

Abstract
The small heat shock protein HSPB1 is a multifunctional, α-crystallin-based protein that has been shown to be neuroprotective in animal models of motor neuron disease and peripheral nerve injury. Missense mutations in HSPB1 result in axonal Charcot-Marie-Tooth disease with minimal sensory involvement (CMT2F) and distal hereditary motor neuropathy type 2 (dHMN-II). These disorders are characterized by a selective loss of motor axons in peripheral nerve resulting in distal muscle weakness and often severe disability. To investigate the pathogenic mechanisms of HSPB1 mutations in motor neurons in vivo, we have developed and characterized transgenic PrP-HSPB1 and PrP-HSPB1(R136W) mice. These mice express the human HSPB1 protein throughout the nervous system including in axons of peripheral nerve. Although both mouse strains lacked obvious motor deficits, the PrP-HSPB1(R136W) mice developed an age-dependent motor axonopathy. Mutant mice showed axonal pathology in spinal cord and peripheral nerve with evidence of impaired neurofilament cytoskeleton, associated with organelle accumulation. Accompanying these findings, increases in the number of Schmidt-Lanterman incisures, as evidence of impaired axon-Schwann cell interactions, were present. These observations suggest that overexpression of HSPB1(R136W) in neurons is sufficient to cause pathological and electrophysiological changes in mice that are seen in patients with hereditary motor neuropathy.
Keywords: Hereditary motor neuropathy, Charcot-Marie-Tooth disease, small heat shock protein, prion protein promoter, axonal degeneration, motor neuron disease
Introduction
Distal hereditary motor neuropathy (dHMN) is clinically similar to axonal forms of Charcot-Marie-Tooth disease (CMT2) but lacks significant clinical or electrophysiological evidence of sensory axon involvement (Pareyson and Marchesi 2009; Reilly and Shy 2009). Missense mutations in HSPB1 (also known as heat shock 27kD protein 1, HSP27) result in CMT disease type 2F (CMT2F; OMIM 606595) and dHMN type II (OMIM 608634) (Evgrafov, Mersiyanova et al. 2004; Chung, Kim et al. 2008; Dimos, Rodolfa et al. 2008; Houlden, Laura et al. 2008; James, Rankin et al. 2008; Ikeda, Abe et al. 2009; Benedetti, Previtali et al. 2010; Luigetti, Fabrizi et al. 2010; Solla, Vannelli et al. 2010). HSPB1 mutations usually result in an adult onset motor neuropathy, however early-onset phenotypes of childhood have also been reported (Kijima, Numakura et al. 2005; Dierick, Baets et al. 2008). Motor neuropathies due to HSPB1 mutations usually present with slowly progressive peroneal muscular atrophy and have electrophysiological evidence of decreased compound muscle action potentials (CMAPs) with normal or slightly reduced sensory nerve action potentials (SNAPs). Few pathological reports from human nerve biopsy exist, however morphological analysis of a sural nerve biopsy from a patient with an HSPB1p.S135C mutation revealed evidence of myelin remodeling with excessively thick myelin and fissures in the setting of normal density of myelinated fibers (Benedetti, Previtali et al. 2010). Ultrastructural analysis revealed atrophic axons with increased microtubule density (Benedetti, Previtali et al. 2010). In a patient with an HSPB1p.T180I mutation, sural nerve biopsy showed mildly reduced myelinated fiber density (Luigetti, Fabrizi et al. 2010).
To study the physiological role of HSPB1 in motor neurons and to investigate the pathogenic mechanisms of mutations in HSPB1, we have developed transgenic PrP-HSPB1 and PrP-HSPB1(R136W) mice. The PrP-HSPB1(R136W) mice did not develop a motor phenotype despite high levels of expression in neurons. These mice did develop electrophysiological and pathological evidence of an axonal motor neuropathy that was age-dependent.
Materials and methods
Transgenic mice
The PrP-HSPB1 vector was generated by cutting out the HSPB1 open reading frame from M13-HSPB1 (GeneCopoeia, Rockville, MD). The fragment was digested with XhoI and ligated into XhoI digested MoPrP.Xho vector (Borchelt, Davis et al. 1996). Vectors containing the insert were mapped by restriction endonuclease digestion and the orientation of the HSPB1 open reading frame within the vector was verified by DNA sequencing (primers: forward: 5′-TACCCGCATAGCCGCCTCTT-3′, reverse: 5′-GATGGTGATCTCGTTGGACT-3′). Prior to injection of FVB/NJ mice (The Jackson Laboratory, Bar Harbor, ME), vector DNA was digested with NotI and purified by agarose gel chromatography. The linearized DNA was injected into fertilized oocytes and transferred the oocytes to foster FVB/NJ females, using standard pronuclear microinjection techniques by the Transgenic Animal Facility at The Ohio State University, Columbus, OH. Transgenic mice were identified by polymerase chain reaction (PCR) using PrP-HSPB1 specific primers: Forward 5′-CACTGGCTGATGACAGACTC-3′ Reverse 5′-CCGGTGTGGACCCCACCCAA-3′ A single nucleotide substitution (C406T) was introduced to produce the R136W mutation using a commercially available kit (Stratagene, La Jolla, CA).
