

Abstract
A variety of neurotransmitters are responsible for regulating neural activity during different behavioral states. Unique responses to combinations of neurotransmitters provide a powerful mechanism by which neural networks could be differentially activated during a broad range of behaviors. Here, we show, using whole-cell recordings in rat hippocampal slices, that group I metabotropic glutamate receptors (mGluRs) and muscarinic acetylcholine receptors (mAChRs) synergistically increase the excitability of hippocampal CA1 pyramidal neurons by converting the post-burst afterhyperpolarization to an afterdepolarization via a rapidly reversible upregulation of Cav2.3 R-type calcium channels. Coactivation of mAChRs and mGluRs also induced a long-lasting enhancement of the responses mediated by each receptor type. These results suggest that cooperative signaling via mAChRs and group I mGluRs could provide a mechanism by which cognitive processes may be modulated by conjoint activation of two separate neurotransmitter systems.
Introduction
Acetylcholine (ACh) and glutamate (Glu) act as neuromodulators to change neural information processing in a variety of ways (Blokland, 1995; Anwyl, 1999; Power et al., 2003; Riedel et al., 2003; Hasselmo, 2006). Muscarinic acetylcholine receptors (mAChRs) and metabotropic glutamate receptors (mGluRs) play important roles in cognitive function, because dysfunction of mAChR and mGluR signaling has been implicated in the pathophysiology of many neurological disorders (Bear et al., 2004; Lee et al., 2004; Ure et al., 2006; Wess et al., 2007). In the hippocampus, ACh and Glu are critically involved in higher brain functions, including learning and memory, but the cellular mechanisms by which these neurotransmitters act are only partially understood and the mechanisms by which they might interact are unexplored (Anwyl, 1999).
In general, the two classes of neuromodulatory mechanisms are modulation of synaptic transmission and modulation of neuronal excitability (Giocomo and Hasselmo, 2007). Among the many effects of activation of mAChRs and mGluRs, the modulation of neuronal excitability has a direct effect on the response of cortical pyramidal neurons to excitatory synaptic input. As with synaptic plasticity, the modulation of excitability can be affected by multiple cellular mechanisms, including changes in the afterhyperpolarization (AHP) after action potentials (APs) (Benardo and Prince, 1982; Greene et al., 1992; Kawasaki et al., 1999; McQuiston and Madison, 1999; Ireland and Abraham, 2002; Young et al., 2004).
The effects of Glu on the modulation of excitability are commonly mediated by group I mGluRs, which are coupled to Gq/11-proteins. Their stimulation triggers phospholipase C (PLC) activation, mobilization of intracellular Ca2+, and ultimately modulation of multiple types of ion channels (Pin and Duvoisin, 1995; Anwyl, 1999). We recently demonstrated that activation of group I mGluRs eliminated the post-burst AHP and produced an afterdepolarization (ADP) through upregulation of Cav2.3 R-type calcium channels (Park et al., 2010). Although multiple studies have reported that activation of mAChRs also induces changes in the AHP, resulting in enhanced excitability (Benardo and Prince, 1982; Cole and Nicoll, 1984a,b; McCormick and Prince, 1986; Kawasaki et al., 1999; McQuiston and Madison, 1999; Lawrence et al., 2006), it is poorly understood which receptor subtypes, signaling mechanisms, and ion channels are responsible for the mAChR-mediated modulation of excitability, particularly in hippocampal CA1 pyramidal neurons. Because these modulatory systems play a vital role in hippocampus-dependent functions, we investigated the effects of activating mAChRs and group I mGluRs on the excitability of hippocampal CA1 pyramidal neurons and sought to reveal the underlying mechanisms for the effects.
We report here that activation of either mAChRs or group I mGluRs using moderate concentrations of agonists or synaptic stimulation results in the conversion of the post-burst AHP into a post-burst ADP. Furthermore, when both receptor types are activated concurrently, these different groups of modulatory systems act synergistically to evoke a robust post-burst ADP, as well as a long-lasting enhancement of the ADP, providing a mechanism by which combined activation of two modulatory systems can cooperatively alter the integrative properties of the neuron.
Materials and Methods
Slice preparation and maintenance.
All experiments were conducted in accordance with a protocol approved by the Animal Care and Use Committee of Northwestern University. Transverse hippocampal slices, 300 μm thick, were prepared from male Wistar rats (25–35 d old) and from either wild-type (C57BL/6J) or Cav2.3 knock-out (22–28 d old) male mice using standard procedures (Park et al., 2010). Animals were deeply anesthetized with halothane or isoflurane, perfused intracardially with ice-cold artificial CSF (ACSF), and decapitated. The brain was then removed rapidly and attached to the stage of a vibrating tissue slicer (Vibratome). Slices were prepared in ice-cold oxygenated ACSF and then allowed to recover for 0.5 h at ∼35°C in a chamber filled with oxygenated ACSF. The slice chamber was subsequently maintained at room temperature, and individual slices were transferred to a submerged chamber in which it was perfused with ACSF (33 ± 2°C) at the rate of 2–3 ml/min. Normal ACSF had the following composition (in mm): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, and 25 dextrose.
Electrophysiology.
Whole-cell current-clamp recordings were made using patch-clamp electrodes pulled from borosilicate glass (1.5 mm outer diameter) and filled with intracellular solution containing the following: 115 mm K-gluconate, 20 mm KCl, 10 mm Na2-phosphocreatine, 10 mm HEPES, 2 mm MgATP, 0.3 mm NaGTP, and 0.1% biocytin. Electrode resistance in the bath was 3–5 MΩ, and series resistance during the recordings was 5–20 MΩ. Recordings were obtained with Dagan BVC-700 amplifiers, using appropriate bridge balance and electrode-capacitance compensation. The membrane potential was held at −65 mV, which required very small current injections (less than −50 pA).
Synaptic stimuli.
To test whether synaptic activation could induce the post-burst ADP, five APs were elicited by brief somatic current injections, either with or without concurrent synaptic stimulation. Responses were monitored once every 5 min (unless otherwise indicated) with a 1 min delay between the two conditions. In all groups, experiments were performed in the presence of blockers of ionotropic glutamate receptors (30 μm CNQX and 50 μm d-AP-5 to block AMPA and NMDA receptors, respectively) and GABA receptors (2 μm SR95531 [2-(3-carboxypropyl)-3-amino-6-(4-methoxyphenyl)pyridazinium bromide] and 1 μm CGP55845 [(2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid hydrochloride] to block GABAA and GABAB receptors, respectively). Bipolar theta glass electrodes filled with ACSF were used in conjunction with a Dagan BSI-950 biphasic stimulus isolator. Stimulating electrodes were placed in proximal stratum radiatum at least 100 μm away from the recorded cell toward CA3. Stimulus intensity was set to produce a 3–6 mV ADP during synaptic stimulation (2–10 mA output).
Analysis.
Data acquisition and analysis procedures were custom programmed in Igor Pro (Wavemetrics). Throughout, reported values are mean ± SEM of data. Statistical tests included the paired or unpaired t test and ANOVA with Tukey's post hoc comparisons. All statistical analyses were performed using Prism 4 software (GraphPad Software). In most experiments, the amplitude of the post-burst potential was quantified at a fixed time, corresponding to the peak of the AHP in normal ACSF (58 ± 3 ms after cessation of current injection; n = 27). Effects were quantified by the change in the post-burst potential (i.e., Δ post-burst potential = ADP in agonists or with synaptic stimulation − AHP in baseline) at this time point in the response.
Pharmacology.
Most drugs were added to the bathing solution. In some experiments, however, drugs were added to the intracellular solution [1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrapotassium salt (BAPTA), guanosine 5′-[β-thio]diphosphate trilithium salt (GDP-β-S)]. For BAPTA-containing internal solution, the K-gluconate concentration was reduced to 100 mm. The following drugs were obtained from Tocris Bioscience: (S)-3,5-dihydroxyphenylglycine (DHPG), LY367385 [(S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid], 2-methyl-6-(phenylethynyl)-pyridine hydrochloride (MPEP), d-AP-5, CNQX disodium salt, U73122 (1-[6[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione), McN-A 343 (McN-A), 4-diphenylacetoxy-N-methylpiperidine methiodide (4-DAMP), cyclopiazonic acid (CPA), and CGP55845. Carbamoylcholine chloride (CCH), oxotremorine M (Oxo), 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrapotassium salt (BAPTA), serotonin hydrochloride (5-HT), (R)(−)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane hydrochloride (DOI), l-(−)-noradrenaline (+)-bitartrate salt monohydrate, methoxamine hydrochloride (MTX), pirenzepine dihydrochloride (PZ), methoctramine hydrate (MCT), tropicamide (Trop), guanosine 5′-[β-thio]diphosphate trilithium salt (GDP-β-S), nickel(II) chloride hexahydrate (Ni2+), SR95531, atropine, dextrose, K-gluconate, sodium phosphocreatine, HEPES, MgATP, NaGTP, and biocytin were purchased from Sigma-Aldrich.
