

Abstract
MMP-9 deficiency protected against photochemical thrombosis induced brain hemorrhagic transformation (HT), but it did not protect against tissue plasminogen activator induced brain hemorrhage. The roles of MMP-2 and/or MMP-9 knockout (KO) in mechanical reperfusion induced HT after ischemia have not been investigated. Here we assessed the effects of MMP-2 KO, MMP-9 KO and MMP-2/9 double KO (dKO) in protecting against mechanical reperfusion induced HT and other brain injuries after the early stages of cerebral ischemia in mice of the same genetic background. Middle cerebral artery occlusion (MCAO) was performed in mice. Reperfusion was started at 1 or 1.5 hours after onset of MCAO. All mice were sacrificed 8 hours after MCAO. We found that both pro- and active MMP-2 and MMP-9 levels were significantly elevated in the early ischemic brain. After the early stages of ischemia and reperfusion, the hemorrhagic incidence was reduced in the cortex of MMP-2 KO mice (p < 0.05 vs. WT). The hemorrhagic volume was significantly decreased in the cortexes of MMP-2 and/or -9 knockout mice (MMP-9 KO vs. WT: p < 0.01, MMP-2 KO and dKO vs. WT: p < 0.001). In the basal ganglia, MMP-2 KO and MMP-2/9 dKO mice displayed a remarkable decrease in hemorrhagic volume (p < 0.01 or 0.05 vs. WT), but MMP-9 KOs did not protect against hemorrhage. MMP-2 and/or -9 knockout mice displayed significantly decreased infarction volume in both the cortex and striatum, in addition to improved neurological function (p < 0.001 vs. WT). The results suggested that MMP-2 deficiency and MMP-2 and MMP-9 double deficiency were more protective than MMP-9 deficiency against HT after the early stages of ischemia and reperfusion. These studies increase our understanding of MMP-2 and MMP-9 in HT development and will help to selectively target MMPs to protect the post-ischemic brain from injury and HT.
Keywords: cerebral ischemia, hemorrhagic transformation, MMP-2 knockout, MMP-9 knockout, MMP-2/9 double knockout, mice
Although it is well known that broad-spectrum matrix metalloproteinase (MMP) inhibitors reduce tissue plasminogen activator (t-PA)-induced hemorrhagic transformation (HT) in the clotstroke model (Lapchak et al., 2000; Sumii and Lo, 2002), a discrepancy about the role of MMP-9 in the development of brain hemorrhage exists. Previous studies indicate that knocking out MMP-9 protects blood-brain-barrier (BBB) function and brain infarct in CD-1 background mice (Asahi et al., 2000; Asahi et al., 2001b). In a stroke model of photochemical thrombosis, MMP-9 deficiency protected against HT after ischemia without t-PA thrombolysis, but it did not protect against t-PA-induced brain hemorrhage in 129SV/CD1mice (Suzuki et al., 2007). So far, it is unknown whether MMP-9 KO will protect mechanical reperfusion induced HT after cerebral ischemia.
Increased proMMP-2 levels coincided with early extravasations of plasma constituents and neuronal injury in non-human primates (Heo et al., 1999), but the relationship between MMP-2 and HT development after ischemia and reperfusion has not been established (del Zoppo, 2010). Little experimental evidence is available about the roles of MMP-2 KO in BBB injuries and HT development. It is also unclear whether MMP-2 KO protects early stage ischemic reperfusion injuries although it was reported that MMP-2 KO did not reduce the infarct volume at 24 hour in C57BL/6J background mice (Asahi et al., 2001a).
Previous study indicated that MMP-2/9 inhibitors rescued laminin from proteolysis and neurons from apoptosis after ischemia (Gu et al., 2005), but it is unknown whether MMP-2 and MMP-9 synergistically contribute to ischemic reperfusion injuries and HT.
The efficacy and safety of t-PA have been firmly established when used within 3 h after the onset of stroke symptoms. New evidence has demonstrated significant improvement in selected patients when t-PA is administered within 4.5 hours after the onset of the symptoms (Stemer and Lyden, 2010). Mechanical recanalization also produced favorable outcomes in patients when used within 8 hours of the onset of stroke symptoms (Smith, 2006). Our previous study indicated that brain hemorrhage and mortality peaked at 3 to 6 hours post-reperfusion after a prolonged ischemia (Lu et al., 2009). Therefore, the early stages of ischemia, reperfusion injuries and HT could be the potential effective window for MMP-2 and/or –9 inhibitions.
The goal of the current study is to investigate the effects of MMP-2 KO, MMP-9 KO and MMP-2/9 double KO (dKO) in protecting against mechanical reperfusion induced HT and other brain injuries after the early stages of ischemia and reperfusion and compare the effectiveness of their effects in mice of the same genetic background (C57BL/6J). A mouse suture middle cerebral artery occlusion model (MCAO) was used to produce the ischemia and mechanical reperfusion. Eight hours after the ischemia and reperfusion, we assessed MMP-2 and -9 activation, injuries to the basal lamina, the incidence and the amount of hemorrhage, along with brain infarcts, edemas and neurological functions.
