

Abstract
A label-free biosensing method for the sensitive detection and identification of bacterial transfer-messenger RNA (tmRNA) is presented employing arrays of silicon photonic microring resonators. Species specific tmRNA molecules are targeted by complementary DNA capture probes that are covalently attached to the sensor surface. Specific hybridization is monitored in near real-time by observing the resonance wavelength shift of each individual microring. The sensitivity of the biosensing platform allowed for detection down to 53 fmol of Streptococcus pneumoniae tmRNA, equivalent to approximately 3.16 × 107 CFU of bacteria. The simplicity and scalability of this biosensing approach makes it a promising tool for the rapid, PCR-free identification of different bacteria via tmRNA profiling.
Keywords: tmRNA, silicon photonics, microring resonator, bacterial detection, label-free biosensing, biomarker
1. Introduction
The ability to rapidly and accurately detect pathogenic bacteria strains is important for the screening of infectious diseases as well as for environmental monitoring and food safety. Unfortunately, traditional microbiological methods for these purposes typically use slow, labor intensive culturing methods. Consequently, numerous biosensing technologies have emerged which address the inadequacies of traditional techniques, offering rapid, selective, and sensitive bacterial detection (Gehring and Tu 2011; Glynn et al. 2006; Lazcka et al. 2007; Velusamy et al. 2010). These techniques frequently rely on the analysis of strain-specific biomarkers which indicate the presence and abundance of specific bacteria strains. Ideally, these biomarkers are present at relatively high copy numbers, while also being sufficiently heterologous at the sequence level to allow for differentiation of the pathogen at both the genus and species levels (O’Connor and Glynn 2010).
An attractive candidate for bacterial biosensing that fulfills aforementioned criteria is transfer-messenger RNA (tmRNA) and its encoding ssrA gene. Specifically, tmRNAs are present in all bacterial species (Keiler et al. 2000; Keiler et al. 1996) at relatively high copy number (Glynn 2007; Lee et al. 1978; Muto et al. 1998) and contains regions of sequence sufficiently unique to unequivocally differentiate between species and genus (McGuinness et al. 2010; O’Grady et al. 2009; Scheler et al. 2011; Schönhuber et al. 2001). In addition, presence of intact RNA molecules can also indicate the condition of bacterial population and help distinguishing viable bacteria from nonviable (Keer and Birch 2003; O’Connor and Glynn 2010). Although the aforementioned properties also apply to widely used biomarker 16S rRNA (and its corresponding gene), several studies have shown that 16S rRNA is not always the most suitable biomarker to differentiate between closely related species (Schönhuber et al. 2001; Yamamoto and Harayama 1995). In particular, 16S rRNA sequence homogeneity between closely related species such as Escherichia coli and some Salmonella species preclude specific identification. Furthermore the discovery of intragenomic heterogeneity between the multiple rRNA genes of some organisms (including Streptococci) may further invalidate use of this target for diagnostics applications (Case et al. 2007; Kilian et al. 2008).
Given the clear diagnostic utility of tmRNAs, previous reports have focused on the fluorescence in situ hybridization (FISH) based detection of specific bacteria (Schönhuber et al. 2001), food analysis by real-time PCR (McGuinness et al. 2010) or nucleic acid sequence based amplification (NASBA) (O’Grady et al. 2009), and pathogen detection using NASBA-microarray combined technology (Scheler et al. 2011). However, the combination of tmRNA-based species identification and emerging biosensing technologies with key attributes (e.g. scalability, time-to-result, sample preparation requirements, etc.) offers the potential for rapid, real-time detection and identification of bacterial contaminants and their abundance in various sample types.
Microring resonators are a relatively new class of photonic microcavity devices that are highly sensitive to changes in the local refractive index, such as those resulting from biomolecular binding events. This sensing modality arises from the discrete wavelengths of light supported by the circular, interferometric structure that is sensitive to changes in the refractive index surrounding the sensor (Armani et al. 2003; Braginsky et al. 1989; Vahala 2003). Since any biomolecular binding event will lead to a measurable shift in optical properties, these sensors have been used to detect a broad array of targets, including both proteins (Luchansky and Bailey 2010; Washburn et al. 2009; Washburn et al. 2010) and nucleic acids (Qavi and Bailey 2010; Qavi et al. 2011a; Qavi et al. 2011b).