Mathematical model.
The mathematical model of synergistic activation of the ADP was produced using hypothetical concentration–response curves generated using the Hill equation:
 |
where E is the of the effector signal (in arbitrary units), [A] is the concentration of agonist, n is the Hill coefficient, and K is the microscopic dissociation constant (i.e., the concentration producing a half-maximal response).
Results
We obtained whole-cell current-clamp recordings from regular-spiking CA1 pyramidal neurons in rat hippocampal slices and determined the effects of cholinergic activation on the spike afterpotentials elicited by intracellular current injection. Similar to previous reports, bath application of 5–10 μm CCH, a nonhydrolyzable cholinergic agonist, produced a small but statistically significant increase in the input resistance (56.4 ± 2.7 MΩ in baseline condition; 62.1 ± 3.1 MΩ after CCH application; n = 27; paired t test, p < 0.001).
In baseline conditions, a burst of APs evoked by five brief current injections (2 nA; 2 ms; 100 Hz) through the recording electrode was followed by a small AHP (Fig. 1; −2.6 ± 0.2 mV; n = 14). Application of CCH caused a conversion of the post-burst AHP to a post-burst ADP (Fig. 1; +7.1 ± 1.2 mV after CCH application; n = 14). At higher concentrations, this ADP was sufficiently large to trigger additional APs firing (data not shown). The effects of CCH in producing the post-burst ADP were reversed during washout of the agonist (Fig. 1).
Figure 1.

Reversible cholinergic modulation of post-burst potentials in CA1 pyramidal neurons. A, Individual responses to five brief current injections (2 nA, 2 ms each) to evoke a burst of APs in the baseline condition (left, gray, t = 0 min in B), after CCH application (middle, black, t = 10 min in B), and after recovery (right, black, t = 25 min in B). Up–down arrow shows the difference between the AHP and the ADP (Δ post-burst potential) measured at the time of the AHP peak in the baseline condition (arrowhead). B, Time course of the post-burst potential amplitudes. Gray-shaded area indicates period of CCH application. n = 14.
To identify the receptor subtype responsible for the CCH-induced post-burst ADP in CA1 pyramidal neurons, we performed a series of pharmacological experiments (Fig. 2). Oxo (1 μm; a specific mAChR agonist) mimicked the effects of CCH (8 μm) on the post-burst potentials, indicating the specific action of CCH on mAChRs. McN-A (100 μm; an agonist specific for M1 receptors) produced a similar effect. We also found that both 4-DAMP (0.1 μm; an antagonist with equal affinity for M1 and M3 receptors) and PZ (0.1 μm; an antagonist specific for M1 receptors) prevented the CCH-induced ADP. In contrast, either MCT (0.2 μm; an antagonist specific for M2 receptors) or Trop (0.2 μm; an antagonist specific for M4 receptors) failed to prevent the effects. Together, these data (summarized in Fig. 2C) suggest that M1 receptor activation is required for the cholinergic conversion of the post-burst AHP into the post-burst ADP in CA1 neurons.
Figure 2.
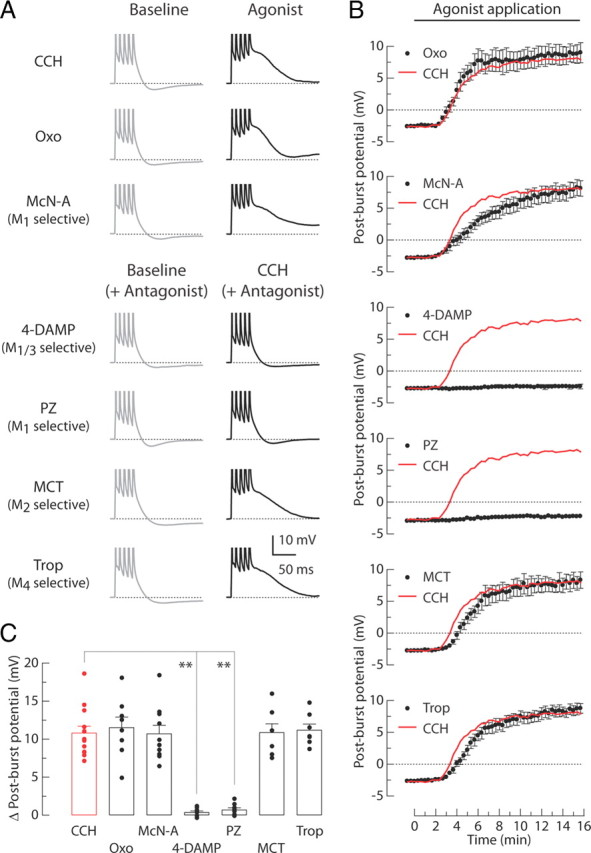
M1 receptors mediate the CCH-induced post-burst ADPs. A, Individual responses to bursts of five APs in baseline condition (left, gray) or 15 min after application of agonists (right, black). Experiments were performed in seven different groups: 8 μm CCH, 1 μm Oxo, or 100 μm McN-A in agonist tests; 0.1 μm 4-DAMP, 0.1 μm PZ, 0.2 μm MCT, or 0.2 μm Trop with 8 μm CCH in antagonist tests. B, Time courses of the post-burst potential amplitudes after application of agonists beginning at t = 0 min. Red lines indicate the averaged response from the CCH-treated group without antagonists. C, Summary of the change in post-burst potentials (t = 15 min minus t = 0 min response) in each condition. Each symbol represents the amplitude of individual experiments. One-way ANOVA, p < 0.0001; post hoc tests versus CCH, **p < 0.001. CCH, n = 13; Oxo, n = 8; McN-A, n = 10; 4-DAMP, n = 8; PZ, n = 8; MCT, n = 7; Trop, n = 7.
Based on our observations suggesting the involvement of the M1 receptor subtype in the CCH-induced ADP, we investigated whether the ADP required G-protein-dependent activation of PLC and calcium signaling (Fig. 3). The effects of CCH were tested with GDP-β-S (0.5 mm; an inhibitor of G-protein signaling), U73122 (20 μm; a PLC inhibitor), CPA (30 μm; a blocker of ATP-dependent calcium uptake into stores), and BAPTA (10 mm; a calcium chelator). All of these manipulations inhibited the induction of the ADP, as indicated by the reduced magnitudes of the change in the post-burst potential (Fig. 3C). We showed previously that activation of group I mGluRs by a selective agonist (DHPG) produced a post-burst ADP with similar pharmacology (Park et al., 2010). Here, we performed additional experiments indicating that this effect was not produced by agonists for other receptors that are also coupled to Gq/11 signaling (α1 adrenergic receptor, MTX; 5-HT2 serotonergic receptor, DOI; data not shown), suggesting that it is somewhat specific to mAChR and group I mGluR activation in these neurons.
Figure 3.