Experimental procedures
Animals
The University of Cincinnati Animal Care Committee approved all experimental procedures. All procedures conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
MMP-2 KO (MMP-2−/−) mice in the C57BL/6 J genetic background were a kind gift from Dr. L. Matrisian (Vanderbilt University, Nashville, TN). The homozygous MMP-2-deficient mice were the progeny of heterozygous breeding pairs of C57BL/6 J mice with a disruption in the MMP-2 gene. These breeding pairs were backcrossed for more than six generations (Itoh et al., 1997). MMP-9 KO (MMP-9−/−) mice in the C57BL/6J genetic background were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and crossed to C57BL/6J mice for more than five generations before being made homozygous. MMP-2 KO (MMP-2−/−) mice were crossed with MMP-9 KO (MMP-9−/−) mice to generate heterozygous (MMP-2+/−MMP-9+/−) mice. These mice were then intercrossed to obtain double knockout (MMP-2−/− MMP-9−/−) and control wild-type (WT, MMP-2+/+MMP-9+/+) mice. MMP-2−/− MMP-9−/− male and MMP-2+/− MMP-9−/− female mice reproduced normally and no abnormal phenotype was observed except that a few mice exhibited hydrocephalus. The genotypes of all mice were verified using PCR. The animals were allowed to access food and water ad libitum and were housed with a 12-hour light–dark cycle.
In all experiments, we used male mice that were 13 to 15 weeks of age with 25 to 27 g body weight.
Cerebrovascular anatomy
An established vascular casting method was used to assess the cerebrovascular angioarchitecture (Maeda et al., 1998). Briefly, the mice were anesthetized before the cerebrovasculature was perfused with Latex (Latex injection solution, ScholAR Chemistry) mixed with a small amount of India ink (American Master Tech). The anastomoses are defined as the narrowest part of the vessels or halfway between the nearest branching points of the middle and anterior cerebral arteries. First, the total number of anastomoses per hemisphere was counted. Next, the identified anastomose points were connected into a line and the distance from midline was measured at 2, 4, and 6 mm from the frontal pole with Axiovision software (Zeiss).
The middle cerebral artery occlusion model (MCAO) and the experimental groups
The standard intraluminal middle cerebral artery occlusion method was used to induce focal ischemia, as previously described (Lu et al., 2008; Engel et al., 2011). Briefly, each mouse was anesthetized with 1.5% isoflurane in 28.5% oxygen and 70% nitrous oxide using a face mask. The rectal temperature of all animals was maintained at 37 ± 0.5 °C with a feedbackcontrolled heating blanket. Mice were placed in the supine position. Following a midline skin incision, the left common carotid artery, the external carotid artery, and the internal carotid artery were exposed. A 6.0 nylon monofilament coated with silicon was introduced into the left internal carotid artery through the external carotid artery to occlude the origin of the middle cerebral artery. The sutures were randomly assigned to the mice in different groups. The wound was then closed, and the suture was kept in place. After 1 h or 1.5 h ischemia, the mice were reanesthetized, the neck skin was reopened and the nylon suture was removed to achieve reperfusion. Intracranial ischemia and reperfusion were confirmed by laser Doppler flowmetry (5 mm lateral and 2 mm posterior to bregma). To prevent hypothermia after surgery, the mice were transferred to a temperature-controlled incubator at 37 °C for 20 min until the animals woke up completely. The mice were then transferred to cages with Delta Phase Isothermal Pads (Braintree scientific, Inc.). Five groups of animals were studied: (1) sham group: a sham operation in WT mice (MMP-2+/+MMP-9+/+); (2) WT group: ischemia–reperfusion in WT mice (MMP-2+/+MMP-9+/+); (3) MMP-2 KO group: ischemia–reperfusion in MMP-2 KO mice (MMP-2−/−); (4) MMP-9 KO group: ischemia–reperfusion in MMP-9 KO mice (MMP-9−/−); and (5) dKO group: ischemia–reperfusion in MMP-2/9 dKO mice (MMP-2−/− MMP-9−/−). The number of animals for each group is listed in the figure legends. The surgical procedure of the sham operation group was the same as the other three groups, but there was no suture insertion and occlusion of the MCA. All mice were sacrificed 8 h after either the sham operation or the onset of ischemia. For gel zymography and immunohistochemistry, animals experienced 1.5 h ischemia. Four died animals (2 in WT group, 1 in MMP-2 KO group and 1 in dKO group) were excluded from data collection. To avoid animal death, the ischemic time was shortened to 1 h for brain infarct, hemorrhage, brain swelling and behavior measurement.
Gelatin zymography
Gelatin zymography was performed as previously described (Suofu et al., 2010). Briefly, brains were quickly removed and sliced into four 2-mm coronal slices at 4 °C. Brain slices were incubated in 2.3.5-triphenyltetrazolium chloride (TTC) (Sigma, St. Louis, MO) for 10 min at 37 °C. The infarction area was then dissected and homogenized. Supernatants obtained from homogenized brains were then recovered and incubated with gelatin-Sepharose 4B. After centrifugation, the pellets were eluted in buffer containing 10% dimethyl sulfoxide. The proteins in the eluent were separated by electrophoresis through 10% polyacrylamide zymogram gels containing gelatin (Invitrogen, Carlsbad, California, USA). After washing in 2.5% Triton X-100 to remove SDS, the gels were washed in 50 mM Tris-HCl, pH 7.5, and incubated overnight in developing buffer (50 mM Tris-HCl, pH 7.5, 10 mM CaCl2, 1μM ZnCl2, and 0.05% BRIJ-35) at 37 °C. The gels were then stained with 0.1% Coomassie blue and then destained. MMP activity was represented by clearly digested regions, which were then photographed and analyzed using the MicroComputer Image Device (MCID) imaging system.
