

Abstract
ATP-binding cassette (ABC) drug efflux transporters in the CNS are predominantly localized to the luminal surface of endothelial cells in capillaries to impede CNS accumulation of xenobiotics. Inflammatory mediators and cellular stressors regulate their activity. Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease of upper and lower motor neurons characterized by extensive neuroinflammation. Here we tested the hypothesis that disease-driven changes in ABC transporter expression and function occur in ALS. Given the multitude of ABC transporters with their widespread substrate recognition, we began by examining expression levels of several ABC transporters. We found a selective increase in only two transporters; P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) both at mRNA and protein levels, in the SOD1-G93A mouse model of ALS, specifically in disease-affected CNS regions. Detailed analysis revealed a similar disease-driven increase in P-gp and BCRP levels in spinal cord microvessels, indicating that their altered expression occurs at the blood spinal cord barrier. Transport activity of P-gp and BCRP increased with disease progression in spinal cord and cerebral cortex capillaries. Finally, P-gp and BCRP protein expression also increased in spinal cords of ALS patients. Preclinical drug trials in the mouse model of ALS have failed to decisively slow or arrest disease progression; pharmacoresistance imparted by ABC transporters is one possible explanation for these failures. Our observations have large implications for ALS therapeutics in humans and suggest that the obstacle provided by these transporters to drug treatments must be overcome to develop effective ALS pharmacotherapies.
Keywords: Amyotrophic Lateral Sclerosis, ABC Transporters, P-glycoprotein, Breast Cancer Resistance Protein, Blood-Brain Barrier, Blood-Spinal Cord Barrier, Pharmacoresistance
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disease that primarily affects motor neurons in the motor cortex, brain stem, and spinal cord. Research on ALS pathogenesis is focused on the contribution of a number of molecular mechanisms including oxidative stress, toxic protein aggregates, defective axonal transport, mitochondrial impairment, neuroinflammation and excitotoxicity (Boillée et al., 2006; Ilieva et al., 2009). However, despite the availability of drugs that can target these mechanisms, preclinical studies using the SOD1-G93A mouse have failed to identify a drug that significantly slows disease progression or delays the onset of symptoms (Traynor et al., 2006). Similarly, research efforts aimed at developing new effective therapeutics have been essentially ineffective in human ALS. Thus far, riluzole is the only FDA-approved drug for treatment of ALS and it exhibits marginal efficacy.
One mechanism contributing to difficulties in treating CNS diseases using pharmacotherapy (e.g., epilepsy and HIV-related neurological conditions) is the obstacle provided by the activity of ATP-binding cassette (ABC) transporters in brain barrier tissues (Löscher and Potschka, 2005). These transporters are ATP-driven efflux pumps that exhibit remarkably broad specificity limits. In the blood-brain and blood-spinal cord barriers (BBB, BSCB), several ATP transporters face the vasculature, including P-glycoprotein (ABCB1/P-gp), breast cancer resistance protein (ABCG2/BCRP), and members of the multidrug-resistance associated protein family (ABCC/MRP) (Miller, 2010). Together, these transporters limit entry of xenobiotics, including many therapeutic drugs, into the CNS (Neuwelt et al., 2011).
We previously reported a disease-driven up-regulation of P-glycoprotein (ABCB1/P-gp) expression in spinal cord homogenates from the SOD1-G93A mice (Boston-howes et al., 2008) and suggested that increased P-gp activity could account for the therapeutic failure of NDGA, a glutamate transport enhancer, which we tested in a preclinical trial in the SOD1-G93A mouse as a potential therapeutic for ALS (Boston-howes et al., 2008). Furthermore, riluzole, which has limited efficacy in ALS patients, is a substrate of P-gp and BCRP (Milane et al., 2007; Milane et al., 2009), implying that these transporters could limit the brain disposition of riluzole and other potential therapeutics (Milane et al., 2010). The link between drug efflux transporters and ALS is just beginning to be investigated, but understanding the contribution of these transporters towards pharmacoresistance in ALS is vital to devise more efficacious therapies for this untreatable disease. Therefore, we tested the hypothesis that ALS drives increased expression and activity of multidrug efflux transporters at the BBB and BSCB thus limiting drug bioavailability. We found a selective increase in expression of P-gp and BCRP during disease progression in two ALS mouse models; mutant SOD1-G93A and mutant TDP43-A315T mice. In the CNS of both models, P-gp and BCRP up-regulation is specific to cortex and spinal cord. Similarly, systemic protein expression of both transporters does not change based on whole liver homogenate. Expression of other drug efflux transporters like Mrp2 as well as other transport proteins important for absorption and disposition of drugs in the brain (e.g. organic anion transporter, OATP2 and glucose transporter, GLUT-1) was unaltered. Functional studies with spinal cord and cerebral cortex capillaries isolated from SOD1-G93A mice paralleled specific increases in P-gp and BCRP expression. Finally, we found increased protein expression of P-gp and BCRP in spinal cord extracts from ALS patients. The selective increase in expression and activity of only two transporters in regions affected by neurodegeneration in ALS suggests a highly regulated, ALS-driven pharmacoresistance rather than a non-specific response of the BBB and BSCB to neuroinflammatory stimuli, and indicate the need to identify strategies specifically targeted to these two transporters to overcome therapeutic failures in ALS.
Materials and Methods
Mouse models of amyotrophic lateral sclerosis
All animals were housed in accordance with Thomas Jefferson University IACUC and the NIH Guide for the Care and Use of Laboratory Animals. Mutant SOD1-G93A mice model of ALS [B6.Cg-Tg(SOD1-G93A)1Gur/J] were purchased from Jackson Laboratories (stock #004435). Male SOD1-G93A mice were bred in house with B6SJL females. Offspring were assessed for the presence of the human SOD1 transgene and copy number by PCR. Mutant SOD1-G93A mice developed disease symptoms with onset at 95–105 days of age, and progressed to an endstage disease state at approximately 150–160 days of age. Mutant TDP-43 mice model of ALS [B6.Cg-Tg(Prnp-TARDBP*A315T)95Balo/J] were purchased from Jackson Laboratories (stock # 010700). Male TDP-43-A315T mice were bred in house with B6SJL females. Offspring were assessed for the presence of the human TDP-43 transgene by PCR. The lifespan of the mutant TDP-43-A315T mice was approximately 75-90 days of age.