Key advantages of this technology include the small footprint and high scalability of the microrings afforded by their genesis in semiconductor fabrication. These characteristics position the platform advantageously for multiplexed biomarker analysis where diagnostic value improves with increasing size of the panel. Additionally, the near real time monitoring capability expedites assay development and allows valuable kinetic information to be directly observed, which is in contrast to endpoint-based biomarker detection methodologies such as blots or ELISAs.
In this paper we demonstrate the label-free detection and identification of tmRNA molecules using arrays of silicon photonic microring resonators. Bacterial analysis via tmRNA detection is a natural application of the platform given the low abundance of tmRNA molecules in bacteria, obviating the need for target amplification steps. Additionally, like all nucleic acid studies, complementary Watson-Crick base pairing is utilized to specifically and sensitively capture tmRNA molecules to the sensor surface for robust bacterial differentiation. Herein, we show the ability to distinguish between closely related bacterial species by monitoring corresponding tmRNA specific probe hybridization in a single, multiplexed assay. As little as 52.4 fmol of Streptococcus pneumoniae tmRNA was specifically detected, equivalent to 3.16 × 107 CFU of corresponding bacteria. Furthermore, the high specificity of this approach is demonstrated by the preferential capture of target tmRNAs in the presence of ~ 100-fold excess of off-target tmRNA molecules.
2. Materials and methods
2.1 In vitro tmRNA synthesis
Streptococcus pneumoniae ATCC 33400 (S.pneumoniae) and Klebsiella pneumoniae ATCC 13883 (K.pneumoniae) were obtained from DSMZ (Braunschweig, Germany). Streptococcus agalactiae (S.agalactiae) and Enterococcus faecium (E.faecium) were obtained from University College Hospital (Galway, Ireland). ssrA genes of each species were cloned into pCR® II-TOPO vectors (Invitrogen, Carlsbad, CA) under the transcriptional control of a T7 promoter sequence (gene sequences shown in SI). Both vector linearization and RNA synthesis were carried out according to the manufacturer’s suggestions with minor customizations. Briefly, the vector was linearized in 1χ buffer R using HindIII restriction endonuclease at 37 °C for 120 min. tmRNA was transcribed in vitro using 250 ng of linearized vector and 40 U of T7 RNA polymerase. The final reaction buffer contained 2 mM ATP, 2 mM CTP, 2 mM GTP and 1 mM UTP; 40 U RiboLock™ ribonuclease inhibitor was added to prevent possible RNA degradation. A final reaction volume of 50 μl was achieved by adding ultrapure H2O. All reagents were purchased from (Fermentas-ThermoFischer, Vilnius, Lithuania). The transcription reaction continued for 180 min at 37°C prior to purification with a RNeasy MinElute cleanup kit (Qiagen Inc., Valencia, CA). Nucleic acids were quantified by UV-Vis measurements (NanoDrop 1000; ThermoFischer, Wilmington, DE).
2.2 Microring resonator sensors and instrumentation
All sensor chips and read-out instrumentation were purchased from Genaltye, Inc. (La Jolla, CA), and have been previously described (Iqbal et al. 2010; Luchansky et al. 2010; Washburn et al. 2009).To summarize, chips containing 32 individually addressable microring sensors were used for each experiment. The chips were functionalized to present ssDNA capture probes using commercially available hydrazone-bond linker chemistry (described below). Functionalized chips were loaded into a microfluidic assembly that interfaced to the sensor scanner system.