Immunohistochemistry
Immunohistochemistry analysis was performed as previously described (Suofu et al., 2010). Briefly, frozen coronal sections (20 μm) were used. After fixing in 2% paraformaldehyde for 10 min, the sections were blocked for 1.5 h in blocking buffer (2% serum, 0.2% Triton X-100, 0.1% bovine serum albumin in 0.1 M PBS). Following incubation with rabbit anti-laminin (1:4000, Sigma, St. Louis, MO), the sections were washed in PBS and incubated with biotinylated secondary antibodies for 1.5 h (Vector Laboratories, Burlingame, CA, USA). Next, ABC reagent (Vector Laboratories) was applied to sections after three 5-min PBS washes. Finally, diaminobenzidine was used to visualize the horseradish peroxidase. Four images were taken in the cortex and the striatum. The percentage of laminin-positive microvascular area in the measured image area (% of stained area) and the average microvascular number in each square millimeter of the image (numbers/mm2) were measured and calculated using the MicroComputer Image Device (MCID) imaging system. The average microvascular size, which is the microvascular area in the image divided by the total microvascular number in the image, was also calculated.
Hemorrhagic rates and volumes
At 8 h, the mice were sacrificed, and seven coronal slices per brain (1 mm thickness) were prepared using a brain matrix. The slices were scanned for the quantification of hemorrhagic areas. The hemorrhagic area on the surface of the slice was quantified with 10-fold magnification using an MCID imaging system, and the hemorrhage volume of each slice was calculated as the hemorrhagic area on each surface times the slice thickness. The incidence of hemorrhage was calculated as the number of mice with hemorrhage divided by the total number of mice in each group (Lu et al., 2009).
Infarction volume, edema and neurological deficits
Following scans of the slices for quantification of hemorrhages, the brain slices were then stained with 2% 2.3.5-triphenyltetrazolium chloride (TTC) for 10 min in a 37 °C water bath (Liu et al., 2009). Areas that were not stained red with TTC were identified as the infarct area. After staining, the brain slices were fixed with 4% paraformaldehyde for 30 min and scanned. The standard indirect method was used for measuring the areas of infarction on each surface of the slice, and ischemic lesion volumes were quantified with the MCID automated imaging system. Brain edemas were calculated as previously described (Lu et al., 2009).
A vibrissae-stimulated forepaw placing test was used to assess deficits in the sensorimotor cortex or the striatum (Belayev et al., 2003). The animals were gently held by their neck skin with their forelimbs hanging freely. Stimulation of the right vibrissae by gently brushing against the edge of a table results in responses by the right forepaw. A successful placement response was determined by the ability of the mice to place a forepaw on the table. The test was conducted immediately before sacrifice. For each mouse, 10 trials were performed in triplicate.
Statistical analysis
Quantitative data were expressed as the mean ± SD. Statistical comparisons were conducted using ANOVA for group comparisons. Hemorrhage rates were analyzed using chisquared test. Differences with p < 0.05 were considered statistically significant.
Results
Cerebrovascular boundaries and post-ischemic changes in regional cerebral blood flow are similar in MMP-2 KO, MMP-9 KO, MMP-2/9 dKO and wild-type mice
Figure 1 shows the MCA cerebrovascular anatomy after the injection of the vascular system with latex in wild-type, MMP-2 KO, MMP-9 KO and MMP-2/9 dKO mice. The vascular boundaries of the MCA were not significantly different between the groups (Table 1).
Figure 1. Dorsal view of the cerebral hemisphere of C57BL/6J wild-type, MMP-9 KO, MMP-2 KO and MMP-2/9 dKO mice after microvascular injection with India ink-stained latex (n = 4 for each group).
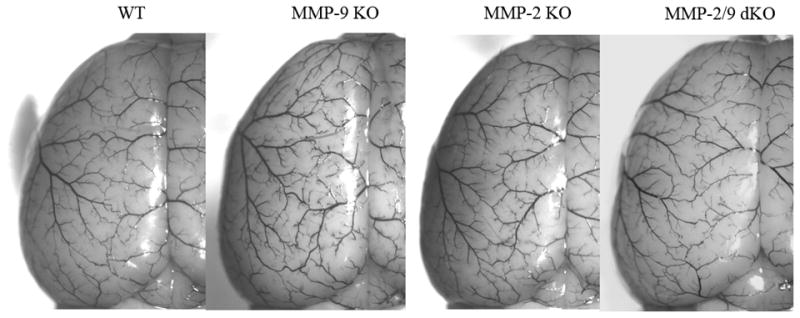
There is a similar distribution of dorsal vessels and boundary zones for the anterior and middle cerebral arteries in all groups.
Table 1.
Vascular boundaries between the anterior and middle cerebral arteries.
| WT | MMP-9 KO | MMP-2 KO | dKO | |
|---|---|---|---|---|
| Distance of the boundary from the midline | ||||
| 2 mm from the frontal pole | 1.7 ± 0.1 | 1.6 ± 0.1 | 1.6 ± 0.1 | 1.6 ± 0.1 |
| 4 mm from the frontal pole | 2.2 ± 0.1 | 2.0 ± 1.7 | 2.2 ± 0.3 | 2.1 ± 0.2 |
| 6 mm from the frontal pole | 2.6 ± 0.3 | 2.3 ± 0.3 | 2.4 ± 0.2 | 2.4 ± 0.3 |
| Number of anastomoses per hemisphere | 9.2 ± 0.5 | 9.2 ± 0.4 | 8.6 ± 0.9 | 8.4 ± 0.5 |
dKO: MMP-2/9 double knockout.
Regional cerebral blood flow (rCBF) was measured in the cortex at the baseline, 10 min after the onset of ischemia, and 10 min before and after reperfusion in all ischemic mice and shown in Table 2. After ischemia, the rCBF decreased in all groups (p < 0.01 vs. baseline value). The degree of ischemia was not significantly different between the groups. After reperfusion, the values of the average rCBF recovered to the pre-ischemic levels (Table 2).