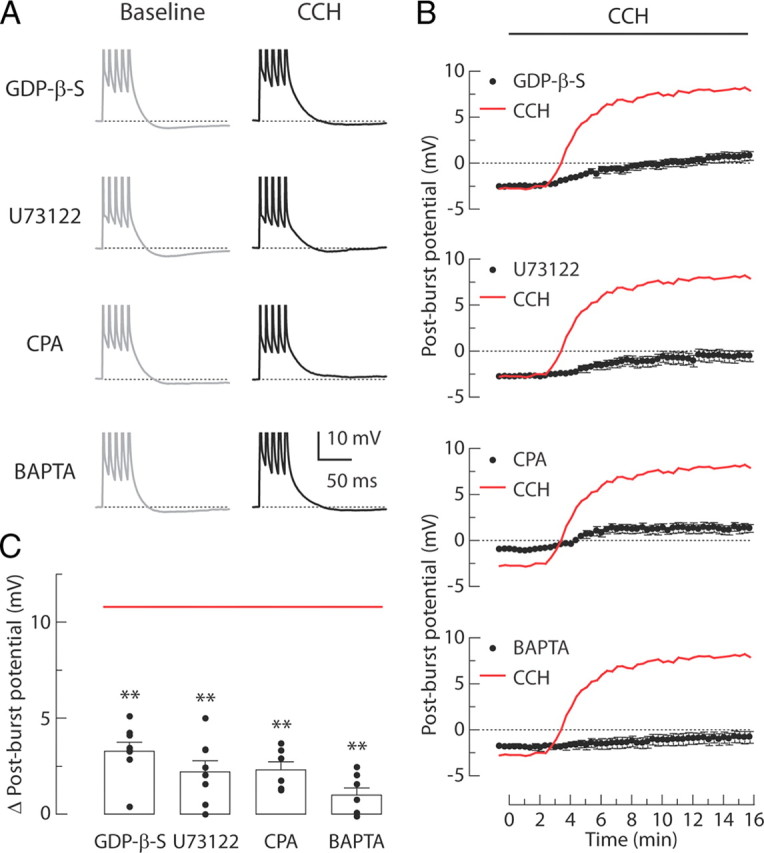
Signaling mechanisms involved in the CCH-induced post-burst ADPs. A, Individual responses to bursts of five APs in baseline condition (left, gray) or 15 min after CCH application (right, black). Experiments are performed in four different groups: 0.5 mm GDP-β-S, 20 μm U73122, 30 μm CPA, or 10 mm BAPTA. B, Time courses of the post-burst potential amplitudes after application of agonists beginning at t = 0 min. Red lines indicate the averaged response from the CCH-treated control group as in Figure 2B. C, Summary of the change in post-burst potentials (t = 15 min minus t = 0 min response) in each condition. Red line indicates the average of change in post-burst potentials from the CCH-treated control group in Figure 2C. Each symbol represents the amplitude of individual experiments. One-way ANOVA, p < 0.0001; post hoc tests versus CCH, **p < 0.001. GDP-β-S, n = 8; U73122, n = 8; CPA, n = 6; BAPTA, n = 7.
The mGluR-mediated post-burst ADP requires upregulation of Cav2.3 R-type calcium channels in CA1 pyramidal neurons (Park et al., 2010). This was also the case for the CCH-induced post-burst ADP, because application of Ni2+ (50 μm, which blocks Cav2.3 and Cav3.2 channels; Soong et al., 1993; Lee et al., 1999) prevented the ADP response in CCH-treated CA1 neurons (Fig. 4A,B) and the ADP was significantly reduced in Cav2.3 knock-out mice (Wilson et al., 2000) compared with wild-type mice (Fig. 4C,D). These results support the idea that activation of mAChRs (M1 subtype) and group I mGluRs both produce a change of the post-burst potential through a mechanism involving G-protein, PLC activation, Ca2+ release, and ultimately upregulation of R-type channels (Tai et al., 2006). Despite the common action of DHPG and CCH, we ruled out the possibility that either of these agonists was acting directly or indirectly to activate the other receptor subtype (Fig. 5).
Figure 4.

Involvement of Cav2.3 calcium channels in the CCH-induced post-burst ADP. A, B, Effects of Ni2+ on the CCH-induced ADPs. A, Individual responses to bursts of five APs in baseline condition (left, gray) or 15 min after application of 8 μm CCH (right, black) in the presence of 50 μm Ni2+. B, Time courses of the post-burst potential amplitudes after application of CCH beginning at t = 0 min (left). Red line indicates the averaged response from the CCH-treated control group without Ni2+ as shown in Figure 2B. Summary of the change in post-burst potentials (right, n = 10). Red line indicates the average of change in post-burst potentials from the CCH-treated control group in Figure 2C. Unpaired t test versus CCH, ***p < 0.0001. C, D, Effects of CCH on the ADP in wild-type (WT) and Cav2.3 knock-out (KO) mice. C, Individual responses in baseline condition (left, gray) or 15 min after application of 30 μm CCH (right, black) in either wild-type or knock-out mice. D, Time courses of the post-burst potential amplitudes after application of CCH beginning at t = 0 min (left). Summary of the change in post-burst potentials (right; wild type, n = 7; knock-out, n = 6). Unpaired t test, **p < 0.001.
Figure 5.
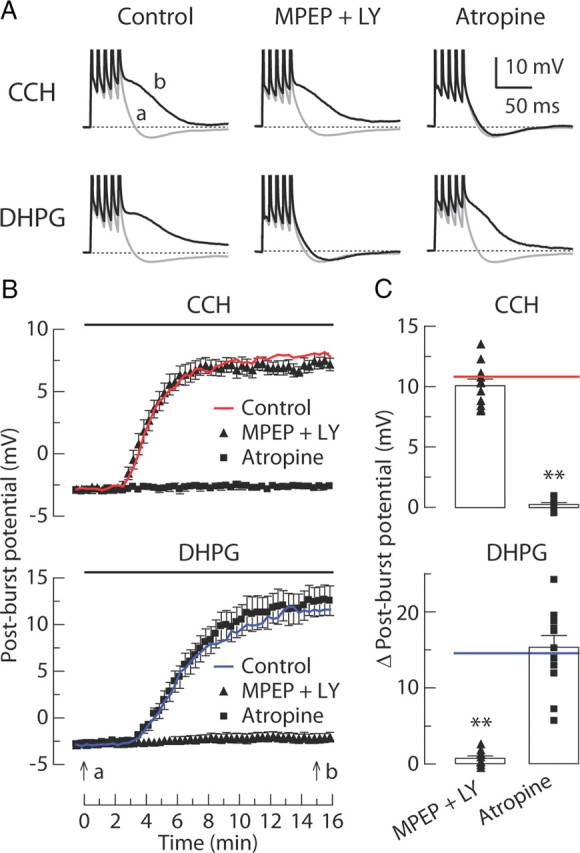
CCH-induced and DHPG-induced ADPs can occur without activation of the other receptor type. A, Individual responses to bursts of five APs in baseline condition (gray, t = 0 min in B) or after application (black, t = 15 min in B) of either 8 μm CCH or 2 μm DHPG. Experiments are performed in six different groups: control, 10 μm MPEP + 100 μm LY367385 (LY), and 10 μm atropine in either CCH groups or DHPG groups. B, Time courses of the post-burst potential amplitudes after application of either CCH or DHPG beginning at t = 0 min. C, Summary of the change in post-burst potential (t = 15 min minus t = 0 min response) in each condition. Each symbol represents the amplitude of individual experiments. Red line and blue line show the average of control in CCH groups and DHPG groups, respectively. In CCH groups: control, n = 13; MPEP + LY367385, n = 11; atropine, n = 9. In DHPG groups: control, n = 14; MPEP + LY367385, n = 9; atropine, n = 12. One-way ANOVA, p < 0.0001; post hoc tests versus control, **p < 0.001.
mGluRs and mAChRs have synergistic effects on hippocampal pyramidal neurons (Moore et al., 2009), so we investigated whether their concurrent activation could nonlinearly enhance the magnitude of the ADP. To examine this possibility, we first applied agonists using low concentrations (3 μm CCH or 0.75 μm DHPG) that reduced the AHP but did not induce an ADP. Doubling these concentrations (6 μm CCH or 1.5 μm DHPG) produced a change in the post-burst potential that was slightly more than double the effect of the low concentrations (Fig. 6C). Because the effects of the two low concentrations produced responses that were similar to each other, a synergistic effect of the low concentrations applied simultaneously would be indicated by an ADP response larger than that produced by the higher concentration of either agonist alone. Consistent with this, when the low concentrations of agonists were applied together, the change in the post-burst potential was 80% larger than the effect of the higher concentration of either agonist applied individually and 160% larger than the sum of the effects of the low concentrations applied individually (Fig. 6C). These results demonstrate that the effect of activating mGluRs and mAChRs is highly synergistic.
Figure 6.