2.3 Design of specific ssDNA capture probes and chaperones
Specific capture probes for S.pneumoniae (5′-CTTAATCGTATCTCGCTAATAATAAG-3′) and S.agalactiae (5′-TTTTTACAGTAGCCAAACGTAGTTTG-3′) were designed with SLICSel software as described previously (Kaplinski et al. 2010). From the array of designed probes, “SP_dom 4_24” specific for S.pneumoniae and “s_a 4_3” specific for S.agalactiae were selected based on their specificity and sensitivity in fluorescent microarray hybridization experiments (Kaplinski et al. 2010; Scheler et al. 2011). A poly-T linker was added to the S.agalactiae probe to match the surface distance of other probe. Human miRNA 24-1 specific DNA capture probe (5′-CTGTTCCTGCTGAACTGAGCCA-3′) was used as the baseline reference probe. Chaperone oligonucleotides, designed to disrupt tmRNA secondary structure, were optimized from previously published data (Chap A-F) and complementary to different regions of S.pneumoniae tmRNA. The chaperone sequences were obtained from Metabion (Martinsried, Germany).
2.4 ssDNA capture probe attachment to the sensor chip surface
All of the capture probes were obtained with 5′ amine functionality from Integrated DNA Technologies (Coralville, IA) and were HPLC purified prior use. All of the probes were next modified with a succinimidyl-4-formylbenzoate linker (Solulink, San Diego, CA) to introduce an aryl aldehyde moiety. In parallel, sensor chips were cleaned and functionalized as described previously (Qavi and Bailey 2010; Qavi et al. 2011a) to introduce reactive hydrazine groups. Small aliquots (1μl) of modified DNA probes were deposited onto the chip surface in a spatially controlled manner prior to overnight incubation in a humidity chamber and the resulting hydrazone bond linkage afforded covalent capture probe immobilization. Both the high DNA probe concentration and overnight incubation ensured the surface achieved a high density of DNA probes as monitored in real-time experiments (data not shown) and consistent with literature reports (Peterson et al. 2001). A minimum of three rings were functionalized with the same capture probe for replicate analysis. Prior to hybridization experiments, the substrates were sonicated in 8 M urea for 7 min to remove any non-covalently immobilized capture probe.
2.5 Hybridization experiments with tmRNA molecules
tmRNA hybridization was performed in a buffer consisting of 30% formamide, 30 mM ethylenediaminetetracetic acid (EDTA), 4× Saline-Sodium-Phosphate-EDTA (SSPE) Buffer, 0.2% SDS, and 2.5× Denhardt’s Solution. All of the hybridization experiments were carried out at room temperature by recirculating tmRNA solutions across the sensor surface at a rate of 24 μl/min for 60 minutes using a P625 peristaltic pump (Instech Labs, Plymouth Meeting, PA). Solutions were directed across the surface via a 0.178 mm thick, U-shaped single channel Mylar gasket sandwiched between a Teflon cartridge and the sensor chip. To avoid complications with hybridization between the capture probes and the secondary structure natively adopted by tmRNA molecules, three different sample preparation methods were compared: thermal tmRNA denaturation, thermal tmRNA denaturation with a 10-fold excess of chaperones, and chemical tmRNA fragmentation. Denaturation consisted of 15 min treatment at 95 °C in 100 μL of hybridization buffer. Chaperones are short DNA sequences added to the sample matrix in order to disrupt extended secondary structures adopted by full length tmRNAs based upon hybridization to specific regions of the tmRNA. Chemical tmRNA fragmentation by metal ion-catalysis (Liu et al. 2007) was performed in 100mM ZnCl2 containing Tris (pH 7.0) buffer for 15 min at 70° C resulting in a mixture of 80-120 nucleotide long RNA fragments according to agarose gel electrophoresis. 10 μl of fragmented tmRNA solution was added to 90 μl of hybridization solution after the fragmentation. All solutions were cooled to room temperature before sample introduction and all of the experiments were carried out using 1.66 pmol of S.pneumoniae tmRNA (equaling to 1×1012 tmRNA molecules or 1×109 CFU of bacteria, respectively), unless stated otherwise.
2.6 Data analysis
All data for tmRNA specific microrings was corrected for temperature and instrument drift by subtracting a series of baseline reference probes. All data was analyzed, fit, and graphed using OriginPro8 (OriginLab, Northampton, MA).