Table 2.
The change in regional cerebral blood flow after ischemia-reperfusion.
| before MCAO | 10 min after MCAO | 10 min before reperfusion | 10 min after reperfusion | |
|---|---|---|---|---|
| WT | 100 | 13.8 ± 3.9** | 15.2 ± 3.9** | 123.5 ± 43.3 |
| MMP-9 KO | 100 | 12.9 ± 4.6** | 18.3 ± 5.4** | 116.6 ± 33.5 |
| MMP-2 KO | 100 | 12.5 ± 3.4** | 16.7 ± 5.1** | 107.9 ± 33.1 |
| dKO | 100 | 11.7 ± 5.2** | 13.4 ± 5.8** | 116.7 ± 49.0 |
dKO: MMP-2/9 double knockout. MCAO: middle cerebral artery occlusion.
p < 0.01 compared with the levels before MCAO.
Both MMP-2 and MMP-9 were upregulated in the early stage of ischemic-reperfusion injuries
Gel zymography detected basal levels of both MMP-2 and MMP-9 in the sham operation mouse brains. After ischemic reperfusion injury, however, both pro- and active MMP-2 and MMP-9 levels were significantly elevated in the wild-type mice compared with sham control (Fig 2). MMP-2 and/or -9 gelatinase expression and activity were eliminated in MMP-2 and/or -9 KO mice, as demonstrated by gel zymography (Fig. 2a). MMP-2 levels in MMP-9 KO or MMP-9 levels in MMP-2 KO mice did not reach statistical significance when compared individually to those in wild-type mice (Fig. 2b and Fig. 2c).
Figure 2. Gel zymogram and quantification of band density in the brains of adult mice after focal MCA ischemia (n = 4).

I, ischemia; R, reperfusion; dKO, double knockout. #p < 0.05, ##p < 0.01 compared with the sham group. a) representative images. b) and c) quantification of MMP-2 and -9 levels. MCA ischemia for 1.5 h followed by reperfusion for 6.5 h leads to increased proMMP-2 and proMMP-9 levels and increased MMP-2 and -9 activation (a, b and c). MMP-9 KO did not significantly increase MMP-2 levels (b), and MMP-2 KO did not significantly increase MMP-9 levels (c) when compared with the WT group after focal MCA ischemia.
MMP-2 KO, MMP-9 KO and MMP-2/9 dKO protected against injuries to the cerebral microvascular basal lamina during the early stage of cerebral ischemia and reperfusion
Laminin immunohistochemistry was performed to assess injuries to the basal lamina in cerebral blood vessels in mice. The loss of the laminin in WT mice after ischemia and reperfusion was significant in the cortex and striatum following 1.5 hours of ischemia and 6.5 hours of reperfusion (a total of 8 h), as determined by the percentage of laminin-stained area compared with sham controls (Fig. 3a–c; p < 0.01). The laminin-positive microvessels were degraded to short segments with a decreased average microvascular size (Table 3; p < 0.01). The microvascular basal lamina were significantly protected from ischemia reperfusion injury in MMP-2 KO (p < 0.01 vs. WT), MMP-9 KO (p < 0.05 vs. WT) and MMP-2/9 dKO (p < 0.01 vs. WT) mice in the cortex but not in the striatum (Fig. 3a, b; Table 3).
Figure 3. Laminin immunohistochemistry and quantification of the laminin staining in adult mice after focal MCA ischemia (n = 5).

The quantification of laminin immunopositive microvasculature (% of laminin-stained area) in the cortex (a) and the striatum (b). Representative figures (10×) in the cortex (c), scale bar = 100 μm; I, ischemia; R, reperfusion; dKO, MMP-2/9 double knockout group; ###p < 0.001 compared with the sham control, **p < 0.01 and ***p < 0.001 compared with the WT ischemia-reperfusion control. MMP-2 and/or -9 KO protected against injuries to the basal lamina of laminin immunopositive microvasculature.
Table 3.
The effects of MMP-2 and/or -9 KOs on the average microvascular size and numbers of laminin-immunoreactive microvessels in the cortex and the striatum.
| Groups | Cortex | Striatum | ||
|---|---|---|---|---|
| Average microvascular size (μm2/profile) | Microvascular numbers/mm2 tissue | Average microvascular size (μm2/profile) | Microvascular numbers/mm2 tissue | |
| Sham | 276.4 ± 29.3 | 332.5 ± 17.0 | 281.5 ± 47.9 | 286.1 ± 11.6 |
| WT | 155.2 ± 10.5## | 307.8 ± 30.7 | 134.5 ± 11.9## | 275.0 ± 25.3 |
| MMP-2 KO | 185.7 ± 30.3## | 347.7 ± 47.1 | 152.9 ± 25.5## | 261.5 ± 34.3 |
| MMP-9 KO | 170.0 ± 24.0## | 344.3 ± 40.2 | 149.4 ± 14.4## | 258.5 ± 28.9 |
| dKO | 180.6 ± 25.7## | 356.5 ± 57.3 | 144.0 ± 21.7## | 285.8 ± 20.4 |
dKO: MMP-2/9 double knockout.
p < 0.01 compared with the sham control.