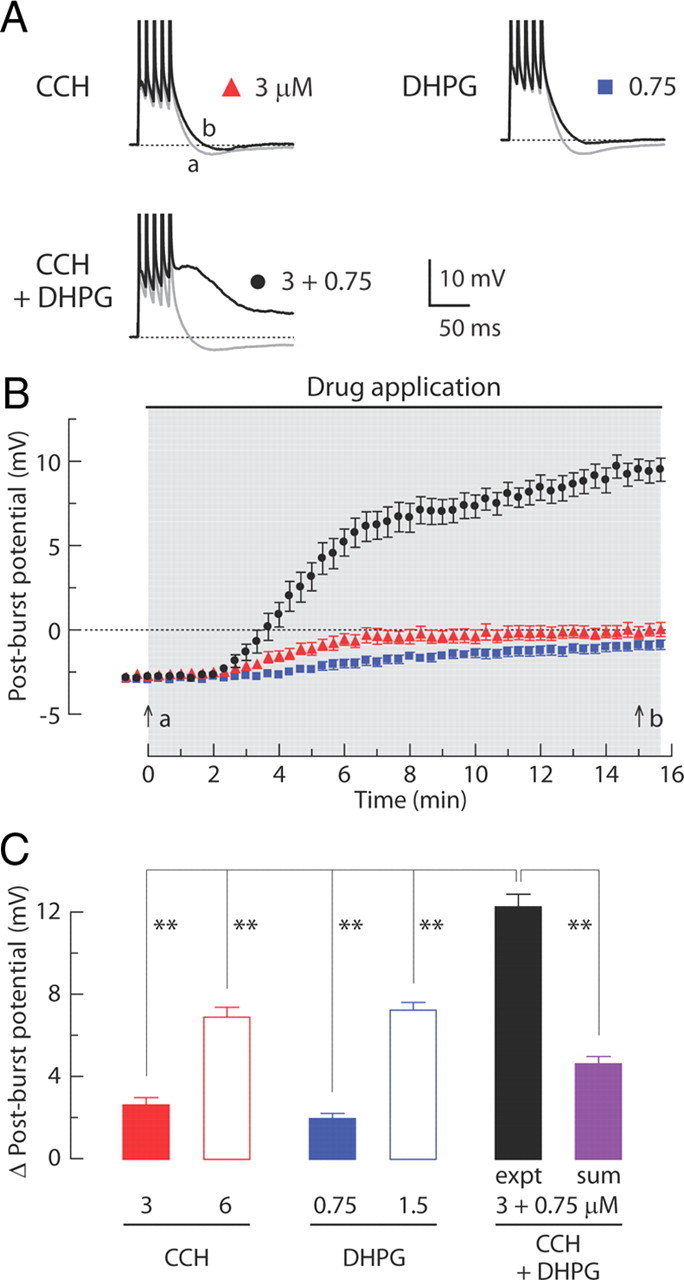
Coapplication of low concentrations of CCH and DHPG induces a post-burst ADP. A, Individual responses to bursts of five APs in baseline condition (gray, t = 0 min in B) or after application (black, t = 15 min in B) of CCH (3 μm), DHPG (0.75 μm), or both (3 μm CCH + 0.75 μm DHPG). B, Time courses of the post-burst potential amplitudes after application of CCH, DHPG, or both. Gray-shaded area indicates period of drug application. C, Summary of the change in post-burst potentials (t = 15 min minus t = 0 min response) in each condition. In CCH groups: 3 μm, n = 12; 6 μm, n = 11. In DHPG groups: 0.75 μm, n = 15; 1.5 μm, n = 11. In CCH + DHPG group: 3 μm CCH + 0.75 μm DHPG; experiment (expt), n = 16. For the linear sum, 16 randomly selected pairs were used from the independent measures in 3 μm CCH and 0.75 μm DHPG. One-way ANOVA, p < 0.0001; post hoc tests versus experiment, **p < 0.001.
Although there are many possible mechanisms for this synergy, one possibility is that both receptors act through a common intracellular effector signal such as PLC or one of its downstream effectors. To test the plausibility of such a mechanism, we developed a mathematical model in which each agonist increased the concentration of an intracellular signal in a manner described by the Hill equation and the ADP was induced by the intracellular signal, also in a manner described by the Hill equation. The concentration–response curves for the intracellular signal are entirely hypothetical but are constrained by a few observations (Fig. 6C): (1) the low concentrations of DHPG and CCH must produce approximately equal intracellular responses because they produce similar ADPs; (2) the high concentrations must produce approximately equal intracellular responses, for the same reason; (3) for the ADP to be explained by the Hill equation (or any other monotonic relationship), the sum of the intracellular signal produced by each low concentration had to be greater than the intracellular signal produced by either of the high concentrations. Using these constraints, we were able to develop a model that was consistent with our data (Fig. 7A–C). Two key features of the model were necessary to fit the data. First, the low and high concentrations that we used for each agonist had to be in a sublinear region of the dose–response curve for activation of the intracellular effector signal. Supralinear relationships would not work because this would result in intracellular signals from the high concentrations that exceeded the sum of the intracellular signal produced by the low concentrations of agonists (Fig. 7D). Second, the relationship between the ADP and the intracellular effector had to be supralinear, such that a small increase in the intracellular effector produced a large increase in the amplitude of the ADP. Although this model was consistent with our data, it by no means rules out more complicated explanations for the synergistic effect of activating both receptor types (see Discussion). For example, if there is a supralinear relationship between the metabotropic receptors and a common intracellular effector signal (Fig. 7D), then the ADP in response to combined activation cannot be explained by the action of this single intracellular effector (Fig. 7E), suggesting a more complex mechanism for the observed synergy (see Discussion).
Figure 7.
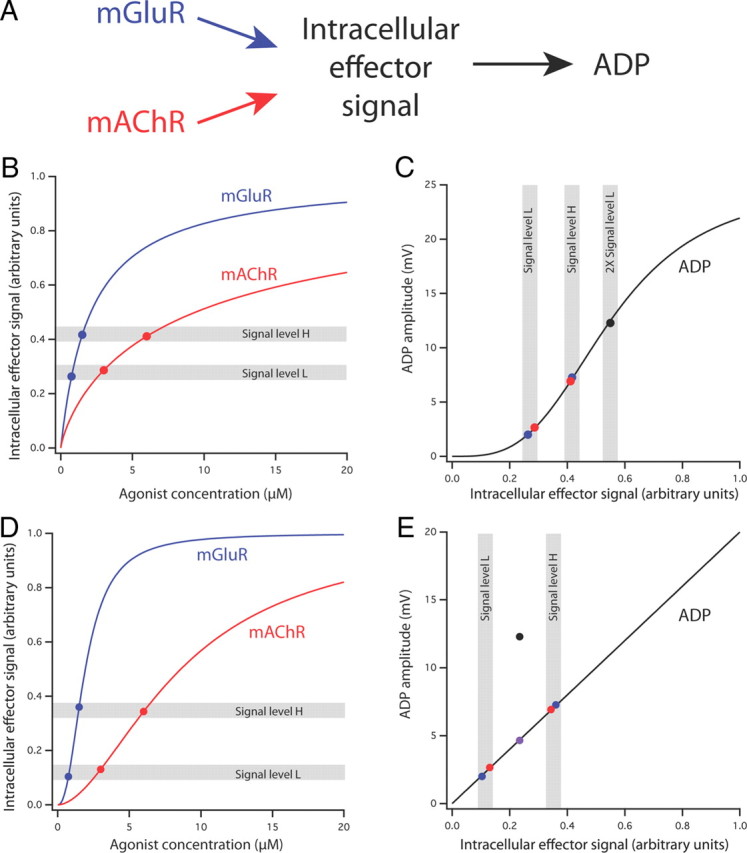
A mathematical model of synergistic activation of the ADP by mGluR and mAChR. A, Schematic representation of the model. Activation of mGluR and mAChR results in nonlinear activation of an intracellular effector signal (e.g., PLC or IP3). This signal in turn results in nonlinear activation of the ADP. B, Hypothetical concentration–response curves of the intracellular effector signal for agonists of mGluR (blue) and mAChR (red). Both curves were generated by the Hill equation (see Materials and Methods; mGluR, Hill coefficient, n = 1, microscopic dissociation constant, K = 2.1 μm; mAChR, n = 0.8, K = 9.4 μm). The two curves were constructed such that each function is sublinear over the range of concentrations used in the experiments (0.75 and 1.5 μm for mGluR; 3 and 6 μm for mAChR), and the low concentrations and high concentrations produced effector signal levels (indicated by gray bands L and H, respectively) that were approximately equal for the two agonists. C, Hypothetical concentration–response curve for the ADP produced by the intracellular effector signal (Hill equation, n = 3.3, K = 0.55 μm). Data points correspond to actual ADP response means shown in Figure 6C (mGluR, blue; mAChR, red; both together, black). Gray bands indicate signal levels shown in B, including two times signal level L. This combination of functions produces a good fit of the data, including the synergistic mGluR/mAChR response. D, Hypothetical concentration–response curves of the intracellular effector signal for agonists of mGluR (blue) and mAChR (red). Both curves were generated by the Hill equation (see Materials and Methods; mGluR, Hill coefficient, n = 2.3, microscopic dissociation constant, K = 1.9 μm; mAChR, n = 1.8, K = 8.6 μm). The two curves were constructed such that each function is supralinear over the range of concentrations used in the experiments. E, Hypothetical linear concentration–response curve for the ADP produced by the intracellular effector signal. This model can explain the separate mGluR and mAChR data, as well as the linear sum (purple point), but not the synergistic mGluR/mAChR response.