Messenger RNA isolation and qRT-PCR
RNA was isolated from the lumbar enlargement of the spinal cord using the RNeasy Lipid Tissue Mini RNA isolation kit (Qiagen). Reverse transcription was performed with the RT2 First Strand Kit (SABiosciences) according to the manufacturer’s protocol. PCR arrays (Drug Transporter, SABiosciences, cat# PAMM-070) and real-time quantitative PCR reactions were performed using RT2 Real-Time SYBR Green/Rox qPCR Master Mix (SABiosciences) and run on an ABI Prism 7500. Real-time PCR was performed for ABCB1B, ABCG2, ABCC3, ACTB (Primer set catalog #, Unigene #; PPM03995B, Mm.146649; PPM03906E, Mm.333096; PPM03395E, Mm.23942; PPM02945A, Mm.328431; Qiagen). The thermocycler parameters were 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec, and 60°C for 1 min. The results were analyzed by the comparative Ct method and normalized with Mouse ACTB.
Western Blot analysis
Spinal cord, cerebellum and cerebral cortex were homogenized as previously described (Boston-howes et al., 2008) or capillaries were isolated according to (Ge and Pachter, 2006). Samples containing equal amounts of protein were separated by SDS-PAGE using 12% or 4-15% Polyacrylamide gels (Bio-Rad) and transferred to PVDF membranes (GE Healthcare). Membranes were blocked (5% milk in TBST) and exposed to primary antibody for 1 hr at room temperature or overnight at 4°C. Following washes (TBST, 3 × 15 min), membranes were incubated with secondary antibody (1:3,000 for anti-mouse and anti-rabbit IgG and 1:5,000 for anti-rat IgG) for 1 hr at room temperature, washed again and developed using ECL Plus reagents for chemiluminescent detection (Bio-Rad Quantity-One Detection System). Protein bands were quantified using Quantity-One software (Biorad).
Antibodies
The following primary antibodies were used: anti-P-glycoprotein (Rabbit; 1:100; Santa Cruz, cat #sc-8313), anti-Bcrp (Rat; 1:200; Santa Cruz, cat #sc-58224), anti-Mrp2 (Goat; 1:200; Santa Cruz, cat #sc-5770), and anti-GAPDH (Mouse; 1:20,000; Fitzgerald Industries International, cat #10R-G109A).
Capillary Transport Assay
The capillary isolation procedure and transport assay have been described previously (Hartz et al., 2010) (Miller et al., 2000). Briefly, spinal cords and cerebral cortices were removed from SOD1-G93A mice at presymptomatic (50 day-old) and diseased stage (130 day-old) and stripped of meninges and surface blood vessels. White matter was dissected from the cerebral cortex. After homogenizing the spinal cords or cortex in ice-cold buffer A containing 2.7 mM KCl, 1.2 mM KH2PO4, 136.9 mM NaCl, 8.1 mM Na2HPO4, 1 mM CaCl2, 0.5 mM MgCl2, 5 mM D-glucose and 1mM sodium pyruvate, an equal amount of 30% Ficoll was added to the homogenate and centrifuged at 5,800 × g for 20 minutes. Pellets containing capillaries were resuspended in the above buffer containing 1% BSA and passed through a glass bead column. Capillaries were bound to the glass beads and released by gentle agitation, washed with buffer A, and used immediately for the transport assay. Isolated capillaries were plated on confocal imaging chamber slides (Lab-Tek Chambered Coverglass #1 borosilicate, Thermo Scientific) in buffer A solution. Capillaries were incubated with or without inhibitors, followed by incubation with specific fluorescent substrates for 1 hour at room temperature with 2 μM NBD-CSA for P-glycoprotein (Miller et al., 2000), 2 μM Texas red for Mrp2 (Bauer et al., 2008), and 2 μM BODIPY-Prazosin for BCRP (Shukla et al., 2009). Luminal substrate accumulation was imaged after 1 hr. For each treatment, images of 10 capillaries were acquired by confocal microscopy using a Zeiss 510 inverted confocal microscope and a 40x oil-immersion objective with a 488-nm or 543-nm excitation laser. For each experiment, pinhole diameter, photomultiplier gain, and laser power were the same for all treatments. A minimum of 10 capillaries per treatment were imaged. Quantification of luminal fluorescence was determined using Image J software and specific transport determined by the difference between total luminal substrate fluorescence and fluorescence in the presence of the inhibitor.
Human Tissue Homogenization
Spinal cord sections of post-mortem human tissues were homogenized in EDTA buffer (2mM EDTA) with protease inhibitors. The homogenate was centrifuged at 39,000 × g for 10 minutes at 4°C. The pellet fraction was resuspended in buffer containing 150mM NaCl, 10mM NaPi, 1% SDS (pH=7.4) with Complete™ Protease Inhibitor and frozen at -80°C for further analysis. ALS tissue samples, two sporadic ALS cases and one familial ALS case, were derived from patients between 50 and 67 years of age with progressive clinical symptoms resulting in respiratory failure.