MMP-2 KO and MMP-2/9 d KO mice displayed better protection against hemorrhagic transformations than MMP-9 KO mice in the early stage of ischemic reperfusion injuries
The early stage of ischemia and reperfusion causes hemorrhagic infarction (HI), showing multiple dots, small petechiae (HI1) and more confluent petechial hemorrhage (HI2) (Fig. 4c). The hemorrhagic rates were reduced in MMP-2 KO (p < 0.05 vs. WT) in the cortex (Table 4). Hemorrhagic volumes were significantly decreased in MMP-2 KO (p < 0.001 vs. WT), MMP-9 KO (p < 0.01 vs. WT) and MMP-2/9 KO mice (p < 0.001 vs. WT) in the cortex (Fig. 4a). In the basal ganglia, MMP-2 KO and MMP-2/9 dKO mice also displayed a remarkable decrease in hemorrhagic volume (p < 0.01 or 0.05 vs. WT, Fig. 4b). However, knocking out MMP-9 did not decrease hemorrhagic volume in the basal ganglia.
Figure 4. Effects of MMP-2 and/or -9 knockouts on brain hemorrhage transformation after 1 h ischemia and 7 h reperfusion(n = 15 in the WT group. n = 11 in the MMP-9 KO group; n = 10 in another two groups).

a) Cortex hemorrhage, b) basal ganglia hemorrhage, c) representative images. dKO: MMP-2/9 double knockout group. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the WT ischemia-reperfusion control. &p < 0.05 and &&p < 0.01 compared with the MMP-9 KO group. MMP-9 KO did not protect against hemorrhage in the basal ganglia.
Table 4.
Rate of cortex and basal ganglia hemorrhage.
| Cortex | Basal ganglia | |||||
|---|---|---|---|---|---|---|
| Groups | Mice with hemorrhage | Mice with no hemorrhage | Hemorrhage rate (%) | Mice with hemorrhage | Mice with no hemorrhage | Hemorrhage rate (%) |
| WT | 15 | 0 | 100 | 15 | 0 | 100 |
| MMP-9 KO | 10 | 1 | 90.9 | 11 | 0 | 100 |
| MMP-2 KO | 7 | 3 | 70* | 10 | 0 | 100 |
| dKO | 9 | 1 | 90 | 10 | 0 | 100 |
DKO, MMP-2/9 double knockout.
p < 0.05 compared with the WT ischemia-reperfusion control.
Brain infarction volume, edema and neurological deficits were reduced in MMP-2 and/or - 9 KO mice in the early stages of ischemic-reperfusion injuries
Fig. 5 illustrates the volumes of infarction. At 8 h after the onset of ischemia, infarction volumes were significantly reduced in MMP-2 KO, MMP-9 KO and MMP-2/9 dKO mice in both the cortex and the basal ganglia (Fig. 5a and b, p < 0.001 vs. WT). The protective effects of the MMP-2/9 dKO mice were not significantly different than the MMP-2 or -9 single KO. There were no observed cortex infarction in two MMP-2 KO mice and one MMP-2/9 dKO mouse. MMP-2 KO mice also displayed decreased brain swelling in the cortex (Fig. 6a, p < 0.05 vs. WT). Compared with wildtype mice, neurological deficits were ameliorated in MMP-2 KO, MMP-9 KO and MMP-2/MMP-9 dKO mice (Fig. 6b, p < 0.001 vs. WT).
Figure 5. Effects of MMP-2 and/or -9 knockouts on brain infarct in mice after 1 h ischemia and 7 h reperfusion (n = 12 in the MMP-2 KO group; n = 13 in other groups).

a) cortex, b) basal ganglia, c) representative images (TTC staining, white area represents infarct area). dKO: MMP-2/9 double knockout group. ***p < 0.001 compared with the WT ischemia-reperfusion control. MMP-2 and/or -9 knockouts had equal protection against brain infarct after 1 h ischemia and 7 h reperfusion.
Figure 6. Effects of MMP-2 and/or -9 knockouts on brain edema and behavioral deficits after 1 h ischemia and 7 h reperfusion (n = 12 in the MMP-2 KO group; n = 13 in other groups).

Behavioral deficits were assessed using the forelimb placing test. Higher forelimb placing scores represent better improvements in function. a) cortex swelling, b) forelimb placing score (%). dKO: MMP-2/9 double knockout group. *p < 0.05, ***p < 0.001 compared with the WT ischemia-reperfusion control.
Discussion
Although both MMP-2 and MMP-9 are upregulated after focal cerebral ischemia, there are controversies concerning the time course of the expression and activation of MMP-2 and MMP-9 in the brain after ischemic stroke. Some studies indicated that MMP-2 levels increased significantly as early as 1 hour and was greater than MMP-9 during the early stage of cerebral ischemia and reperfusion (Heo et al., 1999; Rosenberg et al., 2001; Yang et al., 2007). There was no increase in MMP-2 mRNA and activity, while MMP-9 mRNA and activity markedly increased at 24 hours of reperfusion (Yang et al., 2007). Other studies demonstrated that MMP-9 responses appear to dominate in the acute phase, whereas MMP-2 elevations seem to occur in the delayed phase after stroke (Gasche et al., 1999; Fujimura et al., 1999; Planas et al., 2001). In current studies, we found that both MMP-2 and MMP-9 were significantly upregulated in the early stage of ischemic-reperfusion injuries, suggesting that both of them will contribute to early ischemic brain injuries.