To determine whether synaptically released ACh and Glu could induce an ADP, we stimulated Schaffer collateral inputs to CA1 neurons in the presence of blockers of AMPA, NMDA, GABAA, and GABAB receptors (see Materials and Methods), to eliminate EPSPs and IPSPs, but without blocking mAChRs and mGluRs. The post-burst potential was monitored during a baseline condition (AP only; 2 ms somatic current steps at 100 Hz) and during high-frequency synaptic stimulation using an electrode positioned in proximal stratum radiatum (AP with synaptic stimulation; synaptic stimulation at 50 Hz) (Fig. 8A). In the absence of synaptic stimulation, each burst was followed by an AHP; however, when synaptic stimulation was delivered (on alternate trials), bursts during synaptic stimulation were followed instead by an ADP (Fig. 8A). The change in the post-burst potential was estimated from the difference between the post-burst potentials [Δ post-burst potential = ADP in AP with SYN − AHP in AP only (in which SYN is synaptic stimulation)]. To determine the involvement of synaptically stimulated mAChRs and group I mGluRs in the induction of the post-burst ADP, we applied either MPEP and LY367385 to block group I mGluRs or atropine to block mAChRs. The results of these experiments were compared with a separate group of control experiments performed over the same recording time but without the antagonists. In the control group, the change in the post-burst potential gradually increased over the course of the experiment (Fig. 8B,C; 202 ± 12% of initial value, n = 9). In contrast, antagonism of either receptor type resulted in a decrease in the modulation of the post-burst potential (Fig. 8B,C; MPEP + LY367385, 40 ± 7%, n = 7; atropine, 52 ± 4% of initial value, n = 7). The magnitudes of the change in the post-burst potential at the end of the experiment (t = 60 min) in the presence of antagonists were 20% (MPEP + LY367385) and 26% (atropine) of the control group, consistent with a substantial contribution of mAChRs and group I mGluRs to the induction of the post-burst ADP triggered by synaptically released neuromodulators.
Figure 8.
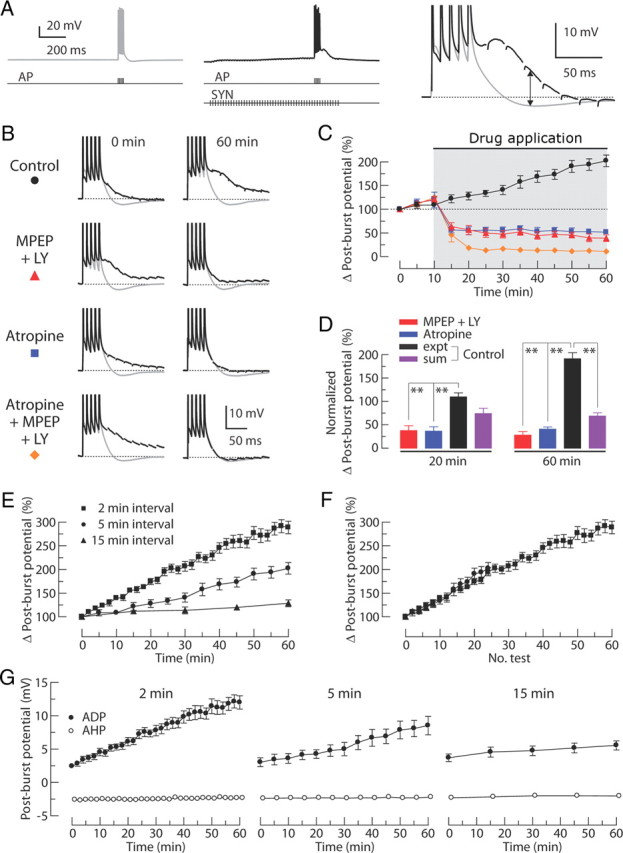
Effects of group I mGluR antagonists and mAChR antagonist on the post-burst ADP induced by synaptic stimulation (SYN) is paired with somatic AP to evoke the post-burst ADPs. Stimuli are five APs (100 Hz) alone or together with synaptic stimulation (50 Hz for 1 s). A, Typical responses from a single cell either without (left, gray) or with (middle, black) synaptic stimulation. Examples for each condition are expanded and overlaid on the right. Arrow indicates the difference between the AHP and the ADP (Δ post-burst potential). B, Alternating responses with and without synaptic stimulation (black and gray, respectively) are superimposed. Representative responses are obtained from the control group, 10 μm MPEP + 100 μm LY367385 (LY) group, 10 μm atropine group, or atropine + MPEP + LY367385 group at the beginning of the experiments (left, t = 0 min in C) and the end of the experiments (right, t = 60 min in C). C, Normalized change in post-burst potentials over time (control, n = 9; MPEP + LY367385, n = 7; atropine, n = 7; atropine + MPEP + LY367385, n = 7). Gray-shaded area indicates period of drug application. D, Summary of the change in post-burst potentials at t = 20 and t = 60 min in C. Responses from the control group, MPEP + LY367385 group, and atropine group are normalized to the magnitude of atropine + MPEP + LY367385 group. For the linear sum, nine randomly selected pairs were used from the independent measures in MPEP + LY367385 group and atropine group. One-way ANOVA, p < 0.0001; post hoc tests versus experiment (expt), **p < 0.001. E, Normalized change in post-burst potentials over time from experiments with different intervals between individual tests (2 min interval, n = 13; 5 min interval, n = 9; 15 min interval, n = 7). F, Data from E are replotted as a function of the number of tests. G, Absolute post-burst potential with or without synaptic stimulation (filled and open circles, respectively). Without synaptic stimulation, the post-burst AHP was stable in all groups.
Although the magnitude of the change in the post-burst potential in the control group gradually increased over the course of the experiment, the long-term increase in the synaptically induced ADP was eliminated in the conditions without either cholinergic or glutamatergic signaling, indicating that this increase requires signaling via both of these receptor types. The long-term effects of synaptic stimulation on the change in post-burst potential in the control group was activity dependent; the rate of growth of the ADP was relatively slow with long intervals between trials (15 min) and faster with shorter stimulation intervals (2 min) (Fig. 8E,F). Without synaptic stimulation, the post-burst AHP was stable over time in all groups (Fig. 8G).
We next examined whether the post-burst ADPs induced by synaptically released neuromodulators resulted from synergistic actions of the cholinergic and glutamatergic receptor types. We applied atropine, MPEP, and LY367385 to block signaling via both receptor types and monitored the resulting changes in the post-burst potential. Under these conditions, the magnitude of the change in the post-burst potential was decreased to 11 ± 3% of the initial value (Fig. 8B,C; n = 7). When normalized to the control group, the change in the post-burst potential was almost fully inhibited (95% inhibition). Because the magnitude of the change in the post-burst potential in the control group was 180% larger than the sum of the changes in the presence of antagonists at the end of the experiment (Fig. 8D; t = 60 min), we conclude that modulation of the post-burst potential in response to synaptically released neurotransmitters results from a synergistic action of mAChRs and group I mGluRs.