Results
P-gp and BCRP transporter expression increases with disease progression in the SOD1-G93A ALS mouse model
We initially used a PCR drug transporter array to compare mRNA levels in extracts from the lumbar spinal cords of presymptomatic (50 day old) and symptomatic (130 day old) SOD1-G93A mice (Fig. 1A). Of the ABC transporters localized to the CNS barrier, P-gp and BCRP were the only ones that significantly increased in expression levels in the symptomatic, diseased mice (1.64-fold and 1.66-fold, respectively). No increase was found for any of the multidrug resistance-associated protein (Mrp) isoforms detected, i.e., Mrp1 (ABCC1), Mrp2 (ABCC2), Mrp4 (ABCC4), and Mrp5 (ABCC5), nor for Glut1, a marker for endothelial cells, or an organic anion-transporting polypeptide (OATP2). Other transporters involved in handling xenobiotics, metabolites or catabolites such as the organic anion transporters (OATs) or organic cation transporters (OCTs) were below detection levels in both presymptomatic and symptomatic groups. We validated the increases in P-gp and BCRP mRNA expression using qRT-PCR (Fig. 1B). P-gp and BCRP expression increased significantly in symptomatic mice compared to presymptomatic mice (2.13-fold and 1.72-fold increase in ΔΔCt, respectively). As in the microarray study, Mrp2 expression was the same in presymptomatic (50 day old) and symptomatic (130 day old) SOD1-G93A mice.
Figure 1. ABC drug efflux transporter expression levels in SOD1-G93A mice.
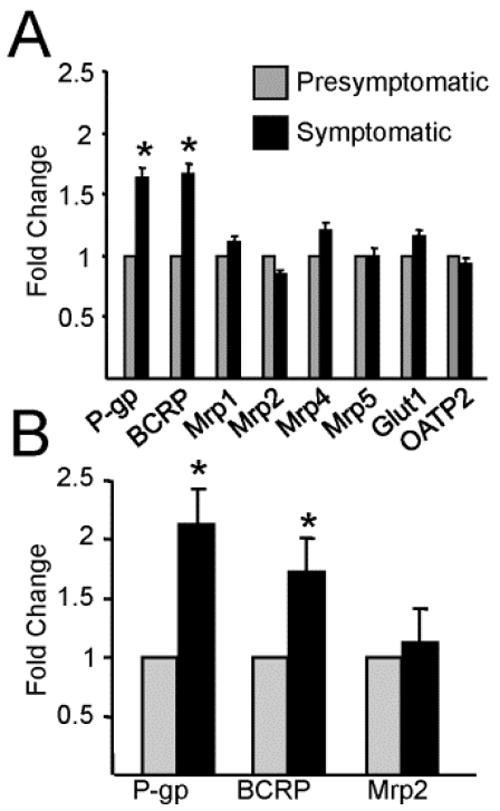
Isolated messenger RNA from the lumbar spinal cord of SOD1-G93A mice shows an increase in ABCG2 (Bcrp) and ABCB1B (P-gp) with no change in other ABC transporters, the endothelial cell marker (Glut1), and a member of the organic anion transporters (OATP2) between presymptomatic (50 days old) and symptomatic mice (130 days old). (A) PCR drug transporter array identifying increases in the mRNA coding for two ABC drug efflux transporters with localization to the blood-spinal cord barrier (P-gp and BCRP) throughout disease progression in the SOD1-G93A mouse model. (B) qRT-PCR validation of ABCB1B, ABCG2, and ABCC2 which confirm results found in the array. Analysis was run as three independent samples in triplicate per time point (n=3) with values represented using the ΔΔCt method as fold-change using an internal control. Statistical analysis was done using Student’s T-test and compared ΔCT values along with the corresponding standard errors. *P < 0.05, data are shown as mean ± SEM.
We used western blotting to measure changes in drug efflux transporter protein expression in whole spinal cord, cerebral cortex and cerebellum from presymptomatic (50 days old), onset (90 days old), and symptomatic (130 days old) SOD1-G93A mice. In spinal cord homogenates, P-gp and BCRP expression increased throughout the course of the disease (Fig. 2A,B). In symptomatic mice P-gp and BCRP expression levels were 196% ± 36% and 169% ± 29% higher than levels in presymptomatic mice (P<0.05 for both transporters). Mrp2 expression did not change with disease progression. Homogenates from the cerebal cortex also showed a significant increase in BCRP protein expression in symptomatic mice (128% ± 10%; Fig. 2C,D). Although P-gp protein expression tended to be higher in symptomatic mice (135% ± 27%), the difference in mean values was not significant (P = 0.18) compared to early diseased mice. If the motor area of the cerebral cortex were the only area affected by the disease (Ozdinler et al., 2011), the increase in transporter expression might have been diluted out in the sample of whole cortex homogenate. Thus, to study whether changes in P-gp and BCRP are specific to areas primarily affected by ALS, we used the cerebellum as a control. There were no significant differences in protein expression of P-gp, BCRP, or Mrp2 in the cerebellum of SOD1-G93A mice throughout the course of ALS pathology (Fig. 2E,F). Importantly, ABC transporter expression did not change with age in spinal cord homogenates from age-matched wild-type mice that overexpressed human SOD1 (Fig. S1).
Figure 2. P-gp and BCRP protein expression increases are specific to the spinal cord and cerebral cortex of the CNS.