Housing and procedures involving animals and their care were conducted in accordance with the Principles of Laboratory Animal Care (National Institutes of Health publication number 86-23) and the guidelines approved by the Institutional Animal Care and Use Committee at the Ohio State University.
Copy number determination
Mice positive for transgene expression were subjected to HSPB1 gene copy number analysis by quantitative PCR (Q-PCR) using 10 ng genomic DNA, 2x TaqMan Gene Expression Master Mix (Applied Biosystems, Warrington, UK), 20x TaqMan probes (Applied Biosystems, Warrington, UK) [the assay identification numbers are human HSPB1 (Hs03044127_g1); human GAPDH (Hs02758991_g1); mouse GAPDH (Mm99999915_g1)], total volume 20 μl for each reaction, on a 7300 Real-Time PCR System (Applied Biosystems) using standard 7300 Fast Real-Time PCR software. Ct values were compared to a reference DNA sample containing only two copies of human HSPB1.
Digital droplet PCR for determination of HSPB1 levels in spinal cord and brain
Ten milligrams of brain and spinal cord tissues from PrP-HSPB(R136W) and PrP-HSPB1 mice were pulverized in liquid N2 with mortar and pestle, and homogenized with 1 ml of Trizol (Invitrogen). Total RNA was isolated according to the manufacturer’s directions (RNAeasy Mini Kit, Qiagen). HSPB1 (Hs03044127_g1, Applied Biosystems, Warrington, UK) and customized cyclophiln (primers: forward: 5′GTCAACCCCACCGTGTTCTT3′, reverse: 5′TTGGAACTTTGTCTGCAAACA3′, probe CTTGGGCCGCGTCT-NED) probes were used to amplify cDNA template by QX100™ Droplet Digital™ PCR System (BioRad). Each PCR reaction (ran in duplicate) was partitioned into 10,467 to 13,369 oil droplets and each droplet is read individually by the ddPCR fluorescence detectors (Hindson, Ness et al. 2011). The distribution of positive and negative droplets conforms to Poisson statistical distributions (Pinheiro, Coleman et al. 2012). Relative HSPB1 levels were determined by calculating the ratio of HSPB1 product to cyclophilin product.
Immunohistochemistry
Anesthetized mice were perfused intracardially with 4% paraformaldehyde in phosphate-buffered saline (PBS). The lumbar segment of spinal cord and sciatic nerve were dissected and postfixed in the same solution overnight at 4°C. 40μm thick transverse free-floating sections were cut using a Vibrotome 3000 plus tissue sectioning machine (Vibotome, St. Louis, MO). Nonspecific binding was blocked using a blocking solution consisting of 10% donkey serum, 0.1% Triton X-100-PBS for 2 hours at room temperature. The sections were then incubated overnight at 4°C with the appropriate dilution of primary antibody in blocking solution. The corresponding secondary antibodies (Alexa Fluor-488; Alexa Fluro-546 secondary fluorescent antibodies, Invitrogen, Carlsbad, CA) were added 1:200 in blocking solution for 2 hour at room temperature. Sections were then rinsed with 0.1 M PBS and placed on coverslips with aqueous non-fluorescing mounting medium (Immu-mount, Thermo Scientific, Pittsburgh, PA). Images were obtained with a Zeiss LSM510-META confocal laser-scanning microscope (Zeiss, Thornwood, NY). Primary antibodies used for these experiments were: anti-choline acetyltransferase (ChAT,1:200 dilution) (Millipore, USA); anti-heat shock protein 27 (human HSPB1,1:500 dilution) (Millipore, USA), anti-heat shock protein 25 (mouse HSPB1, 1:500 dilution) (Millipore, USA), anti-glial fibrillary acidic protein (GFAP, 1:1000 dilution) (Santa Cruz, USA) and anti-neurofilament 200 (NF-200, 1:500 dilution) (Sigma-Aldrich, USA)
Western blot Analysis
Brain and spinal cord tissue were flash frozen in liquid nitrogen, crushed using a mortar and pestle and then homogenized in ice cold buffer [T-PER reagent (Thermo Scientific, Rockford, IL), 10X protease inhibitor (Roche Diagnostic, Mannheim, Germany)] with 20 strokes of a hand held glass homogenizer. The homogenate was then subjected to centrifugation at 20,000g for 15 min and 4°C. The supernatant was then transferred to a separate tube and protein concentration was determined spectrographically (SmartSpec Plus Spectrophotometer, Bio-Rad). Protein electrophoresis was performed using the NuPAGE electrophoresis pre-cast gel system (Novex Mini-Cell, Invitrogen). Protein from 20 μg of cytoplasmic extracts were separated on a NuPAGE® 4-12% Bis-Tris gel (Invitrogen) and transferred to nitrocellulose membranes. The membrane was incubated with rabbit polyclonal anti-HSPB1 (1:1000, Abcam, Cambridge, MA) and mouse monoclonal anti-GAPDH (1:4000, Applied Biosystems, USA) primary antibodies for 1hour at room temperature. After washing in PBS + 0.1%Tween 20, membrane was incubated with IRDye 680 goat anti-rabbit (1:10000; Li-Cor Biotechnology, Nebraska) and IRDye 680LT goat anti-mouse (1:10000; Li-Cor Biotechnology, Nebraska) secondary antibodies for 1 hour at room temperature. The membrane was scanned by an Odyssey® infrared imaging system (Li-Cor), and intensities were analyzed using the software provided by the manufacturer.