Previous studies also revealed a discrepancy about the role of MMP-9 in HT development after stroke. Elevated MMP-9 levels correlate with HT severity in stroke patients (Ramos-Fernandez et al., 2011). Knocking out MMP-9 protected against photochemical thrombosis induced HT, but it did not protect against t-PA-induced brain hemorrhage, the mechanism of which is unknown (Suzuki et al., 2007). Our results demonstrated that MMP-9 KO decreased hemorrhagic volumes in the cortex, but it did not protect against hemorrhage in the striatum after the early phase of ischemia and reperfusion. This may be because, at least in part, MMP-9 KO decreased the injuries to the basal lamina in the cortex and not in the striatum. Previous study indicated that MMP-9 deficiency preserved blood-brain-barrier function (Asahi et al., 2001b). This effect will contribute to its protection against HT. However, previous studies also demonstrated that MMP-9 deficiency increased hemorrhage, mortality and neurological defects in a collagenase-induced intracerebral hemorrhagic (ICH) model. The compensatory increases of other MMPs, including MMP-2 and MMP-3, may account for the worsened phenotype in the MMP-9 deficient mice (Tang et al., 2004). In our study, gel zymography did not detect a statistically significant compensatory increase in MMP-2 activity in MMP-9 KO mice. These results suggest that there are additional mechanisms for the development of HT in MMP-9 KO mice. Previous studies demonstrated that MMP-9 cleaved a tissue factor pathway inhibitor (Belaaouaj et al., 2000). Overexpression of MMP-9 in porcine coronary arteries promoted intravascular thrombus formation by reducing TFPI and increasing tissue factor (TF) (Morishige et al., 2003). MMP-9 was also documented to cleave protease nexin-1 (PN-1), which is the major endogenous inhibitor of thrombin (Wagner et al., 1989; Choi et al., 1990; Xu et al., 2010). It is possible that knocking out MMP-9 may cause increased TFPI and PN-1 and decreased TF and thrombin activity, as well as jeopardize haemostatic functions in an ischemic brain. These interactions could explain why deletion of MMP-9 does not protect against ischemia/reperfusion-induced hemorrhage. Further studies are needed to clarify these possible mechanisms in ischemic brain.
In current study, we also provided the first evidence that MMP-2 deficiency decreased the hemorrhage incidence in the cortex, reduced the hemorrhagic volume and brain infarct in both the cortex and the striatum after the early phase of ischemia and reperfusion. Previous studies indicated that elevated MMP-2 was highly related to the degradation of tight junction (TJ) proteins and basal lamina, BBB disruption and neuronal injury after ischemia (Rosenberg et al., 1998; Heo et al., 1999; Yang et al., 2007). The treatment with the MMP inhibitor BB-1101 reversed the degradation of the TJ proteins and blocked the opening of the BBB in the early stage of ischemia and reperfusion (Yang et al., 2007). These studies are in line with our results. Our data showed that MMP-2 KO did not reduce laminin breakdown in striatum but reduced hemorrhage in basal ganglia. Previous studies indicated that MMP-2 proteolytically processed fibrin and fibrinogen. The fibrinogen processing brought about the formation of a product unable to form fibrin clots (Hiraoka et al., 1998; Monaco et al., 2007). Thus, MMP-2 KO could mediate hemostasis at least in part via decreased fibrinogen proteolysis. In current study, MMP-2 KO reduced cortex swelling, but MMP-9 KO and MMP-2/9 double KO did not. Previous studies revealed that MMP-9 inactivated chemokine CXC1/Gro-α, CXCL4/PF-4, CXCL5//ENA-78, CXCL6/GCP-2, CXCL9/MIG, CXCL10/IP-10, CXCL11/ITAC and CXCL12/SDF-1 (Rodriguez et al., 2010). MMP-9 KO could increase these chemokine activity and leukocyte influx into ischemic tissues and attenuate its protection against BBB injuries and brain edema. Activated MMP-2 hydrolyzed and released fibroblast growth factor (FGF) receptor type 1 (FGFR1) extracellular domain (Levi et al., 1996). It also cleaved CXCL12/SDF-1 (Zhang et al., 2003), which is constitutively expressed in adult brain (Banisadr et al., 2003). The cleaved form of CXCL12/SDF-1 is highly neurotoxic, and induces neuronal apoptosis and inflammation in mice (Zhang et al., 2003). MMP-2 KO could better protect brain edema and protect brain infarct through increasing FGF function and eliminating the cleaved product of CXCL12/SDF-1. MMP-9 and MMP-2 inhibition prevented the degradation of extracellular matrix and BBB injuries (Asahi et al., 2001b; Heo et al., 1999; Yang et al., 2007). These effects may also be related to the brain edema reduction in MMP-2 KO mice and less infarct in MMP-9 KO, MMP-2 KO and MMP-2/9 double KO mice. Future studies are needed to clarify these possible mechanisms. Our results also indicated that MMP-2 KO, MMP-9 KO and MMP-2/9 double KO mice all decreased infarct volume in the striatum after ischemia, but MMP-9 KO mice did not display decreased HT in the striatum. These suggest that infarct reduction may not be a main reason for HT protection in MMP-9 KO mice although it could contribute to HT protection in MMP-2 KO and MMP-2/9 double KO mice.
Current study indicates that the early stages of ischemia-reperfusion injuries and HT are an effective treatment window for MMP-2 and/or -9 inhibitions. Other authors reported that an MMP-2 KO did not protect against brain infarct when ischemia was severe (2 h post-ischemia and 22 h post-reperfusion) (Asahi et al., 2001a). This suggests that the brain protection via MMP-2 inhibition needs to be potentiated after stroke. We also demonstrated that simultaneously knocking out MMP-2 and MMP-9 protects against acute ischemic brain injury, especially better protect HT than MMMP-9 deficiency, but the protections in this double knockout mice was not different from those in the MMP-2 KO mice. It is unclear whether MMP-9 KO protects or potentiates protective effects of MMP-2 KO against mechanical reperfusion induced HT and other brain injuries after prolonged ischemia and reperfusion. The studies to answer these questions are currently underway in our laboratory. Further studies are needed to find the best combination of MMP-2 KO with other MMP subtype inhibition for protecting post-ischemic brains from injuries and HT development and responsible mechanisms to help develop selective and effective treatment for stroke.