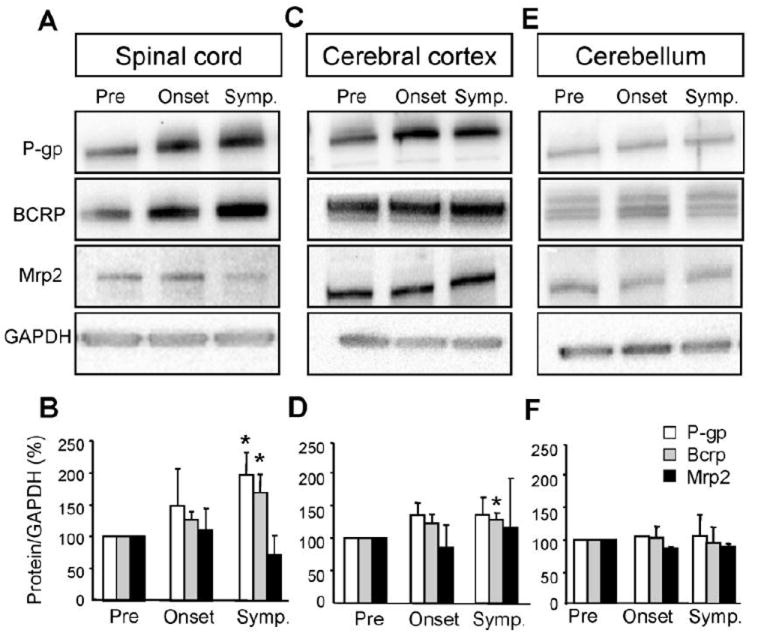
Disease-driven increases of P-gp and Bcrp protein expression in SOD1-G93A mice are specific to the spinal cord (A,B) and cerebral cortex (C,D) compared to the cerebellum (E,F). The band intensity of transporter expression levels were normalized to loading controls (GAPDH) and quantifications include a minimum of three independent experiments (n = 3). Data analysis was performed using two-tailed Student’s T-tests. *P < 0.05, data are shown as mean ± SEM. Abbreviations: Pre = Presymptomatic mice at 50 days; Onset = Mice at onset of symptoms at 90 – 100 days; Symp = Symptomatic mice at 130 days.
To test the systemic expression of P-gp and BCRP, we used whole liver homogenates from SOD1-G93A mice. There were no significant differences throughout disease course in protein expression for either transporter in the liver, indicating that, at least in the SOD1-G93A mice, systemic circulation is not the primary mediator of altered ABC transporter expression and the potential ensuing decreased bioavailability of CNS therapeutics (Fig. 3).
Figure 3. Systemic levels of P-gp and BCRP protein expression are unchanged in SOD1-G93A mice.
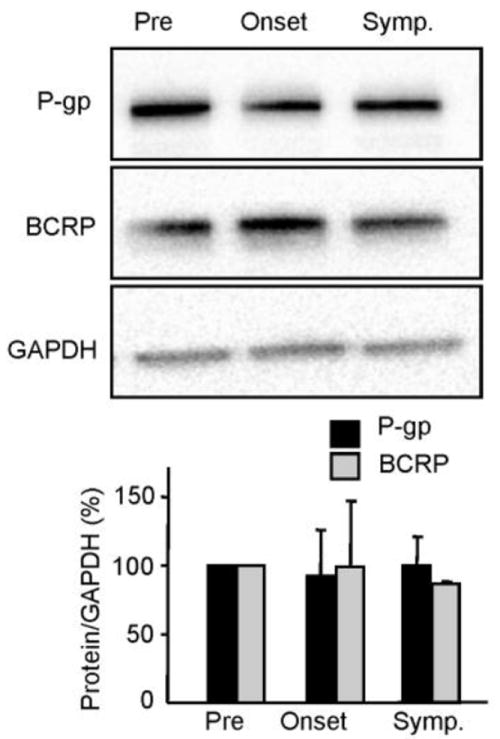
Whole liver homogenate does not show a protein expression increase in the drug transporters, P-gp and BCRP, throughout disease progression in SOD1-G93A mice. The band intensity of transporter expression levels were normalized to loading controls (GAPDH) and quantifications include a minimum of three independent experiments (n = 3). Data analysis was performed using two-tailed Student’s T-tests. *P < 0.05, data are shown as mean ± SEM. Abbreviations: Pre = Presymptomatic mice at 50 days; Onset = Mice at onset of symptoms at 90 – 100 days; Symp = Symptomatic mice at 130 days.
Recently, another mouse model of ALS carrying a mutation in the TARDBP gene was developed (Wegorzewska et al., 2009). Our initial experiments show that homogenate of spinal cord and cerebral cortex from symptomatic TARDBP mice also exhibit a tissue-specific increase in P-gp and BCRP expression, not seen in the cerebellum (Fig. 4).
Figure 4. P-glycoprotein and BCRP increase in spinal cord and cerebral cortex homogenates of another ALS mouse model, TDP-43 (A315T).
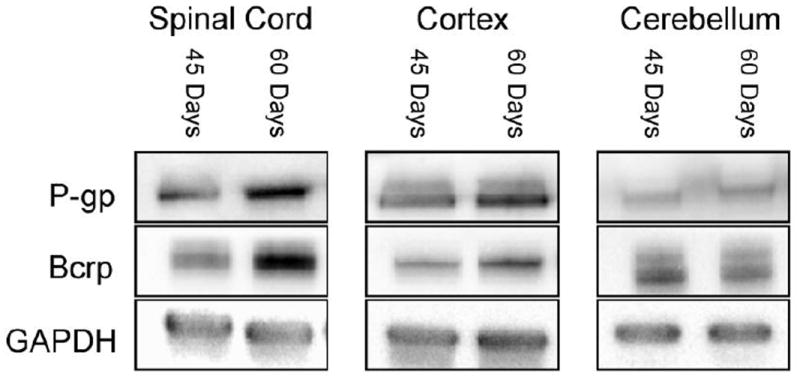
Disease-driven increases of P-gp and Bcrp protein expression in TDP-43 (A315T) mice are specific to the spinal cord and cerebral cortex compared to the cerebellum. The band intensity of transporter expression levels were compared to loading controls (GAPDH).
In the CNS, P-gp and BCRP are mainly localized in the endothelial cells of the vasculature (Miller, 2010). In order to further identify these transporter changes at the BSCB, microvessels were isolated from the spinal cord of presymptomatic, onset, and symptomatic SOD1-G93A mice. The microvessel isolation showed enrichment of the endothelial cell marker, von Willebrand factor, and a marked decrease in Iba1 and Map2, microglial and neuronal markers respectively (Fig S2). Consistent with the data from whole spinal cord homogenates (above), protein expression of P-gp and BCRP was significantly higher (P<0.05) in spinal cord capillaries of symptomatic mice (187% ± 48% increase for P-gp; 202% ± 31% increase for BCRP; Fig. 5).