To utilize the curves for quantitative analysis of binding, we fit a 1:1 Langmuir kinetic binding isotherm, described by:
The initial slope of this function was determined by evaluating its derivative at t = t0. At concentrations below 1.66 pmol, a linear fit was utilized to determine the initial slope response.
3. Results and discussion
In this study, we demonstrate the quantitative detection of specific tmRNA molecules for bacterial biosensing using arrays of silicon photonic microring resonators. We decided to examine tmRNA from the respiratory tract pathogen S.pneumoniae due to its widespread negative impact on human health and also the complications that are linked with current detection technologies, such as a lack of sufficient specificity, rapidness, or robust quantitative capability (Blaschke 2011). Recently published work has established the utility of tmRNA for distinguishing between S.pneumoniae and five other closely related respiratory tract pathogens in a highly sensitive manner (Scheler et al. 2011). In this study, tmRNA molecules from three other pathogens (K.pneumoniae, E. faecium and S.agalactiae) are used for comparison purposes.
A schematic of the tmRNA hybridization assay is shown in Figure 1. In this assay, a DNA probe complementary to the target tmRNA of interest is covalently attached to the microring surface, after which a solution containing the target tmRNA is flowed across the sensor. The hybridization of tmRNA onto the probe-modified sensor surface results in a change in the wavelength of light that is resonantly coupled into the microring, resulting in an easily measured shift.
Figure 1.

Schematic demonstrating the principle of microring optical resonator based detection of tmRNA hybridization, including the tmRNA binding curve response of four separate microrings. The asterisk (*) denotes the time at which the solution containing 320 ng of fragmented target tmRNA is introduced to the sensor chip.
Unlike small nucleic acids such as siRNAs and miRNAs, tmRNAs frequently possess significant secondary and tertiary structures (Burks et al. 2005) that can complicate simple hybridization based detection. This is of particular concern when making measurements near room temperature, which is convenient from a sensor operation perspective. Early observations reinforced these complications as hybridization responses of tmRNA molecules to the sensor surface were extraordinarily slow. To address this challenge, three RNA pre-treatment methods were investigated to determine the optimal conditions for tmRNA detection at room temperature. These methods included: chemical fragmentation of the tmRNAs using ZnCl2, denaturation of the target tmRNAs by heating them to 95°C before cooling back to room temperature, and thermal denaturation of the targets in the presence of chaperone oligonucleotides designed to assist in unfolding the tmRNA. These chaperone sequences were designed and demonstrated to bind to predicted secondary structure regions in S.pneumoniae tmRNA, prevent refolding of tmRNA after denaturation and therefore enhance tmRNA hybridization in previous work (Kaplinski et al. 2010).
As shown in Figure 2, fragmentation of the tmRNA was the most effective method in order to enhance both the binding kinetics and overall net response magnitude. We attribute this primarily to the reduced secondary structure present in the shorter (80-120 nucleotides) tmRNA fragments. Our results agree with the previous report by Wu and co-workers in which RNA fragmentation was also found to be the most effective strategy to improve hybridization efficiency and sensitivity in a fluorescent microarray analysis (Liu et al. 2007). Consequently, tmRNA molecules were fragmented in all subsequent experiments to improve the sensor performance.
Figure 2.
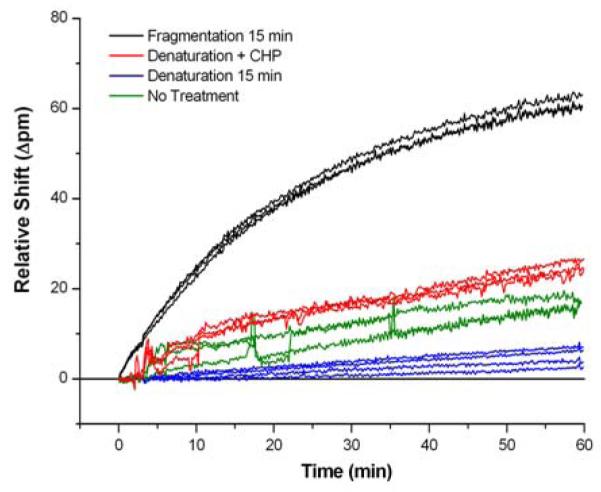
Comparison of the hybridization-adsorption response of untreated tmRNA with different pre-treatment methods. The fragmentation methodology yielded the highest response and was used for all experiments.
