Vasulka C, Johnson JD. Ca2+, caldesmon, and myosin light chain kinase exchange with calmodulin. J Biol Chem. 1993;268:7958–7964. [PubMed] [Google Scholar]
- 31.Davis JP, Tikunova SB, Walsh MP, Johnson JD. Characterizing the response of calcium signal transducers to generated calcium transients. Biochemistry. 1999;38:4235–4244. doi: 10.1021/bi982495z. [DOI] [PubMed] [Google Scholar]
- 32.Murphy AM, Kogler H, Marban E. A mouse model of myocardial stunning. Mol Med Today. 2000;6:330–331. doi: 10.1016/s1357-4310(00)01732-9. [DOI] [PubMed] [Google Scholar]
- 33.Tardiff JC, Hewett TE, Palmer BM, Olsson C, Factor SM, et al. Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J Clin Invest. 1999;104:469–481. doi: 10.1172/JCI6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Juan F, Wei D, Xiongzhi Q, Ran D, Chunmei M, et al. The changes of the cardiac structure and function in cTnTR141W transgenic mice. Int J Cardiol. 2008;128:83–90. doi: 10.1016/j.ijcard.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 35.Kreutziger KL, Piroddi N, McMichael JT, Tesi C, Poggesi C, et al. Calcium binding kinetics of troponin C strongly modulate cooperative activation and tension kinetics in cardiac muscle. J Mol Cell Cardiol. 2011;50:165–174. doi: 10.1016/j.yjmcc.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 37.Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. 1997;336:267–276. doi: 10.1056/NEJM199701233360407. [DOI] [PubMed] [Google Scholar]
- 38.Davis J, Wen H, Edwards T, Metzger JM. Thin filament disinhibition by restrictive cardiomyopathy mutant R193H troponin I induces Ca2+-independent mechanical tone and acute myocyte remodeling. Circ Res. 2007;100:1494–1502. doi: 10.1161/01.RES.0000268412.34364.50. [DOI] [PubMed] [Google Scholar]
- 39.Rarick HM, Tu XH, Solaro RJ, Martin AF. The C terminus of cardiac troponin I is essential for full inhibitory activity and Ca2+ sensitivity of rat myofibrils. Journal of Biological Chemistry. 1997;272:26887–26892. doi: 10.1074/jbc.272.43.26887. [DOI] [PubMed] [Google Scholar]
- 40.Kass DA, Solaro RJ. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation. 2006;113:305–315. doi: 10.1161/CIRCULATIONAHA.105.542407. [DOI] [PubMed] [Google Scholar]