There are two possible explanations for the mAChR/mGluR-dependent growth of the ADP in response to repeated synaptic stimulation: it could be caused by a sensitization of the signaling that leads to an ADP in response to activation of mAChR or mGluR alone or it could be caused by a sensitization of the synergy between these two receptors and their respective signaling pathways. To distinguish between these possibilities, we allowed the ADP to be enhanced by repeated synaptic stimulation (in the absence of receptor antagonists) and then applied MPEP and LY367385 to block group I mGluRs, atropine to block mAChRs, or the combined antagonists to block both receptor types. We compared the ADP resulting from activation of one receptor type (i.e., mediated by mAChRs in the presence of MPEP and LY367385 or mediated by group I mGluRs in the presence of atropine) after short (10 min) and long (40 min) periods of sensitization by synaptic stimulation (2 min intervals). The longer sensitization period resulted in an enhancement of the ADP mediated by a single receptor type (Fig. 9A,B), and the sum of the enhanced mAChR-mediated ADP and the mGluR-mediated ADP was equivalent to the total sensitization observed with both signaling pathways intact (Fig. 9C). These results indicate that the sensitization produced by synaptic stimulation can be accounted for by summation of the enhanced ADP mediated by each individual receptor type, with no enhancement of their synergistic interactions. In a related experiment, we found that bath application of CCH and DHPG similarly enhanced the ADP in response to bath application of CCH or DHPG alone (Fig. 10).
Figure 9.
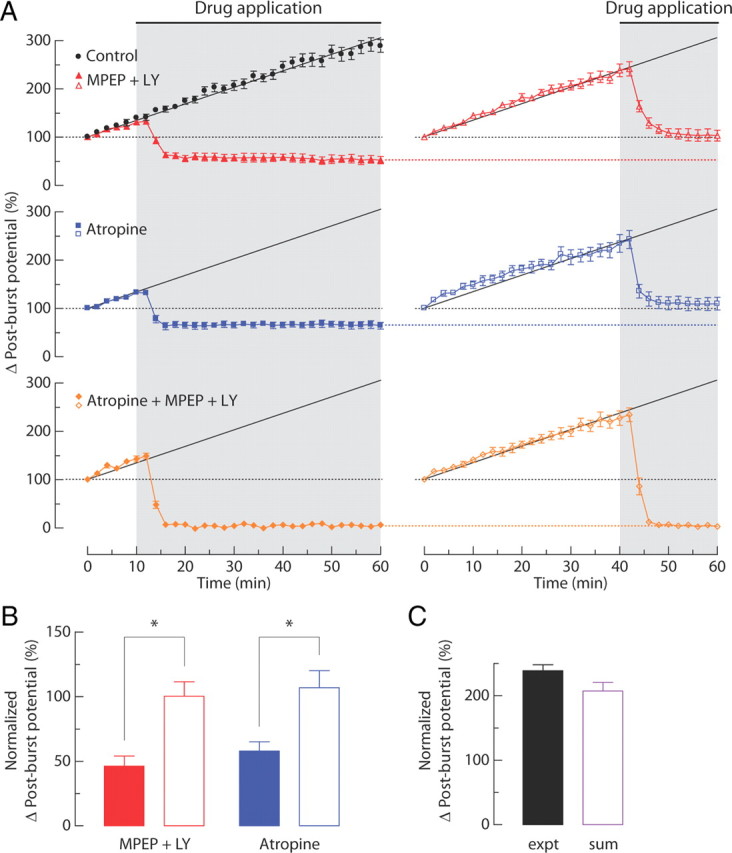
Effects of repeated synaptic stimulation on the post-burst potential induced in the presence of group I mGluR antagonists and mAChR antagonist. A, Normalized changes in post-burst potentials over time. Gray-shaded area indicates period of drug application (filled symbol, from t = 10 min; open symbol, from t = 40 min). A linear regression line to the control group is shown by a black line. B, Summary of the change in post-burst potentials at the end of experiments in MPEP + LY367385 (LY) and atropine group (filled bars, t = 60 min after drug application at t = 10 min; open bars, t = 60 min after drug application at t = 40 min). C, Change in post-burst potentials in the control at t = 42 min and the sum of MPEP + LY367385 and atropine group (open symbol in A) at t = 60 min. For the linear sum, 13 randomly selected pairs were used from the independent measures in MPEP + LY367385 group and atropine group. Unpaired t test, *p < 0.01. Control, n = 13; MPEP + LY367385, n = 7 each; atropine, n = 8 each; atropine + MPEP + LY367385, n = 7 each. expt, Experiment.
Figure 10.

Application of both CCH and DHPG increases the change of the post-burst potential induced by either CCH or DHPG. A, CCH-induced ADP. Time courses of the post-burst potential amplitudes induced by either CCH (left; n = 11) or CCH after CCH + DHPG (middle; n = 12). Summary of the change post-burst potentials at the end of experiments (right; unpaired t test, *p < 0.01). B, DHPG-induced ADP. Time courses of the post-burst potential amplitudes induced by either DHPG (left; n = 12) or DHPG after CCH + DHPG (middle; n = 10). Summary of the change in post-burst potentials at the end of experiments (right; unpaired t test, *p < 0.01).
Discussion
Together, our results suggest that concurrent activation of the M1 subtype of muscarinic receptors (mAChRs) and group I mGluRs (mGluR1 and mGluR5) activates signaling that is sufficient to modulate the post-burst potential in hippocampal CA1 pyramidal neurons. This modulation has both rapidly reversible and longer-lasting components, both of which could be induced by either bath application of receptor agonists or axon stimulation, resulting in release of ACh and Glu from presynaptic terminals. Both the rapidly reversible and the longer-lasting effects of activating these receptors displayed synergistic actions—effects that were much greater when mAChRs and mGluRs were activated together than when either type was activated on its own.
Although both the M1 mAChR and group I mGluR systems are well known to be capable of contributing to neuronal excitability in many brain areas (Benardo and Prince, 1982; McCormick and Prince, 1986; Greene et al., 1992; Kawasaki et al., 1999; McQuiston and Madison, 1999; Ireland and Abraham, 2002; Young et al., 2004; Lawrence et al., 2006; Pressler et al., 2007; Gulledge et al., 2009), very little is known about interactions between the two systems. A previous study identified long-lasting changes in intrinsic excitability associated with these two metabotropic receptor systems (Moore et al., 2009). This new work identifies an additional, acute effect, as well as an interaction between the acutely induced ADP and longer-lasting sensitization of the ADP by synergistic activation of M1 mAChRs and group I mGluRs.
There are several other neurotransmitter systems modulating neuronal excitability in the hippocampus. For example, activation of adrenergic receptors has been known to increase or decrease AP firing depending on the receptor subtypes (Pang and Rose, 1987; Mynlieff and Dunwiddie, 1988). We found that application of adrenergic or serotonergic agonists induced little or no change of the post-burst potential in hippocampal CA1 neurons (data not shown). In addition, synaptically released ACh and Glu were necessary and sufficient for the long-lasting sensitization of the ADP, suggesting that no other neurotransmitters were involved in these effects in our conditions.
Similar to our previous report showing that group I mGluR activation induces an ADP in CA1 neurons (Park et al., 2010), the M1 mAChR-mediated effects required activation of Gq/11-coupled receptors, PLC activation, intracellular Ca2+ release, and Cav2.3 R-type calcium channels. How can receptors that engage similar signaling pathways have synergistic effects that exceed increased activation of a single receptor type? One simple possibility would be that each receptor achieves limited activation of a single intracellular effector (e.g., PLC), whereas activation of both receptor types could produce more robust activation of the effector. We were able to establish the plausibility of this model mathematically, but other more complex mechanisms seem equally likely.
One such alternative mechanism is that optimal modulation of the afterpotential engages multiple signaling pathways, including pathways that are differentially activated by the two metabotropic receptors. It is well known that receptors coupling to Gq/11-protein can have other effects in addition to activation of PLC. In fact, there is a growing body of evidence that heptahelical receptors can signal via associations with intracellular signal molecules other than G-proteins (Hall et al., 1999). For example, in CA1 pyramidal neurons, a slow component of the post-burst ADP is not blocked by G-protein inhibitors (Park et al., 2010), and in oriens/alveus interneurons, group I mGluR-induced depolarization is not blocked by G-protein or PLC inhibitors (Gee and Lacaille, 2004). In addition to these examples suggesting the existence of G-protein-independent signaling, mAChR and group I mGluR systems may also signal with distinct molecules on the pathway downstream from Gq/11-proteins. For example, in isolated hippocampal CA1 neurons, mAChR and group I mGluR inhibition of IGIRK have been shown to be mediated by PLC/PKC and phospholipase A2/arachidonic acid signaling, respectively (Sohn et al., 2007).