Figure 5. P-gp and BCRP expression levels increase at the blood-spinal cord barrier.
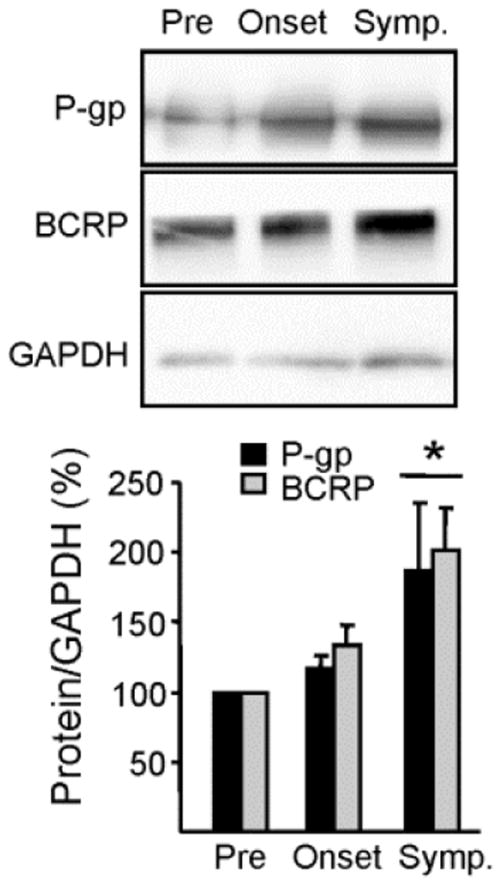
Isolated spinal cord microvessels show a protein expression increase in the drug transporters, P-gp and Bcrp, throughout disease progression in SOD1-G93A mice (*p < 0.05). Transporter expression levels were normalized to loading controls (GAPDH) and quantifications include a minimum of three separate experiments. Data analysis was performed using two-tailed Student’s T-tests. Data are shown as mean ± SEM. Abbreviations: Pre = Presymptomatic mice at 50 days; Onset = Mice at onset of symptoms at 90 – 100 days; Symp = Symptomatic mice at 130 days.
Increased P-gp and BCRP transport activity follows disease progression in SOD1-G93A mice
The BSCB resides within the spinal cord capillary endothelium (Bartanusz et al., 2011). To determine whether increases in expression were also paralleled by an increase in the overall activity of these transporters, we measured specific transport activity of P-gp, BCRP and Mrp2 in capillaries from spinal cord and cerebral cortex of wild-type mice and presymptomatic and symptomatic SOD1-G93A mice. Transport activity was determined using a series of assays previously validated for brain capillaries capillaries (Wang et al., 2010). The assay involves exposing capillaries to fluorescent substrates (NBD-CSA for P-glycoprotein, bodipy-prazosin for BCRP and TR for Mrp2) and using confocal microscopy to measure luminal substrate accumulation in the absence and presence of specific transport inhibitors (PSC833 for P-glycoprotein, Ko143 for BCRP, and MK571 for Mrp2). For each transporter, the difference between the two measurements is taken as specific transport activity.
Both the representative capillary images and the graphs illustrating measured luminal fluorescence demonstrate P-gp and BCRP transport activity in capillaries from symptomatic SOD1-G93A mice was significantly higher than corresponding activity in non-transgenic littermates (130 days) and presymptomatic SOD1-G93A mice (Fig. 6). This was the case for capillaries from spinal cord and cerebral cortex. Mrp2 activities in spinal cord and cerebral cortex did not change with disease state. This data suggests that, as for expression, alteration in ABC transporter activity is specific for P-gp and BCRP.
Figure 6. P-gp and BCRP activity is increased in symptomatic SOD1-G93A mice.
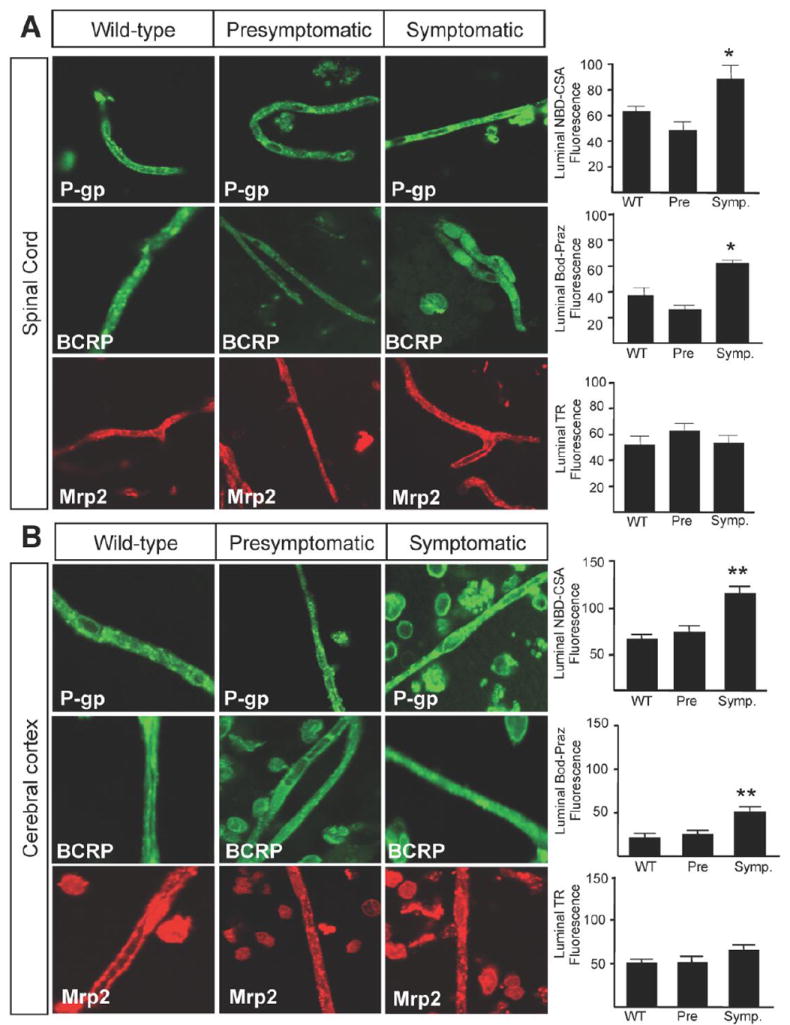