Electrophysiology
For electrophysiological experiments 5 mice were examined in each group [PrP-HSB1(R136W), PrP-HSB1 and age matched, non-transgenic, littermate controls] at 6 month and 1 year of age. Mice were anesthetized with 100 mg/kg ketamine + 10 mg/kg xylazine (Sigma-Aldrich, St. Louis, Missouri) delivered intraperitoneally. Surface temperature was maintained between 30-32°C. Nerve conduction studies were performed by a single, blinded, electromyographer (WDA) using a portable electrodiagnostic system (Synergy EMG machine version 9.1, Oxford Instruments, Abingdon, UK). Waveforms were displayed on an LCD monitor (Toshiba North America, Houston, Texas). Distance for electrode placement was measured using a compass with a needle edge and a millimeter-graduated tape measure. The amplitude and conduction velocity was determined for each nerve studied.
Sciatic motor nerve conduction studies were performed as previously described (Xia, Yosef et al. 2010). The low pass filter was set at 20 Hz, and the high-pass filter was set at 10 kHz. Compound muscle action potential amplitude and conduction velocity were recorded using surface recording electrodes (Alpine Biomed, Skovlunde, Denmark) placed over the gastrocnemius muscle while stimulating the sciatic nerve with a needle electrode (Ambu USA, Glen Burnie, Maryland) at proximal and distal sites (5 mm proximal to the recording site at the midline of the posterior thigh and then 10 mm more proximal at the medial gluteal region). Orthodromic sensory and motor conduction studies of the tail nerve were performed using a non-invasive technique similar to that previously described in the rat (Kurokawa, de Almeida et al. 2004). For the orthodromic tail sensory studies the filter settings were set at 20 Hz for the low pass and 2 kHz for the high pass. Two pairs of ring electrodes (Alpine Biomed, Skovlunde, Denmark) were used for stimulation and recording. The recording ring electrodes were placed at the base of the tail. The stimulating ring electrodes were placed 30 mm distally. During the tail motor conduction studies the low pass filter was set at 3Hz and the high pass filter at 10 kHz. The recording ring electrodes were placed 30 mm distal to the base of the tail. For distal stimulation the stimulating electrodes were placed 10 mm proximal to the recording electrodes. For the proximal stimulation the stimulating electrodes were placed 20 mm proximally. For the sensory and motor conductions a disposable disk electrode (Carefusion, Middleton, Wisconsin) was placed on the hindlimb, tail or sacrum so that the artifact was minimized.
Neuropathology and morphometry
Semi thin and ultrastructural analysis were conducted in 6 PrP-HSB1(R136W) mice at the age of 6 months (n=1), 1 year (n=4) and 1.5 year of age (n=1), 4 PrP-HSB1 mice and 4 non transgenic, littermate control mice (1 at 6 months and 3 at 1 year of age in each group). Mice were given xylazine/ketamine anesthesia and killed by cardiac perfusion, with 4% paraformaldehyde followed by 5% glutaraldehyde both in 0.1 M phosphate buffer. Lumbar and sacral segments of spinal cord, dorsal root ganglia from L4-S1 levels, sciatic nerves, soleus, lumbrical and intrinsic foot muscles were removed under a dissecting microscope. These tissues were dissected into small blocks and were processed for plastic embedding for light microscope thick sections and electron microscopy using standard methods established (Sahenk and Mendell 1979).
Quantitative morphology
Quantization of Schmidt-Lanterman incisures (SLI) density
Sciatic nerves from 3 mice at 1 year of age in each group were used for quantitative analysis. Three randomly selected areas of the 1 μm-thick toluidine blue-stained cross sections from mid-sciatic nerves were photographed using 60x objective and printed at 1500x final magnification. Myelin profiles with putative SLI (i.e. with a clear circular band separating two dark myelin bands) were marked with a ‘S’ as described previously (Gould, Byrd et al. 1995). For each animal, counts from all 3 randomly selected representative unit areas (total area analyzed 0.7mm2) were included and the SLI density histograms were generated as number per total unit area analyzed.
G ratio of the myelinated fibers
The g ratio refers to the ratio of axonal diameter/fiber diameter and lower g ratios represent axons with thicker myelin (Beuche and Friede 1985). For g ratio determinations, images from 3 representative areas from 3 PrP-HSPB1(R136W) mice, 3 PrP-HSPB1 and 3 non-transgenic, littermate controls at 1 year of age were captured at x100 magnification, and the shortest axial lengths as axon diameters and fiber diameters were recorded with a calibrated micrometer, using the AxioVision, 4.2 software (Zeiss) as described previously(Sahenk, Galloway et al. 2010). The g ratio distribution histograms were generated as a percent of total fibers analyzed. Measurements from 1023 myelinated fibers in PrP-HSPB1(R136W), 1191 in PrP-HSPB1 and 1089 in non-transgenic, littermate controls at 1 year of age were included in each group. In addition, the myelinated fiber densities (number/mm2 of endoneurial area) were estimated in each group.