Once fragmentation was established as the most effective pre-treatment for tmRNA samples, we sought to optimize fragmentation time. As shown in Supplementary Information Figure S2, the time in which the sample was exposed to the ZnCl2 fragmentation solution was systematically varied from zero to 60 minutes, and the resulting hybridization responses measured using identically prepared sensors. These experiments indicated that 10 min of treatment was sufficient for optimal sensor performance, and a standard 15 minute fragmentation period was used for all subsequent experiments. Interestingly, we did not observe any significant change in the non-specific sensor response as a function of fragmentation time.
In order for a microbial diagnostic technology to be useful, it must respond quantitatively and specifically to low levels of target marker molecules in a high background of non-target nucleic acid sequences. This is due to the diversity of bacterial species that may be present in clinical or other types of samples. Addressing specificity first, we functionalized a single sensor array with ssDNA capture probes targeting bacterial tmRNAs from S.pneumoniae and S.agalactiae. We subsequently introduced a series of tmRNAs from four bacterial species (K.pneumoniae, E. faecium, S.pneumoniae, and S.agalactiae) sequentially across the sensor surface. Each tmRNA solution had 1.66 pmoles of target. As seen in Figure 3, K.pneumoniae and E.faecium tmRNA did not elicit a response while subsequent hybridization steps with both S.agalactiae and S.pneumoniae tmRNA demonstrated strong and specific responses from the microrings modified with complementary DNA capture probes. The different response magnitudes from S.pneumoniae- and S.agalactiae-specific microrings can be attributed to differences in the probes’ length and hybridization properties (duplex melting temperatures (Tm), binding affinity, nucleotide composition and positioning) of the different DNA capture probe-RNA target pairs. Additionally, targeting complementary regions in fragmented tmRNA molecules can still be unevenly hindered by any remaining secondary structure. In the future, probe sequences could be redesigned to normalize for these differences, or higher Tm forming locked nucleic acids (LNAs) could be employed. Nonetheless, results at this stage clearly demonstrate the potential of the microring resonator platform to directly detect bacterial tmRNAs and discriminate between unique strains on the basis of differential hybridization.
Figure 3.
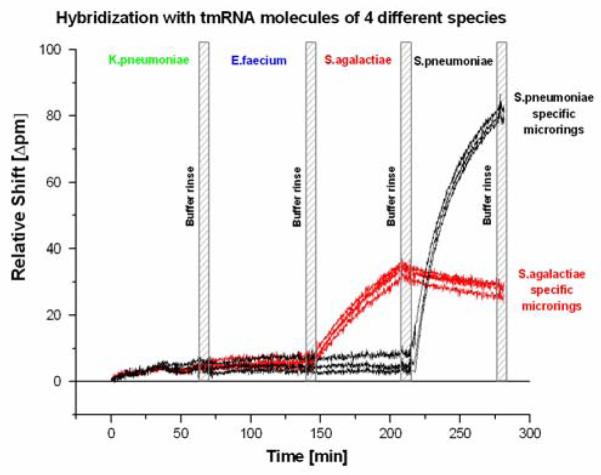
The specific detection of two tmRNA species on a continuous single chip assay. Microrings were functionalized with either Streptococcus pneumonia (black) or Streptococcus agalactiae (red) tmRNA-specific DNA oligonucleotide capture probes. tmRNA from four different bacterial species (Klebsiella pneumoniae, Enterococcus faecium, S.agalactiae, and S.pneumoniae) were sequentially introduced into the flow chamber, and a specific response observed for each tmRNA. Washing steps with neat buffer solution were carried out between hybridization steps.