How might these alternate signaling pathways get engaged preferentially by synergistic activation of mAChRs and mGluRs? One intriguing mechanistic possibility is that the two different G-protein-coupled receptors form a macromolecular complex (Smith and Milligan, 2010). The resulting protein–protein interactions in such receptor heteromers have been shown to result in a variety of effects on receptor function, including alterations in ligand binding affinity and downstream signaling (Rozenfeld and Devi, 2010), which could cause the signaling cascade involving Gq/11, PLC, and IP3 to be activated to a greater extent in the presence of both agonists than in the presence of only one agonist or cause the activation of some of the additional signaling pathways discussed above. Another possible mechanism is that synergistic effects arise from the activation of M1 mAChRs or group I mGluRs in adjacent cells, such as in presynaptic terminals or glia, thus leading to release of neurotransmitters from these structures and ultimately resulting in activation of other receptors and signaling pathways in pyramidal neurons.
We found that the induction of an ADP is dependent on modulation of Cav2.3 channels, but other channels may also be modulated—perhaps as a result of a synergistic effect of mAChRs and mGluRs—to reduce the AHP and/or enhance the ADP in concert with the R-type channel effect. One possible candidate is calcium-activated nonselective cation channels, which have been suggested to play a role in the modulation of the spike afterpotential (Greene et al., 1994; Guérineau et al., 1995; Fraser and MacVicar, 1996; Congar et al., 1997; McQuiston and Madison, 1999; Shalinsky et al., 2002; Lawrence et al., 2006). Another possibility is that K+ channels may be modulated in concert with Ca2+ channels (Constanti and Bagetta, 1991; Constanti et al., 1993; Greene et al., 1994). These possibilities are supported by the observation that changes in the post-burst potential are reduced but not eliminated in the Cav2.3 knock-out mice (Fig. 4). Although the long-lasting alterations responsible for the long-term sensitization of the ADP modulation we describe are unknown, there may be some overlap with the long-lasting enhancement of burst firing caused by synergistic activation of mAChRs and group I mGluRs after theta-burst synaptic stimulation of CA1 neurons (Moore et al., 2009).
The facilitatory effects of simultaneous activation of two neuromodulatory systems on neuronal activity have been described previously for the induction of LTP and LTD (Bröcher et al., 1992; Watabe et al., 2000; Scheiderer et al., 2008) as well as plasticity of bursting (Moore et al., 2009). Our study extends this concept to include synergistic effects that more rapidly modulate neuronal function. The ability of ACh and Glu, two major neuromodulators in the hippocampus, to modulate post-burst potentials synergistically provides a mechanism for hippocampal neurons to respond uniquely under conditions in which there is convergent activation of both systems, whereas activation of only one of the two systems would produce little or no response. These conditions can lead to both short-term and long-term changes in intrinsic excitability that may be responsible for ongoing modulation of hippocampal function during different behavioral states.
What novel properties might be conferred on the animal by engaging synergistic actions of two modulatory neurotransmitters? ACh is part of the reticular activating system, which plays a key role in regulating sleep–wake cycles. Although it is well known that the reticular activating system involves several neurotransmitters, the mechanisms by which multiple transmitters achieve changes in behavioral states are not known. The most obvious mechanism is that different neurotransmitters target different cell types. We show here, however, that two transmitters—ACh and Glu—can act synergistically to modulate excitability in one population of neurons.
In the hippocampus, ACh acts via multiple mechanisms, broadly regulating activity during the active-awake and rapid-eye-movement-sleep states (Hasselmo, 1999), whereas Glu is released selectively onto neurons that are part of the active network. Thus, the requirement for both transmitters could ensure that active neurons are modulated differently from silent neurons during appropriate behavioral states. For example, the modulation we describe might contribute to the well-documented reduction of the AHP that occurs during some forms of hippocampus-dependent learning (Zhang and Linden, 2003; Disterhoft et al., 2004). Another possibility is that enhanced excitability via activation of mAChRs and mGluRs could contribute to the plasticity required for place fields to form during active exploration of new environments. In both of these examples, the requirement for mGluR activation would restrict the plasticity to activated neurons, whereas the requirement for mAChR activation would ensure that the plasticity occurred only during appropriate behavioral states. Although such scenarios are entirely speculative, it is generally true that different combinations of modulatory transmitter receptors will be activated during different behavioral states, thus highlighting the importance of studying the interactions that occur between these neuromodulatory systems.
Footnotes
This work was supported by National Institutes of Health Grants NS-35180 and MH-074866. We thank Brett Mensh, Gabrielle Edgerton, Yujin Kim, Austin Graves, and Robert John Heuermann for helpful discussions and comments on this manuscript and Richard Miller for providing the Cav2.3 knock-out mice.
References
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Benardo LS, Prince DA. Cholinergic excitation of mammalian hippocampal pyramidal cells. Brain Res. 1982;249:315–331. doi: 10.1016/0006-8993(82)90066-x. [DOI] [PubMed] [Google Scholar]
- Blokland A. Acetylcholine: a neurotransmitter for learning and memory? Brain Res Rev. 1995;21:285–300. doi: 10.1016/0165-0173(95)00016-x. [DOI] [PubMed] [Google Scholar]
- Bröcher S, Artola A, Singer W. Agonists of cholinergic and noradrenergic receptors facilitate synergistically the induction of long-term potentiation in slices of rat visual cortex. Brain Res. 1992;573:27–36. doi: 10.1016/0006-8993(92)90110-u. [DOI] [PubMed] [Google Scholar]
- Cole AE, Nicoll RA. The pharmacology of cholinergic excitatory responses in hippocampal pyramidal cells. Brain Res. 1984a;305:283–290. doi: 10.1016/0006-8993(84)90434-7. [DOI] [PubMed] [Google Scholar]
- Cole AE, Nicoll RA. Characterization of a slow cholinergic post-synaptic potential recorded in vitro from rat hippocampal pyramidal cells. J Physiol. 1984b;352:173–188. doi: 10.1113/jphysiol.1984.sp015285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congar P, Leinekugel X, Ben-Ari Y, Crépel V. A long-lasting calcium-activated nonselective cationic current is generated by synaptic stimulation or exogenous activation of group I metabotropic glutamate receptors in CA1 pyramidal neurons. J Neurosci. 1997;17:5366–5379. doi: 10.1523/JNEUROSCI.17-14-05366.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constanti A, Bagetta G. Muscarinic receptor activation induces a prolonged post-stimulus afterdepolarization with a conductance decrease in guinea-pig olfactory cortex neurones in vitro. Neurosci Lett. 1991;131:27–32. doi: 10.1016/0304-3940(91)90329-r. [DOI] [PubMed] [Google Scholar]
- Constanti A, Bagetta G, Libri V. Persistent muscarinic excitation in guinea-pig olfactory cortex neurons: involvement of a slow post-stimulus afterdepolarizing current. Neuroscience. 1993;56:887–904. doi: 10.1016/0306-4522(93)90135-3. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Wu WW, Ohno M. Biophysical alterations of hippocampal pyramidal neurons in learning, ageing and Alzheimer's disease. Ageing Res Rev. 2004;3:383–406. doi: 10.1016/j.arr.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Fraser DD, MacVicar BA. Cholinergic-dependent plateau potential in hippocampal CA1 pyramidal neurons. J Neurosci. 1996;16:4113–4128. doi: 10.1523/JNEUROSCI.16-13-04113.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee CE, Lacaille JC. Group I metabotropic glutamate receptor actions in oriens/alveus interneurons of rat hippocampal CA1 region. Brain Res. 2004;1000:92–101. doi: 10.1016/j.brainres.2003.11.046. [DOI] [PubMed] [Google Scholar]