Neurofilament packing density determination
The ultrastructural morphometric study was performed using cross sectional images at x52,000 final magnification. Neurofilament density histograms were generated by determining the number of neurofilaments per unit hexagonal area in randomly selected axons from PrP-HSPB1(R136W) and non-transgenic, littermate controls at 1 year of age as previously described (Sahenk, Galloway et al. 2010). Ten randomly selected myelinated fibers with axon diameters ranging from 4 to 6 μm were analyzed.
Behavioral analysis
Mice were weighed and inspected for signs of muscle weakness or gross behavioral alterations weekly for 12 months. All behavioral testing was performed by a blinded evaluator. Digital open field testing was performed in 6 month old PrP-HSPB1(R136W) (n=27), PrP-HSPB1 (n=21) and non-transgenic, littermate control mice (n=24) using an open field Plexiglas chamber equipped with multiple photocell receptors and emitters [Digiscan Activity Monitor (Accuscan, Columbus, OH)], for 2 hours. Ladder test [PrP-HSPB1(R136W) (n=7), PrP-HSPB1 (n=9) and non-transgenic, littermate control mice (n=9)] and foot tracing analysis [PrP-HSPB1(R136W) (n=10), PrP-HSPB1 (n=10) and non-transgenic, littermate controls (n=10)] were performed as previously described in 12 month old mice (Butchbach, Edwards et al. 2007; Yuan, Shen et al. 2010). For the foot tracing analysis the plantar surface of the mouse hind paws were painted with red ink and the mouse walked along a corridor of 6 cm×40 cm lined with white paper. From these tracings, the paw length (PL), distance between the 1st and 5th toes (TS) and between the 2nd and 4th toes (IT) were measured manually by a blinded observer.
On a separate day, motor performance was further evaluated in 12 month old mice [PrP-HSPB1(R136W) (n=7), PrP-HSPB1(n=7) and non-transgenic, littermate control mice (n=7)] with a Rotarod apparatus (7650 accelerating model, Ugo Basile, Varese, Italy). Mice were placed on the accelerating rod at a starting speed of 4 rpm, reaching a final speed of 40 rpm in 5 minutes. All the mice were tested for three trials. The mice were allowed to stay on the rod for a maximum of 600 seconds and the time of hold on the rod was scored.
Lastly, grip strength was performed in 12 month old mice [PrP-HSPB1(R136W) (n=5), PrP-HSPB1(n=5) and non-transgenic, littermate control mice (n=5)]. Total peak force (grams) was determined using an electronic Grip Strength Meter, Columbus Instruments (Columbus, OH, USA) with five measurements done on each fore and hind limb from each animal. The three highest measurements were averaged to give the strength score. The mice were allowed to rest for 10 minutes between fore and hindlimb measurements.
Statistical analyses
For all the analyses, comparison among groups was made using one-way analysis of variance (ANOVA) followed by post hoc comparison (Newman Keuls multiple) test. Differences between the means were considered significant at two tailed. Significance level was set at p < 0.05. Statistical analysis was done using the Graphpad Prism (3.0 software).
Results
Creation and characterization of human HSPB1-mutant mice
We created two lines of transgenic mice expressing the human HSPB1 gene under the control of the PrP promoter (PrP-HSPB1) and two lines expressing mutant HSPB1 (PrP-HSPB1(R136W)). We chose one line of each transgenic line that expressed the highest levels of transgene for further study. The PrP-HSPB1 mice contained 2 copies of the transgene while the PrP-HSPB1(R136W) mice contain 10 copies of the transgene.
Expression of HSPB1 transgenes in mouse central and peripheral nervous systems
HSPB1 protein was expressed throughout the nervous system (brain, spinal cord and peripheral nerve) including ChAT positive neurons in the ventral horn of spinal cord of both transgenic lines but not in the non-transgenic, littermate controls as detected by an anti-HSPB1 antibody that is specific for human HSPB1 and does not detect the mouse ortholog, Hsp25 (Figure 1A). There was no difference between the HSPB1 and HSPB1(R136W) expression patterns as seen by immunohistochemical staining throughout the brain and spinal cord. No expression of the transgene was observed in GFAP positive cells of either of transgenic animal line. Transgene expression was cytoplasmic in neurons from wild type (WT) or mutant mice and no subcellular aggregates were noted at 64x magnification (Figure 1A inset). Relative quantitative differences in expression levels of WT and mutant transgenes were not apparent with immunohistochemical analysis. HSPB1 mRNA levels were 2.2 fold higher in the brain of PrP-HSPB1(R136W) animals compared to PrP-HSPB1. Western blot analysis of brain and spinal cord extracts confirmed that expression level of the mutant protein was higher than the expression levels of WT protein in these tissues (Figure 1B).
Figure 1.