Having demonstrated the high specificity of the method, we then focused on establishing the quantitative utility of the platform towards tmRNA detection. DNA-functionalized microring sensors were exposed to different quantities of S.pneumoniae tmRNA, ranging from 52.4 fmol to 16.6 pmol. A cocktail of control tmRNAs from three other bacteria (1.66 pmol each of: K.pneumoniae, E.faecium and S.agalactiae) were also added to the hybridization mixture as a background to mimic the complex matrix in which tmRNA analytes are found naturally. The concentration dependent responses, shown in Figure 4, gave a limit of detection of 52.4 fmol (or 100 μl of 524 pM tmRNA solution). This limit of detection corresponds to roughly 3.16 × 1010 tmRNA molecules or 3.16 × 107 CFU of S.pneumoniae, with a dynamic range of nearly three orders of magnitude.
Figure 4.

a) Concentration dependent responses from the binding of S. pneumonia tmRNA (from 16.6 pmol to 0 pmol, decreasing top-to-bottom). b) Calibration plot of tmRNA induced sensor response. Quantitation was performed by fitting the initial slope.
The limit of detection reported herein surpasses previous reports on the direct, label-free detection of microRNAs and DNA using the same measurement technology (Qavi and Bailey 2010; Qavi et al. 2011a; Qavi et al. 2011b). This increased sensitivity is due to the larger size of the tmRNA targets. Even after fragmentation, the detectable targets are still 80-120 nucleotides in length, compared to the previously investigated 22 nucleotide microRNA sequences. Furthermore, this detection limit is comparable to that achieved using surface plasmon resonance imaging (SPRi), in which the detection of 2nM of Escherichia coli 16S rRNA was reported in a similar direct hybridization assay (Nelson et al. 2001).
Extrapolating beyond this initial report, the potential multiplexing capability of this silicon photonic platform is attractive for more informative bacterial diagnostics, whereby the presence of a larger number of targeted bacteria can be simultaneously probed using a relatively simple and rapid assay protocol. Although the obtained level of sensitivity is far from that achieved with regular culture-based methods and molecular methods, the incorporation of signal and/or target amplification technologies should allow for sensitivity levels comparable to real-time NASBA or real-time RT-PCR based detection of tmRNA molecules, albeit with additional sample and/or assay manipulation steps. Recently an antibody specific to DNA-RNA duplexes was incorporated onto this microring resonator platform and a ~3 order of magnitude improvement in limit of detection was achieved for miRNA analysis (Qavi et al. 2011a). Also, larger refractive index tags can be introduced to dramatically increase limits of detection, as compared to solely label-free analyses (Luchansky et al. 2011). Importantly, both these additional amplification steps could be implemented in two hours or less and thus the assay would retain its quick time-to-result as compared to traditional microbiology-based analysis schemes. Aside from the sensor platform itself, increases in sensitivity can also be accomplished by applying culture enrichment methods (McGuinness et al. 2009; O’Grady et al. 2008) or tmRNA amplification using isothermal NASBA technology (Compton 1991) prior to hybridization.
4. Conclusions
In this manuscript we present a promising approach for bacterial diagnostics and microbial analysis utilizing arrays of silicon photonic microring resonators. The capability of this analysis platform for multiplexed measurements allowed for the rapid discrimination of tmRNA from multiple, distinct bacterial species. As little as 53 fmol of Streptococcus pneumoniae tmRNA, equivalent to roughly 3.16 × 107 CFU of the corresponding bacteria, was easily detected on this platform using a label-free direct hybridization assay. Using the described approach different patient samples, food products, or analyte solutions might be tested and quickly screened for multiple pathogenic contaminants in a highly parallel manner. In particular, this methodology holds significant promise in the fields of environmental monitoring, bio-threat detection, industrial process monitoring, and clinical microbiology—application areas where specificity and time-to-result are of critical importance. Although this work focused on early stage studies designed to show applicability to in vitro synthesized target tmRNA molecules, this platform has been applied to significantly more complex RNA-containing samples (Qavi and Bailey 2010; Qavi et al. 2011a) and thus the assays described herein should be applicable to these samples with only minor modifications to hybridization conditions or minor pre-analysis sample preparation.