The functional transport increases in the drug transporters, P-gp and Bcrp, in isolated spinal cord (A) and cerebral cortex (B) capillaries throughout disease progression in SOD1-G93A mice (*p < 0.01; **p < 0.001). There was no change in the transport activity of Mrp2 in spinal cord (A) or cerebral cortex (B) capillaries. Specific transport activity was determined by quantification of luminal fluorescence in presence of inhibitor subtracted from the luminal fluorescence in the absence of inhibitor for each transporter. Data analysis was performed using one-way analysis of variance (ANOVA). Data are shown as mean ± SEM. Data are shown as mean ± SEM. Abbreviations: Pre = Presymptomatic mice at 50 days; Onset = Mice at onset of symptoms at 90 – 100 days; Symp = Symptomatic mice at 130 days; NBD-CSA = [N-(4-nitrobenzofurazan-7-yl)-D-Lys8]-cyclosporin A; Bod-Praz = BODIPY FL Prazosin; TR = Texas Red.
Increased P-gp and BCRP expression in the lumbar spinal cord of ALS patients
Next, we looked at whether increases in P-gp and BCRP expression could also be detected in the lumbar spinal cord of patients with ALS. We analyzed post-mortem tissues of lumbar spinal cords from 3 ALS patients and 2 non-neurological controls. For both P-gp and BCRP, western blots showed consistently higher protein expression in the tissue of ALS patients than in control subjects (Fig.7). Using the Mrp2 polyclonal goat antibody, we could not detect Mrp2 protein in our samples.
Figure 7. P-gp and BCRP protein expression is increased in ALS patients.
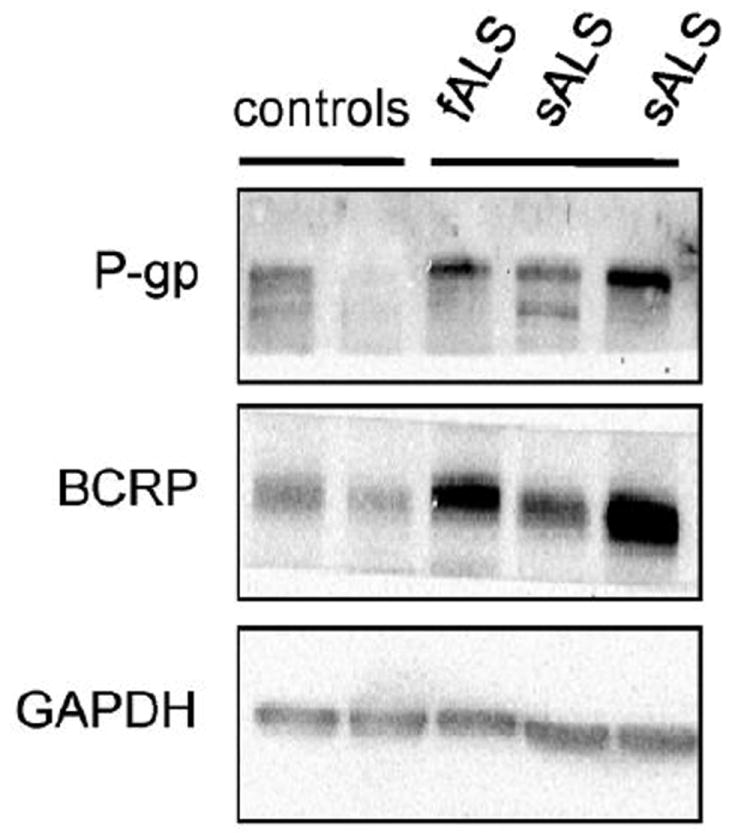
ALS patients demonstrate an increase in P-gp and BCRP protein expression compared to control patients. Homogenates isolated from the lumbar spinal cord of human ALS specimens, one familial ALS patient (fALS) and two sporadic ALS patients (sALS), and control patients.
Discussion
More than 400 membrane transporters that have been annotated in the human genome belong to two major superfamilies, the ATP-binding cassette and the solute carrier. Particular attention has been paid to transporters expressed in epithelial cells in many organs and endothelial barriers for their role in drug disposition, therapeutic efficacy and adverse drug reactions in many diseases. We have identified P-gp and BCRP as the two ABC transporters that increase in expression and function at the BBB and BSCB in mouse models of ALS. The increases were specific to the CNS as protein expression in the liver was unchanged. We reported increases for P-gp and BCRP in mRNA expression, protein expression, and in transport activity in spinal cord and cerebral cortex capillaries, while other transporters were unchanged. This positive correlation between expression levels and activity implies that P-gp and BCRP may be active players in ALS pharmacoresistance. Furthermore, we found that P-gp and BCRP expression levels are higher in post-mortem, spinal cord specimens of ALS patients compared to controls.