Transgenic mice express neuronal-specific HSPB1 transgenes in the ventral horn of the spinal cord. Representative fluorescent micrograph of spinal cord of 6 month old transgenic mice (A) HSPB1 protein (green) expresses in ChAT positive (red) motor neurons but not in the glial cells (blue) suggesting the neuronal specificity of the transgenes. The non-transgenic, age matched control does not show expression of the HSPB1 transgene in neuronal or glial cells. Scale bar 200μm, inset 50μm. (B) HSPB1 protein expression was confirmed by Western blot analysis in spinal cord and brain homogenates.
There was excellent co-localization of HSPB1 with neurofilament-200 (marker for both phosphorylated and non-phosphorylated forms of neurofilament heavy protein) in PrP-HSPB1 and PrP-HSPB1(R136W) transgenic mice (Figure 2).
Figure 2.
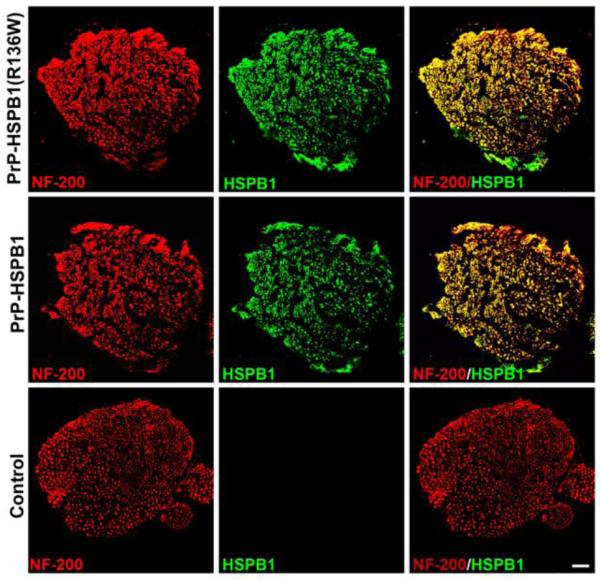
Expression of transgenes in the peripheral nerve. Representative fluorescent micrograph of sciatic nerves of the 6 month old transgenic animals show expression of HSPB1 protein (green) in the NF-200 positive (red) axons. Scale bar, 50μm.
Neuropathological changes consistent with axonal pathology and repair are present in PrP-HSPB1(R136W) mice
Semi-thin sections of the spinal cord, dorsal root ganglia (DRG) neurons, ventral and dorsal roots, sciatic and intramuscular nerves of PrP-HSPB1(R136W) mice at 6 months were found to be normal. In these mice, the number of Schmidt-Lanterman incisures (SLIs) was higher on the cross sections of ventral and dorsal roots as well as the sciatic nerves from PrP-HSPB1(R136W) mice, PrP-HSPB1 and non-transgenic, age-matched littermate control mice compared to a younger age group as reported previously (Gould, Byrd et al. 1995). The percentage of myelinated fibers displaying SLI profiles was estimated to be around 12% in each group and considered to be an age-related change (data not shown).
At 1 year of age, a relative increase in the number of SLIs was seen in the nerves of PrP-HSPB1(R136W) mice as illustrated in cross and longitudinal sections from mid-sciatic nerves compared to age matched representative sections from PrP-HSPB1 and non-transgenic, age-matched littermate control mice (Figure 3A). Quantification of the number of myelinated fibers displaying SLI profiles in PrP-HSPB1(R136W) mice showed that this increase was highly significant (p < 0.001; Figure 3B). In addition, other myelin alterations such as increased myelin corrugation, infoldings and outfoldings and fibers with increased myelin thickness for axon size (axonal atrophy) were commonly seen in all 3 groups. A few fibers undergoing Wallerian degeneration were also present in all specimens examined although myelinated fiber densities for PrP-HSPB1(R136W) (15301 ± 1440 /mm2), PrP-HSPB1 (14252 ± 607/mm2) and controls (15194 ± 1387/mm2) did not differ statistically. Overall, these alterations were present in the entire length of the sciatic system including the ventral and dorsal roots.
Figure 3.

One μm-thick, toluidine blue stained longitudinal and cross sections from mid-sciatic nerves of 1 year old PrP-HSPB1(R136W), PrP-HSPB1 and non-transgenic controls. (A) SLI is marked by arrows on the longitudinal sections; ‘S’ donates axons with SLI on the cross sections. Numerous SLIs are present in PrP-HSPB1(R136W). Bar = 10 μm for all longitudinal and 10 μm for all cross sections. Density of SLI on the cross sections of sciatic nerves from PrP-HSPB1(R136W), PrP-HSPB1 and non-transgenic controls is shown in panel B. The number of SLI determined from a total area of 0.712 mm2 is significantly increased in the PrP-HSPB1(R136W) mice compared to PrP-HSPB1 and non-transgenic controls (***p <0.001).