Supplementary Material
Hihglights.
tmRNAs are promising markers for distinguishing between pathogenic bacterial species.
Quantitation of multiple tmRNAs is demonstrated using a silicon photonic detection platform.
Scalable tmRNA detection is needed for biodefense, food monitoring, and health care, among others.
Table 1.
DNA capture probes used in experiments. Thermodynamic calculations performed using IDT OligoAnalyzer (www.idtdna.com) with default settings.
| sequence | GC% | Tm° | |
|---|---|---|---|
| S.pneumoniae specific | 5′-CTTAATCGTATCTCGCTAATAATAAG-3′ | 30.8 | 42.2 |
| S.agalactiae specific | 5′-TTTTTACAGTAGCCAAACGTAGTTTG-3′ | 42.9 | 47.2 |
| Baseline reference probe | 5′-CTGTTCCTGCTGAACTGAGCCA-3′ | 54.5 | 59.3 |
Acknowledgements
Financial support for this work was provided by the National Institutes of Health (NIH) Director’s New Innovator Award Program, part of the NIH Roadmap for Medical Research, Grant Number 1-DP2-OD002190-01, the Center for Advanced Study at the University of Illinois at Urbana–Champaign, and the Camille and Henry Dreyfus Foundation. This research was also supported by European Social Fund’s Doctoral Studies and Internationalisation Programme DoRa and targeted financing SF0180026s09 and SF0180027s10 from the Estonian Ministry of Education and Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Armani DK, Kippenberg TJ, Spillane SM, Vahala KJ. Nature. 2003;421(6926):925–928. doi: 10.1038/nature01371. [DOI] [PubMed] [Google Scholar]
- Blaschke AJ. Clinical Infectious Diseases. 2011;52:S331–S337. doi: 10.1093/cid/cir048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braginsky VB, Gorodetsky ML, Ilchenko VS. Physics Letters A. 1989;137(7-8):393–397. [Google Scholar]
- Burks J, Zwieb C, Müller F, Wower I, Wower J. BMC molecular biology. 2005;6:14. doi: 10.1186/1471-2199-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case RJ, Boucher Y, Dahllof I, Holmstrom C, Doolittle WF, Kjelleberg S. Applied and environmental microbiology. 2007;73(1):278–288. doi: 10.1128/AEM.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton J. Nature. 1991;350:91–92. doi: 10.1038/350091a0. [DOI] [PubMed] [Google Scholar]
- Gehring AG, Tu S-I. Annual review of analytical chemistry. 2011;4:151–172. doi: 10.1146/annurev-anchem-061010-114010. [DOI] [PubMed] [Google Scholar]
- Glynn B. Research Journal of Biological Sciences. 2007;2:564–570. [Google Scholar]
- Glynn B, Lahiff S, Wernecke M, Barry T, Smith TJ, Maher M. International Journal of Dairy Technology. 2006;59:126–139. [Google Scholar]
- Iqbal M, Gleeson MA, Spaugh B, Tybor F, Gunn WG, Hochberg M, Baehr-jones T, Bailey RC, Gunn LC. Quantum. 2010;16:654–661. [Google Scholar]
- Kaplinski L, Scheler O, Parkel S, Palta P, Toome K, Kurg A, Remm M. BMC biotechnology. 2010;10:34. doi: 10.1186/1472-6750-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keer JT, Birch L. Journal of Microbiological Methods. 2003;53:175–183. doi: 10.1016/s0167-7012(03)00025-3. [DOI] [PubMed] [Google Scholar]
- Keiler KC, Shapiro L, Williams KP. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:7778–7783. doi: 10.1073/pnas.97.14.7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiler KC, Waller PR, Sauer RT. Science. 1996;271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- Kilian M, Poulsen K, Blomqvist T, Havarstein LS, Bek-Thomsen M, Tettelin H, Sorensen UBS. Plos One. 2008;3(7) doi: 10.1371/journal.pone.0002683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazcka O, Del Campo FJ, Muñoz FX. Biosensors & bioelectronics. 2007;22:1205–1217. doi: 10.1016/j.bios.