- Giocomo LM, Hasselmo ME. Neuromodulation by glutamate and acetylcholine can change circuit dynamics by regulating the relative influence of afferent input and excitatory feedback. Mol Neurobiol. 2007;36:184–200. doi: 10.1007/s12035-007-0032-z. [DOI] [PubMed] [Google Scholar]
- Greene C, Schwindt P, Crill W. Metabotropic receptor mediated afterdepolarization in neocortical neurons. Eur J Pharmacol. 1992;226:279–280. doi: 10.1016/0922-4106(92)90073-5. [DOI] [PubMed] [Google Scholar]
- Greene CC, Schwindt PC, Crill WE. Properties and ionic mechanisms of a metabotropic glutamate receptor-mediated slow afterdepolarization in neocortical neurons. J Neurophysiol. 1994;72:693–704. doi: 10.1152/jn.1994.72.2.693. [DOI] [PubMed] [Google Scholar]
- Guérineau NC, Bossu JL, Gähwiler BH, Gerber U. Activation of a nonselective cationic conductance by metabotropic glutamatergic and muscarinic agonists in CA3 pyramidal neurons of the rat hippocampus. J Neurosci. 1995;15:4395–4407. doi: 10.1523/JNEUROSCI.15-06-04395.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Bucci DJ, Zhang SS, Matsui M, Yeh HH. M1 receptors mediate cholinergic modulation of excitability in neocortical pyramidal neurons. J Neurosci. 2009;29:9888–9902. doi: 10.1523/JNEUROSCI.1366-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall RA, Premont RT, Lefkowitz RJ. Heptahelical receptor signaling: beyond the G protein paradigm. J Cell Biol. 1999;145:927–932. doi: 10.1083/jcb.145.5.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME. Neuromodulation: acetylcholine and memory consolidation. Trends Cogn Sci. 1999;3:351–359. doi: 10.1016/s1364-6613(99)01365-0. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME. The role of acetylcholine in learning and memory. Curr Opin Neurobiol. 2006;16:710–715. doi: 10.1016/j.conb.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland DR, Abraham WC. Group I mGluRs increase excitability of hippocampal CA1 pyramidal neurons by a PLC-independent mechanism. J Neurophysiol. 2002;88:107–116. doi: 10.1152/jn.2002.88.1.107. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Palmieri C, Avoli M. Muscarinic receptor activation induces depolarizing plateau potentials in bursting neurons of the rat subiculum. J Neurophysiol. 1999;82:2590–2601. doi: 10.1152/jn.1999.82.5.2590. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, Statland JM, Grinspan ZM, McBain CJ. Cell type-specific dependence of muscarinic signalling in mouse hippocampal stratum oriens interneurones. J Physiol. 2006;570:595–610. doi: 10.1113/jphysiol.2005.100875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HG, Zhu X, O'Neill MJ, Webber K, Casadesus G, Marlatt M, Raina AK, Perry G, Smith MA. The role of metabotropic glutamate receptors in Alzheimer's disease. Acta Neurobiologiae Experimentalis. 2004;64:89–98. doi: 10.55782/ane-2004-1494. [DOI] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E. Nickel block of three cloned T-type calcium channels: low concentrations selectively block alpha1H. Biophys J. 1999;77:3034–3042. doi: 10.1016/S0006-3495(99)77134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Prince DA. Mechanisms of action of acetylcholine in the guinea-pig cerebral cortex in vitro. J Physiol. 1986;375:169–194. doi: 10.1113/jphysiol.1986.sp016112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuiston AR, Madison DV. Muscarinic receptor activity induces an afterdepolarization in a subpopulation of hippocampal CA1 interneurons. J Neurosci. 1999;19:5703–5710. doi: 10.1523/JNEUROSCI.19-14-05703.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SJ, Cooper DC, Spruston N. Plasticity of burst firing induced by synergistic activation of metabotropic glutamate and acetylcholine receptors. Neuron. 2009;61:287–300. doi: 10.1016/j.neuron.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mynlieff M, Dunwiddie TV. Noradrenergic depression of synaptic responses in hippocampus of rat: evidence for mediation by alpha 1-receptors. Neuropharmacology. 1988;27:391–398. doi: 10.1016/0028-3908(88)90148-7. [DOI] [PubMed] [Google Scholar]
- Pang K, Rose GM. Differential effects of norepinephrine on hippocampal complex-spike and theta-neurons. Brain Res. 1987;425:146–158. doi: 10.1016/0006-8993(87)90493-8. [DOI] [PubMed] [Google Scholar]
- Park JY, Remy S, Varela J, Cooper DC, Chung S, Kang HW, Lee JH, Spruston N. A post-burst after depolarization is mediated by group i metabotropic glutamate receptor-dependent upregulation of Ca(v)2.3 R-type calcium channels in CA1 pyramidal neurons. PLoS Biol. 2010;8:e1000534. doi: 10.1371/journal.pbio.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- Power AE, Vazdarjanova A, McGaugh JL. Muscarinic cholinergic influences in memory consolidation. Neurobiol Learn Mem. 2003;80:178–193. doi: 10.1016/s1074-7427(03)00086-8. [DOI] [PubMed] [Google Scholar]
- Pressler RT, Inoue T, Strowbridge BW. Muscarinic receptor activation modulates granule cell excitability and potentiates inhibition onto mitral cells in the rat olfactory bulb. J Neurosci. 2007;27:10969–10981. doi: 10.1523/JNEUROSCI.2961-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel G, Platt B, Micheau J. Glutamate receptor function in learning and memory. Behav Brain Res. 2003;140:1–47. doi: 10.1016/s0166-4328(02)00272-3. [DOI] [PubMed] [Google Scholar]
- Rozenfeld R, Devi LA. Receptor heteromerization and drug discovery. Trends Pharmacol Sci. 2010;31:124–130. doi: 10.1016/j.tips.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiderer CL, Smith CC, McCutchen E, McCoy PA, Thacker EE, Kolasa K, Dobrunz LE, McMahon LL. Coactivation of M1 muscarinic and α1 adrenergic receptors stimulates extracellular signal-regulated protein kinase and induces long-term depression at CA3–CA1 synapses in rat hippocampus. J Neurosci. 2008;28:5350–5358. doi: 10.1523/JNEUROSCI.5058-06.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalinsky MH, Magistretti J, Ma L, Alonso AA. Muscarinic activation of a cation current and associated current noise in entorhinal-cortex layer II neurons. J Neurophysiol. 2002;88:1197–1211. doi: 10.1152/jn.2002.88.3.1197. [DOI] [PubMed] [Google Scholar]
- Smith NJ, Milligan G. Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol Rev. 2010;62:701–725. doi: 10.1124/pr.110.002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JW, Lee D, Cho H, Lim W, Shin HS, Lee SH, Ho WK. Receptor-specific inhibition of GABAB-activated K+ currents by muscarinic and metabotropic glutamate receptors in immature rat hippocampus. J Physiol. 2007;580:411–422. doi: 10.1113/jphysiol.2006.125914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong TW, Stea A, Hodson CD, Dubel SJ, Vincent SR, Snutch TP. Structure and functional expression of a member of the low voltage-activated calcium channel family. Science. 1993;260:1133–1136. doi: 10.1126/science.8388125. [DOI] [PubMed] [Google Scholar]
- Tai C, Kuzmiski JB, MacVicar BA. Muscarinic enhancement of R-type calcium currents in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:6249–6258. doi: 10.1523/JNEUROSCI.1009-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ure J, Baudry M, Perassolo M. Metabotropic glutamate receptors and epilepsy. J Neurol Sci. 2006;247:1–9. doi: 10.1016/j.jns.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Watabe AM, Zaki PA, O'Dell TJ. Coactivation of beta-adrenergic and cholinergic receptors enhances the induction of long-term potentiation and synergistically activates mitogen-activated protein kinase in the hippocampal CA1 region. J Neurosci. 2000;20:5924–5931. doi: 10.1523/JNEUROSCI.20-16-05924.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Toth PT, Oh SB, Gillard SE, Volsen S, Ren D, Philipson LH, Lee EC, Fletcher CF, Tessarollo L, Copeland NG, Jenkins NA, Miller RJ. The status of voltage-dependent calcium channels in α1E knock-out mice. J Neurosci. 2000;20:8566–8571. doi: 10.1523/JNEUROSCI.20-23-08566.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SR, Chuang SC, Wong RK. Modulation of afterpotentials and firing pattern in guinea pig CA3 neurones by group I metabotropic glutamate receptors. J Physiol. 2004;554:371–385. doi: 10.1113/jphysiol.2003.051847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci. 2003;4:885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]