In the central nervous system of PrP-HSPB1(R136W) mice, occasional swollen axons with increased density of axoplasm in the white matter of the sacral and lumbar cords were detected with increasing axonal degeneration, which became most prominent at 1.5 years of age (Supplementary figure 1). The majority of axonal swellings and degeneration were scattered into the external regions of the anterior and lateral funiculus and were also present but to a lesser extent in the posterior column. No abnormalities were seen in the cell bodies of motor or sensory neurons. At the distal end of the neuroaxis, in the semithin sections from soleus and lumbrical muscles rare intramuscular nerves appeared swollen with increased density of axoplasm (Supplementary figure 2A). Occasional severely atrophic denervated fibers were present (Supplementary figure 2B). These features were not seen in non-transgenic or PrP-HSPB1 mice.
Ultrastructural analysis of axons in the sciatic nerve of 1 year old PrP-HSPB1(R136W) mice showed an evident increase in neurofilament density per given area of axon and an increase in the number of membranous organelle profiles and mitochondria (Figure 4). Morphometric analysis of the neurofilament cytoskeleton confirmed these observations by illustrating a shift toward increased neurofilament packing density (number per unit hexagonal area) compared to control (Figure 4). G ratio (axon diameter/fiber diameter) determinations in sciatic nerves showed a shift towards increased myelin thickness both in PrP-HSPB1(R136W) and PrP-HSPB1 mice compared to non-transgenic littermate controls (Figure 5). In addition, compound adaxonal Schwann cell cytoplasmic invaginations segregating axoplasm containing dense core vesicles and membrane bound dense bodies, reminiscent of secondary lysosomes were seen. These alterations were seen more frequently in the 1 year old PrP-HSPB1(R136W) mice compared to age-matched non-transgenic controls (Figure 6).
Figure 4.

Electron micrograph showing a myelinated axon, cross-sectioned through a SLI (asterisk) (A) from the sciatic nerve of a PrP-HSPB1(R136W) mouse at 1 year of age. Axoplasm seems densely packed with neurofilaments and the membranous organelle profiles and mitochondria content appear increased. (B) Morphometric analysis of the neurofilament cytoskeleton confirmed these observations by illustrating a shift toward increased neurofilament packing density (number per unit hexagonal area) in PrP-HSPB1(R136W) compared to non-transgenic controls. Lower left panel shows the PrP-HSPB1(R136W) mouse axon at higher magnification. Increases in the packing density of neurofilaments and in the number of cross sectional profiles of axonal smooth endoplasmic reticulum (arrow) are evident in PrP-HSPB1(R136W) mouse axon compared to an age-matched non-transgenic control axon with same magnification in. Bar = 2μm in A, 0.5μm in lower panels.
Figure 5.

G ratio distribution of fibers in the sciatic nerves from non-transgenic, PrP-HSPB1 and PrP-HSPB1(R136W) mice at 1 year of age. A shift towards increased percent of fibers with smaller g ratio (thicker myelin) is more pronounced in the 1 year old PrP-HSPB1(R136W) mouse. p < 0.001 for control vs. all groups. PrP-HSPB1 vs. PrP-HSPB1(R136W) at 1 year of age, not significant. Number of animals in each group is 3. Values are mean ± S.E.
Figure 6.

Ultrastructure of the adaxonal Schwann cell cytoplasmic invaginations in a sciatic nerve axon from PrP-HSPB1(R136W) mouse at 1year of age. (A) An adaxonal membrane invagination (arrow) is seen to encircle and segregate a collection of axonal membranous organelles. Bar = 2 μm. (B) At higher magnification, a large sequestered area of axoplasm (a) contains dense membranous bodies, vesicles with dense cores (arrow) and large clear vesicles (v) or multivesicular bodies (mvb) Bar = 0.5μm.
Electrophysiological evidence of axonopathy in PrP-HSPB1(R136W) mice
Sciatic motor amplitudes measured in PrP-HSPB1(R136W), PrP-HSPB1 and non-transgenic, littermate control mice were equivalent at the age of 6 months (data not shown). At 1 year, there was a significant decrease in sciatic motor amplitude (p<0.05) and no difference in conduction velocity in PrP-HSPB1(R136W) mice compared to PrP-HSPB1 and non-transgenic, age matched controls (Figure 7). This finding is consistent with distal motor axon dysfunction of the distal sciatic nerves in the PrP-HSPB1(R136W) mice. There were no significant differences in the tail motor and orthodromic tail sensory amplitudes or conduction velocities.
Figure 7.

Electrophysiological evidence of axonopathy in PrP-HSPB1(R136W) mice. (A) Representative sciatic compound muscle action potential (CMAP) responses for control, PrP-HSPB1(R136W), and PrP-HSPB1 mice (Sweep speed: 1ms per division; Amplitude: 20 mV per division). Mean sciatic CMAP amplitudes and conduction velocities for each group. (B) There is a significant difference (p <0.05) between PrP-HSPB1(R136W) and controls in sciatic CMAP amplitude. Number of animals in each group is 5. Values are mean ± S.E.
No phenotypic changes in PrP-HSPB1(R136W) mice
Both lines of transgenic mice developed normally and at weaning show size, weight, and a home cage behavior grossly comparable to that of non-transgenic control littermates. Mortality rates at 1 year of age were similar (PrP-HSPB1(R136W): 6.11%; PrP-HSPB1: 6.53%; non-transgenic controls: 9.31%). There was no change in the body weight of PrP-HSPB1(R136W) transgenic mice compared to PrP-HSPB1 and non-transgenic control mice (data not shown). There were no differences noted between male and female transgenic mice.