Pharmacokinetic studies have indicated that ABC transporters often work together with drug-metabolizing enzymes in drug absorption and elimination. Our findings in two mouse models of ALS as well as in patients are of particular relevance to ALS therapeutics. P-gp and BCRP mediate the ATP-dependent export of drugs from cells, hence they have an important role in limiting entry of various drugs into the CNS. P-gp is expressed in the luminal membrane of endothelial cells in the small intestine and BBB, and in the apical membrane of excretory cells such as hepatocytes. BCRP is a “half ABC transporter” expressed in many peripheral organs and in the brain endothelium whose function is to limit oral bioavailability and transport across the BBB, blood-testis barrier and the maternal-fetal barrier of some selected substrates (Mao and Unadkat, 2005).
P-gp and BCRP increases are driven by pathological conditions in the ALS mouse models. CNS areas that are normally spared by the disease such as the cerebellum showed similar expression levels of P-gp and BCRP throughout the disease. Interestingly, P-gp and BCRP were not up-regulated in organs like the liver in which they are normally highly expressed, suggesting that the up-regulation at the BBB/BSCB is selective and specific to local pathological changes and not driven by systemic and peripheral alterations that could occur non-specifically in the ALS mouse. As seen in animal models and patients with epilepsy, it is plausible that in ALS patients, disease-treating drugs could also exacerbate these increases, further limiting the success of ongoing clinical trials. This hypothesis should be further explored.
Our study focused on profiling differences in expression and functional levels occurring over the course of disease progression in two ALS mouse models as well as the transporter expression differences between ALS patients and control subjects. These comparisons are particularly relevant as we attempt to develop effective pharmacotherapeutics for ALS treatment. There is a potential for other drug transporters to be involved. While the ABC transporters are primarily responsible for development of the multidrug resistance phenotype, there is ongoing debate as to the localization of the ABC transporters in the CNS. Furthermore, the SLC family of transporters, which include the Organic Anion Transporters (OATs), the Organic Cation Transporters (OCTs), the Organic Anion Transport Polypeptides (OATPs), the Nucleosides Transporters (NTs) and Peptide Transporters (PEPTs), are capable of transporting various molecules at the cell membrane.
Previous research detected an increase in P-gp expression of SOD1-G86R mice compared to wild-type mice, but failed to detect any changes in BCRP expression (Milane et al., 2010). There are multiple reasons for this finding. First, Milane et al. (2010) isolated cerebral capillaries and analyzed RNA isolated from the whole brain, which is not the primary area of degeneration in the ALS mouse model. Studying these transporter changes at the spinal cord level proved more sensitive in detecting both mRNA and protein expression. Furthermore, Milane et al. (2010) compared wild-type age-matched littermates to presymptomatic mutant SOD1-G86R mice, which did not allow analysis of changes in transporter levels during the disease. While this comparison is important, we identified transporter changes throughout the progression of ALS providing relevance to drug efflux changes during disease.
Riluzole, the only FDA-approved drug for ALS, was shown to be a P-gp substrate and pharmacological inhibition of P-gp increased the brain disposition of riluzole in a mouse model (Milane et al., 2007). With the recent development of more specific dual P-gp and BCRP inhibitors (elacridar, tariquidar, etc.), combining ALS-treating drugs with drug efflux transporter inhibitors could prove effective in prolonging disease course or delaying symptom progression. As a proof of principle to this idea, transgenic mutant SOD1-G93A/P-gp knockout mice were treated with cyclosporine A, which extended the life of these mice by 12% (Kirkinezos et al., 2004). This experiment is not without fault as there is a potential for compensation from other transporters that may work to exclude compounds with overlapping substrate recognition in the absence of P-gp. Moreover, cyclosporine A was used as the therapeutic agent, which was later shown to be toxic to neuronal cells expressing mutant SOD1 (Maxwell et al., 2004).
In light of recent data linking impairment of the BBB/BSCB to ALS (Zhong et al., 2008; Garbuzova-Davis et al., 2007; Nicaise et al., 2009), understanding the alterations in drug efflux transporters, which are a constitutive part of these barriers, becomes increasingly relevant. Various groups have identified potential BBB/BSCB disruption at various disease states, while others have not detected permeation in the barriers. As the ABC drug efflux transporters directly contribute to the permeability of these barrier properties, our findings of increased transporter activity could explain some of these discrepancies.