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Lee SY, Bailey SC, Apirion D. Journal of bacteriology. 1978;133:1015–1023. doi: 10.1128/jb.133.2.1015-1023.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W-T, Guo H, Wu J-H. Applied and environmental microbiology. 2007;73:73–82. doi: 10.1128/AEM.01468-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchansky MS, Bailey RC. Anal Chem. 2010;82(5):1975–1981. doi: 10.1021/ac902725q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchansky MS, Washburn AL, Martin TA, Iqbal M, Gunn LC, Bailey RC. Biosensors & bioelectronics. 2010;26(4):1283–1291. doi: 10.1016/j.bios.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchansky MS, Washburn AL, McClellan MS, Bailey RC. Lab on a chip. 2011;11(12):2042–2044. doi: 10.1039/c1lc20231f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness S, Barry T, O’Grady J. Food Research International. 2010:12–15. [Google Scholar]
- McGuinness S, McCabe E, O’Regan E, Dolan A, Duffy G, Burgess C, Fanning S, Barry T, O’Grady J. Meat science. 2009;83:555–562. doi: 10.1016/j.meatsci.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Muto A, Ushida C, Himeno H. Trends in biochemical sciences. 1998;23:25–29. doi: 10.1016/s0968-0004(97)01159-6. [DOI] [PubMed] [Google Scholar]
- Nelson BP, Grimsrud TE, Liles MR, Goodman RM, Corn RM. Analytical chemistry. 2001;73:1–7. doi: 10.1021/ac0010431. [DOI] [PubMed] [Google Scholar]
- O’ Grady J, Lacey K, Glynn B, Smith TJ, Barry T, Maher M. FEMS microbiology letters. 2009;301:218–223. doi: 10.1111/j.1574-6968.2009.01822.x. [DOI] [PubMed] [Google Scholar]
- O’ Grady J, Sedano-Balbás S, Maher M, Smith T, Barry T. Food microbiology. 2008;25:75–84. doi: 10.1016/j.fm.2007.07.007. [DOI] [PubMed] [Google Scholar]
- O’Connor L, Glynn B. Expert review of medical devices. 2010;7:529–539. doi: 10.1586/erd.10.22. [DOI] [PubMed] [Google Scholar]
- Peterson AW, Heaton RJ, Georgiadis RM. Nucleic Acids Res. 2001;29(24):5163–5168. doi: 10.1093/nar/29.24.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qavi AJ, Bailey RC. Angewandte Chemie (International ed. in English) 2010;49:4608–4611. doi: 10.1002/anie.201001712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qavi AJ, Kindt JT, Gleeson M.a., Bailey RC. Analytical chemistry. 2011a doi: 10.1021/ac201340s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qavi AJ, Mysz TM, Bailey RC. Analytical chemistry. 2011b doi: 10.1021/ac201659p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheler O, Kaplinski L, Glynn B, Palta P, Parkel S, Toome K, Maher M, Barry T, Remm M, Kurg A. BMC biotechnology. 2011;11:17. doi: 10.1186/1472-6750-11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönhuber W, Bourhis GL, Tremblay J, Amann R, Kulakauskas S. BMC Microbiology. 2001 doi: 10.1186/1471-2180-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahala KJ. Nature. 2003;424(6950):839–846. doi: 10.1038/nature01939. [DOI] [PubMed] [Google Scholar]
- Velusamy V, Arshak K, Korostynska O, Oliwa K, Adley C. Biotechnology advances. 2010;28:232–254. doi: 10.1016/j.biotechadv.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Washburn AL, Gunn LC, Bailey RC. Analytical chemistry. 2009;81:9499–9506. doi: 10.1021/ac902006p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn AL, Luchansky MS, Bowman AL, Bailey RC. Analytical chemistry. 2010;82(1):69–72. doi: 10.1021/ac902451b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Harayama S. Applied and environmental microbiology. 1995;61:3768. doi: 10.1128/aem.61.10.3768-3768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.