We did not observe any functional or motor deficit in PrP-HSPB1(R136W) up to 1 year of age. All the behavioral changes in both transgenic lines were similar to the changes in the non-transgenic controls at 6 months of age. The data is summarized in Table 1. To assess distal muscle weakness that is a hallmark of dHMN, we performed a ladder test, foot print analysis, rotarod test, and a grip strength test. There was no evidence of muscle weakness in 1 year old PrP-HSPB1 or PrP-HSPB1(R136W) (Figure 8).
Table 1.
Behavioral changes in PrP-HSPB1, PrP-HSPB1 (R136W) and age matched, non-transgenic control mice at the age of 6 months
| PrP-HSPB1 | PrP-HSPB1(R136W) | Non-transgenic controls | p-value | |
|---|---|---|---|---|
| Total distance (cm.) | 9330 ± 1352 | 20718± 9088 | 26540 ± 8302 | 0.81 |
| Rest time (sec.) | 5376 ± 306 | 5378± 226 | 5255 ± 261 | 0.93 |
| Movement time (sec.) | 1823 ± 306 | 1820 ± 226 | 1944 ± 261 | 0.93 |
| Sterotypy count | 565 ± 101 | 560 ± 73 | 547 ± 80 | 0.99 |
| Vertical activity time (sec.) | 1031 ± 188 | 573 ± 71 | 559 ± 82 | 0.3 |
| Vertical activity count | 2110 ± 267 | 1481 ± 176 | 1538 ± 232 | 0.72 |
| Wheel rotations | 1130 ± 215 | 408 ± 83 | 667 ± 157 | 0.17 |
Average locomotor activities of transgenic and non-transgenic mice in an open field activity monitor for 2 hours. PrP-HSPB1: n=21, PrP-HSPB1(R136W): n=27 and non-transgenic, littermate controls: n=24. Data expressed as mean ± S.E.
Figure 8.

High level of HSPB1(R136W) protein in neurons results in a subclinical motor neuropathy. No difference in toe spread (TS), intermediate toe spread (ITS) and paw length (PL) in transgenic mice compared to non-transgenic controls. Decrease in latency of fall (n = 7 in each group), fore limb strength and hind limb strength in PrP-HSPB1(R136W) mice compared to PrP-HSPB1 and non-transgenic control mice (n = 5 in each group). Values are mean ± S.E.
Discussion
The age of onset of dHMN and CMT2 resulting from mutations in HSPB1 ranges from 4 to 65 years of age and progression of disease occurs over decades and, to date, 10 missense mutations have been reported (Evgrafov, Mersiyanova et al. 2004; Kijima, Numakura et al. 2005; Tang, Liu et al. 2005). We selected the p.R136W mutation to model in mice because it was reported to have an age of onset in adolescence, one of the more severe phenotypes of the group. In addition, the arginine at p.136 of the HSPB1 gene aligns with a lysine at p.141 of the homologous HSPB8 gene, and mutations at HSPB8p.K141N also result in dHMN type II (Irobi, Van Impe et al. 2004). Patients with dHMN/CMT2 present with progressive weakness, wasting and atrophy of the distal muscles of both upper and lower limbs (Irobi, De Jonghe et al. 2004; Jordanova, Irobi et al. 2006; Bienfait, Baas et al. 2007; Houlden, Laura et al.; Solla, Vannelli et al. 2010). dHMN/CMT2 patients have normal lifespans, are usually able to walk, but often require assisted mobility in early adulthood (Bienfait, Baas et al. 2007; Pareyson and Marchesi 2009).
Our data show that neuronal expression of human HSPB1 with a dHMN-causing mutation R136W in transgenic mice produces a subclinical axonal motor neuropathy. Our findings are in agreement with a recently published study by d’Ydewalle et al. that characterized a transgenic mouse line expressing the S135F mutation (within the central α-crystalline domain) and a line expressing the P182L mutation (within the C-terminal domain) in neurons using Thy1.2 promoter (d’Ydewalle, Krishnan et al. 2011). Remarkably, the expression of each mutation resulted in a distinct phenotype that mimicked the human phenotype associated with that mutation: CMT2 for the S135F mutation and dHMN for the P182L mutation. Motor coordination, muscle strength and CMAP amplitude were most effected in HSPB1(P182L) expressing mice, however both lines had a detectable motor phenotype. We were unable to detect a motor phenotype in the PrP-HSPB1(R136W) mice despite the presence of clear electrophysiological and pathological evidence of an axonal neuropathy. It is possible that differences related to the promoters (PrP vs. Thy1.2) in the two studies play a role, however the mutant protein is expressed at high levels in neurons in both models. The background strain used in both studies is FVB, however it is possible that they were not identical. A more likely possibility is that distinct HSPB1 mutations yield distinct clinical phenotypes in mice, which parallels the phenotypic diversity seen in patients with mutant HSPB1-mediated neuropathies
Additional explanations for the subclinical axonal pathology observed in our 1 year old PrP-HSPB1(R136W) mice may include, 1) insufficient expression levels of the HSPB1(R136W) transgene in neurons, 2) compensatory mechanisms present in mice such as the expression of the HSPB1 mouse homolog, Hsp25, 3) a requirement for expression of the transgene in non-neuronal cells and 4) insertion site effects of the transgene. Mutant HSPB1-mediated dHMN/CMT2 is a dominantly inherited disease requiring only a single mutant allele. The PrP-HSPB1(R136W) animals have multiple copies of the transgene under the control of a highly constitutive promoter in neurons, and so it is unlikely that expression levels in neurons, by itself, are inadequate.