Drug efflux continues to be a problem for a successful treatment of a vast array of disorders. The high expression of these transporters at barrier tissues, including the BBB/BSCB, warrants further investigation into overcoming the acquired pharmacoresistance in multiple pathologies, including neurodegenerative disorders like ALS. With the identification of which transporters are altered in the ALS mouse models and ALS patients (P-gp and BCRP), the increase in expression and function as the disease worsens provides a basis for pharmacological treatment. Our research indicates that potential ALS-treating compounds should be analyzed for their ability to be transported by P-gp and BCRP. Furthermore, as there are specific disease-driven increases in the activity of these transporters, drug disposition changes as the disease progresses should be explored. Various inhibitors (i.e. elacridar and tariquidar) block the actions of both P-gp and BCRP, which would be particularly attractive in association with an effective ALS-treating drug to increase bioavailability and efficacy. Due to potential toxicity of blocking the entire family of ABC transporters, it becomes essential to target particular transporters alongside therapeutics targeting disease rather than an indiscriminate approach of drug efflux transporter inhibition.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health grants R21-NS065481 and RO1-NS074886 to DT, RO1-NS051488 to PP. DAJ is the recipient of an MDA developmental award. The Weinberg Unit for ALS research is also supported by the Farber Family Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartanusz V, Jezova D, Alajajian B, Digicaylioglu M. The blood-spinal cord barrier: Morphology and Clinical Implications. Annals of neurology. 2011;70:194–206. doi: 10.1002/ana.22421. [DOI] [PubMed] [Google Scholar]
- Bauer B, Hartz AMS, Lucking JR, Yang X, Pollack GM, Miller DS. Coordinated nuclear receptor regulation of the efflux transporter, Mrp2, and the phase-II metabolizing enzyme, GSTpi, at the blood-brain barrier. J Cereb Blood Flow Metab. 2008;28:1222–1234. doi: 10.1038/jcbfm.2008.16. [DOI] [PubMed] [Google Scholar]
- Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Boston-howes W, Williams EO, Bogush A, Scolere M, Pasinelli P, Trotti D. Nordihydroguaiaretic acid increases glutamate uptake in vitro and in vivo: Therapeutic implications for amyotrophic lateral sclerosis. Exp Neurol. 2008;213:229–237. doi: 10.1016/j.expneurol.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbuzova-Davis S, Haller E, Saporta S, Kolomey I, Nicosia SV, Sanberg PR. Ultrastructure of blood-brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS. Brain res. 2007;1157:126–137. doi: 10.1016/j.brainres.2007.04.044. [DOI] [PubMed] [Google Scholar]
- Ge S, Pachter JS. Isolation and culture of microvascular endothelial cells from murine spinal cord. J Neuroimmunol. 2006;177:209–214. doi: 10.1016/j.jneuroim.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Hartz AMS, Miller DS, Bauer B. Restoring Blood-Brain Barrier P-glycoprotein Reduces Brain Aβ in a Mouse Model of Alzheimer’s Disease. Mol Pharmacol. 2010;77:715–735. doi: 10.1124/mol.109.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non – cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. Cell. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkinezos IG, Hernandez D, Bradley WG, Moraes CT. An ALS mouse model with a permeable blood – brain barrier benefits from systemic cyclosporine A treatment. J Neurochem. 2004:821–826. doi: 10.1046/j.1471-4159.2003.02181.x. [DOI] [PubMed] [Google Scholar]
- Löscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005;6:591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- Mao Q, Unadkat JD. Role of the breast cancer resistance protein (ABCG2) in drug transport. AAPS journal. 2005;7:E118–E133. doi: 10.1208/aapsj070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell MM, Pasinelli P, Kazantsev AG, Brown RH. RNA interference-mediated silencing of mutant superoxide dismutase rescues cyclosporin A-induced death in cultured neuroblastoma cells. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3178–3183. doi: 10.1073/pnas.0308726100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milane A, Fernandez C, Dupuis L, Buyse M, Loeffler J-philippe, Farinotti R, Meininger V, Bensimon G. Neuroscience Letters P-glycoprotein expression and function are increased in an animal model of amyotrophic lateral sclerosis. Neurosci Lett. 2010;472:166–170. doi: 10.1016/j.neulet.2010.01.078. [DOI] [PubMed] [Google Scholar]
- Milane A, Fernandez C, Vautier S, Bensimon G, Meininger V, Farinotti R. Minocycline and riluzole brain disposition: interactions with p-glycoprotein at the blood-brain barrier. J Neurochem. 2007;103:164–173. doi: 10.1111/j.1471-4159.2007.04772.x. [DOI] [PubMed] [Google Scholar]
- Milane A, Vautier S, Chacun H, Meininger V, Bensimon G, Farinotti R, Fernandez C. Interactions between riluzole and ABCG2/BCRP transporter. Neurosci Lett. 2009;452:12–16. doi: 10.1016/j.neulet.2008.12.061. [DOI] [PubMed] [Google Scholar]
- Miller DS. Regulation of P-glycoprotein and other ABC drug transporters at the blood-brain barrier. Trends Pharmacol Sci. 2010;31:246–254. doi: 10.1016/j.tips.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DS, Nobmann SN, Gutmann H, Toeroek M, Drewe J, Fricker G. Xenobiotic transport across isolated brain microvessels studied by confocal microscopy. Mol Pharmacol. 2000;58:1357–1367. doi: 10.1124/mol.58.6.1357. [DOI] [PubMed] [Google Scholar]
- Neuwelt Ea, Bauer B, Fahlke C, Fricker G, Iadecola C, Janigro D, Leybaert L, Molnár Z, O’Donnell ME, Povlishock JT, et al. Engaging neuroscience to advance translational research in brain barrier biology. Nature reviews. Neuroscience. 2011;12:169–182. doi: 10.1038/nrn2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicaise C, Mitrecic D, Demetter P, De Decker R, Authelet M, Boom A, Pochet R. Impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat. Brain res. 2009;1301:152–162. doi: 10.1016/j.brainres.2009.09.018. [DOI] [PubMed] [Google Scholar]
- Ozdinler PH, Benn S, Yamamoto TH, Güzel M, Brown RH, Macklis JD. Corticospinal motor neurons and related subcerebral projection neurons undergo early and specific neurodegeneration in hSOD1G93A transgenic ALS mice. The Journal of neuroscience. 2011;31:4166–4177. doi: 10.1523/JNEUROSCI.4184-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Zaher H, Hartz A, Bauer B, Ware Ja, Ambudkar SV. Curcumin inhibits the activity of ABCG2/BCRP1, a multidrug resistance-linked ABC drug transporter in mice. Pharm Res. 2009;26:480–487. doi: 10.1007/s11095-008-9735-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor BJ, Bruijn L, Conwit R, Beal F, O’Neill G, Fagan SC, Cudkowicz ME. Neuroprotective agents for clinical trials in ALS: a systematic assessment. Neurology. 2006;67:20–27. doi: 10.1212/01.wnl.0000223353.34006.54. [DOI] [PubMed] [Google Scholar]
- Wang X, Sykes DB, Miller DS, Carolina N. Constitutive Androstane Receptor-Mediated Up-Regulation of ATP-Driven Xenobiotic Efflux Transporters at the Blood-Brain. Mol Pharmacol. 2010;78:376–383. doi: 10.1124/mol.110.063685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106:18809–18814. doi: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O’Banion MK, Stojanovic K, Sagare A, Boillee S, Cleveland DW, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11:420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.