Conclusion
In this study, we have reported that both MMP-2 and MMP-9 are upregulated and activated in the early ischemic brain. We also provided the first evidence that MMP-2 deficiency decreased the hemorrhage incidence in the cortex. MMP-2 deficiency and MMP-2 and MMP-9 double deficiency reduced hemorrhagic volume and brain infarct in both the cortex and the striatum after the early phase of ischemia and reperfusion. This is also first report to compare the effects of MMP-2 KO, MMP-9 KO and MMP-2/ 9 dKO on post-ischemic brain injuries and HT in mice with the same genetic background. The protective effects of an MMP-2 KO or MMP-2/9 dKOs against HT are better than an MMP-9 KO. MMP-2/9 double KOs did not show potentiation in the protection compared with MMP-2 KO alone. These studies increase our understanding of MMP-2 and -9 effects on ischemic injuries and HT development and will help to selectively target MMPs to protect the post-ischemic brain from injury and HT.
Highlight.
Both MMP-2 and MMP-9 were upregulated and activated in the early ischemic brain.
MMP-9 KO decreased hemorrhagic volume in the cortex, but not in the striatum.
MMP-2 KO and MMP-2/9 dKO reduced hemorrhagic volume in both cortex and striatum.
MMP-2 KO and MMP-2/9 dKO was more protective than MMP-9 KO against brain HT.
MMP-2 and/or -9 KO protected against brain infarct and neurological deficits.
Acknowledgments
This study was supported by the following NIH Grants; NS44283 (Broderick J and Clark J), NS50569 (Clark J), and NS57367 (Lu A). The authors would like to thank Professor Lynn M. Matrisian from Vanderbilt University and the laboratory manager Kathy Carter in Dr. Matrisian’s Lab for kindly providing us with breeding pairs of MMP-2 KO mice.
Abbreviations
- MMP
matrix metalloproteinase
- HT
hemorrhagic transformation
- t-PA
tissue plasminogen activator
- KO
knockout
- WT
wild-type
- MCAO
Middle cerebral artery occlusion
- MCID
microcomputer image device
- rCBF
Regional cerebral blood flow
- BBB
blood brain barrier
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681–1689. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- Asahi M, Sumii T, Fini ME, Itohara S, Lo EH. Matrix metalloproteinase 2 gene knockout has no effect on acute brain injury after focal ischemia. Neuroreport. 2001a;12:3003–3007. doi: 10.1097/00001756-200109170-00050. [DOI] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001b;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banisadr G, Skrzydelski D, Kitabgi P, Rostene W, Parsadaniantz SM. Highly regionalized distribution of stromal cell-derived factor-1/CXCL12 in adult rat brain: constitutive expression in cholinergic, dopaminergic and vasopressinergic neurons. Eur J Neurosci. 2003;18:1593–1606. doi: 10.1046/j.1460-9568.2003.02893.x. [DOI] [PubMed] [Google Scholar]
- Belaaouaj AA, Li A, Wun TC, Welgus HG, Shapiro SD. Matrix metalloproteinases cleave tissue factor pathway inhibitor. Effects on coagulation. J Biol Chem. 2000;275:27123–27128. doi: 10.1074/jbc.M004218200. [DOI] [PubMed] [Google Scholar]
- Belayev L, Saul I, Curbelo K, Busto R, Belayev A, Zhang Y, Riyamongkol P, Zhao W, Ginsberg MD. Experimental intracerebral hemorrhage in the mouse: histological, behavioral, and hemodynamic characterization of a double-injection model. Stroke. 2003;34:2221–2227. doi: 10.1161/01.STR.0000088061.06656.1E. [DOI] [PubMed] [Google Scholar]
- Choi BH, Suzuki M, Kim T, Wagner SL, Cunningham DD. Protease nexin-1. Localization in the human brain suggests a protective role against extravasated serine proteases. Am J Pathol. 1990;137:741–747. [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ. The neurovascular unit, matrix proteases, and innate inflammation. Ann N Y Acad Sci. 2010;1207:46–49. doi: 10.1111/j.1749-6632.2010.05760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel O, Kolodziej S, Dirnagl U, Prinz V. Modeling stroke in mice - middle cerebral artery occlusion with the filament model. J Vis Exp. 2011;2423 doi: 10.3791/2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842:92–100. doi: 10.1016/s0006-8993(99)01843-0. [DOI] [PubMed] [Google Scholar]
- Gasche Y, Fujimura M, Morita-Fujimura Y, Copin JC, Kawase M, Massengale J, Chan PH. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab. 1999;19:1020–1028. doi: 10.1097/00004647-199909000-00010. [DOI] [PubMed] [Google Scholar]
- Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, Lipton SA. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401–6408. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:624–633. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
- Hiraoka N, Allen E, Apel IJ, Gyetko MR, Weiss SJ. Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell. 1998;95:365–377. doi: 10.1016/s0092-8674(00)81768-7. [DOI] [PubMed] [Google Scholar]
- Itoh T, Ikeda T, Gomi H, Nakao S, Suzuki T, Itohara S. Unaltered secretion of betaamyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J Biol Chem. 1997;272:22389–22392. doi: 10.1074/jbc.272.36.22389. [DOI] [PubMed] [Google Scholar]
- Lapchak PA, Chapman DF, Zivin JA. Metalloproteinase inhibition reduces thrombolytic (tissue plasminogen activator)-induced hemorrhage after thromboembolic stroke. Stroke. 2000;31:3034–3040. doi: 10.1161/01.str.31.12.3034. [DOI] [PubMed] [Google Scholar]
- Levi E, Fridman R, Miao HQ, Ma YS, Yayon A, Vlodavsky I. Matrix metalloproteinase 2 releases active soluble ectodomain of fibroblast growth factor receptor 1. Proc Natl Acad Sci U S A. 1996;93:7069–7074. doi: 10.1073/pnas.93.14.7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Schafer DP, McCullough LD. TTC, fluoro-Jade B and NeuN staining confirm evolving phases of infarction induced by middle cerebral artery occlusion. J Neurosci Methods. 2009;179:1–8. doi: 10.1016/j.jneumeth.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Clark JF, Broderick JP, Pyne-Geithman GJ, Wagner KR, Khatri P, Tomsick T, Sharp FR. Mechanical reperfusion is associated with post-ischemic hemorrhage in rat brain. Exp Neurol. 2009;216:407–412. doi: 10.1016/j.expneurol.2008.12.020. Epub 2009 Jan 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Clark JF, Broderick JP, Pyne-Geithman GJ, Wagner KR, Ran R, Khatri P, Tomsick T, Sharp FR. Reperfusion activates metalloproteinases that contribute to neurovascular injury. Exp Neurol. 2008;210:549–559. doi: 10.1016/j.expneurol.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Hata R, Hossmann KA. Differences in the cerebrovascular anatomy of C57black/6 and SV129 mice. Neuroreport. 1998;9:1317–1319. doi: 10.1097/00001756-199805110-00012. [DOI] [PubMed] [Google Scholar]
- Monaco S, Gioia M, Rodriguez J, Fasciglione GF, Di Pierro D, Lupidi G, Krippahl L, Marini S, Coletta M. Modulation of the proteolytic activity of matrix metalloproteinase-2 (gelatinase A) on fibrinogen. Biochem J. 2007;402:503–513. doi: 10.1042/BJ20061064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishige K, Shimokawa H, Matsumoto Y, Eto Y, Uwatoku T, Abe K, Sueishi K, Takeshita A. Overexpression of matrix metalloproteinase-9 promotes intravascular thrombus formation in porcine coronary arteries in vivo. Cardiovasc Res. 2003;57:572–585. doi: 10.1016/s0008-6363(02)00710-1. [DOI] [PubMed] [Google Scholar]
- Planas AM, Sole S, Justicia C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis. 2001;8:834–846. doi: 10.1006/nbdi.2001.0435. [DOI] [PubMed] [Google Scholar]
- Ramos-Fernandez M, Bellolio MF, Stead LG. Matrix metalloproteinase-9 as a marker for acute ischemic stroke: a systematic review. J Stroke Cerebrovasc Dis. 2011;20:47–54. doi: 10.1016/j.jstrokecerebrovasdis.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Rodriguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803:39–54. doi: 10.1016/j.bbamcr.2009.09.015. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Cunningham LA, Wallace J, Alexander S, Estrada EY, Grossetete M, Razhagi A, Miller K, Gearing A. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res. 2001;893:104–112. doi: 10.1016/s0006-8993(00)03294-7. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- Smith WS. Safety of mechanical thrombectomy and intravenous tissue plasminogen activator in acute ischemic stroke. Results of the multi Mechanical Embolus Removal in Cerebral Ischemia (MERCI) trial, part I. AJNR Am J Neuroradiol. 2006;27:1177–1182. [PMC free article] [PubMed] [Google Scholar]
- Stemer A, Lyden P. Evolution of the thrombolytic treatment window for acute ischemic stroke. Curr Neurol Neurosci Rep. 2010;10:29–33. doi: 10.1007/s11910-009-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumii T, Lo EH. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke. 2002;33:831–836. doi: 10.1161/hs0302.104542. [DOI] [PubMed] [Google Scholar]
- Suofu Y, Clark J, Broderick J, Wagner KR, Tomsick T, Sa Y, Lu A. Peroxynitrite decomposition catalyst prevents matrix metalloproteinase activation and neurovascular injury after prolonged cerebral ischemia in rats. J Neurochem. 2010;115:1266–1276. doi: 10.1111/j.1471-4159.2010.07026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Nagai N, Umemura K, Collen D, Lijnen HR. Stromelysin-1 (MMP-3) is critical for intracranial bleeding after t-PA treatment of stroke in mice. J Thromb Haemost. 2007;5:1732–1739. doi: 10.1111/j.1538-7836.2007.02628.x. [DOI] [PubMed] [Google Scholar]
- Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, Zhang JH. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004;24:1133–1145. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- Wagner SL, Geddes JW, Cotman CW, Lau AL, Gurwitz D, Isackson PJ, Cunningham DD. Protease nexin-1, an antithrombin with neurite outgrowth activity, is reduced in Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:8284–8288. doi: 10.1073/pnas.86.21.8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, McKee CM, Cao Y, Ding Y, Kessler BM, Muschel RJ. Matrix metalloproteinase-9 regulates tumor cell invasion through cleavage of protease nexin-1. Cancer Res. 2010;70:6988–6998. doi: 10.1158/0008-5472.CAN-10-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinasemediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Zhang K, McQuibban GA, Silva C, Butler GS, Johnston JB, Holden J, Clark-Lewis I, Overall CM, Power C. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci. 2003;6:1064–1071. doi: 10.1038/nn1127. [DOI] [PubMed] [Google Scholar]