Motor neurons constitutively express the HSPB1 mouse ortholog, Hsp25 (Maatkamp, Vlug et al. 2004). Hsp25 is not, however, required for motor neuron development or maintenance (Huang, Min et al. 2007). In a knock-in mouse model, where the coding sequence of Hsp25 was replaced by a lacZ reporter gene, the endogenous Hsp25 expression was most prominent in skeletal and cardiac muscle, and the mice had normal survival and behavior (Huang, Min et al. 2007). These animals were not studied to determine whether subclinical neuropathic changes were present to our knowledge. There were no changes in Hsp25 protein expression in the PrP-HSPB1 or PrP-HSPB1(R136W) in spinal cord extracts (data not shown), nevertheless, we cannot rule out the possibility that the presence of Hsp25 attenuated the development of a more severe motor neuropathy in the PrP-HSPB1(R136W) mice.
The characteristic electrophysiological findings of axonal degeneration are amplitude loss with relatively normal conduction velocity. The finding of reduced sciatic motor amplitude in the PrP-HSPB1(R136W) mice correlates with the pathological changes in peripheral nerve and confirms the presence of an axonopathy involving the motor axons. Although myelinated fiber densities of mid-sciatic nerves from transgenic mice did not differ from age matched controls, we found evidence of distal axonopathy as illustrated with the presence of swollen terminal axons and denervation atrophy in muscle fibers in support of the decreased motor amplitudes. As expected, these findings were not seen widespread in all tissue blocks examined. It should be noted that electrophysiology is more reliable screening tool for detecting early distal axonal disease compared to relatively rare occurrence of intramuscular nerves within 1μm thick sections for quantification. The lack of a significant difference of the tail motor and orthodromic sensory responses is consistent with the expected findings of a dHMN electrophysiological phenotype with disproportionate involvement of distal motor axons.
One intriguing possibility to account for the subclinical neuropathic changes in the PrP-HSPB1(R136W) mice is that expression of the HSPB1(R136W) transgene in non-neuronal cells, such as astrocytes and microglia, is required at high levels for the development of a more severe phenotype. Indeed, the role of non-neuronal cells in the progression of motor neuron death in ALS mouse models is an area of interest (Bruijn, Becher et al. 1997; Di Giorgio, Carrasco et al. 2007; Nagai, Re et al. 2007). Future work to explore the role of HSPB1(R136W) expression in astrocytes, Schwann cells, and/or microglia in the development of axonal motor neuropathy in vivo may lead to insights that may be generalized to other forms of motor neuron disease.
At 12 months of age, we observed an increased number of SLIs and increased neurofilament packing density associated with an increase in the number of membranous organelle profiles and mitochondria in peripheral nerve of PrP-HSPB1(R136W) mice suggestive of impaired transport in the axon. This is consistent with the recent report by d’Ydewalle and colleagues. These investigators demonstrated disrupted axonal transport in dorsal root ganglion cell cultures derived from their symptomatic HSPB1(S135F) transgenic mice (d’Ydewalle, Krishnan et al. 2011). They went on to find decreased acetylated α-tubulin levels in these cultures and in sciatic nerve (and not in spinal cord) of the mutant HSPB1 expressing mice which, when restored by treatment with histone deacetylase 6 (HDAC6) inhibitors, restored many of the behavioral and electrophysiological deficits in these mice (d’Ydewalle, Krishnan et al. 2011). In our R136W mice, alterations in neurofilament packing density and abnormalities in membranous organelles were observed as in other disease models. Disruption of neurofilament assembly due to CMT mutant neurofilaments [NF-L(Pro8Arg) and NF-L(Gln333Pro)] leads to impaired axonal transportation in SW13 mammalian cells and rat cortical neurons (Brownlees, Ackerley et al. 2002). Similarly, accumulation of neurofilament is a recognized pathological finding in ALS, Alzheimer’s disease, Parkinson’s disease, dementia with Lewy bodies and diabetic neuropathy (Cleveland and Rothstein 2001; Miller, Ackerley et al. 2002). In addition, adaxonal Schwann cell cytoplasmic invaginations segregating axoplasm containing dense core vesicles and membrane bound dense bodies reminiscent of secondary lysosomes, were also common features of this axonopathy. These Schwann cell-axon networks are known to have a predominant occurrence adjacent to SLIs and at the paranodes and that these sequestered structures by adaxonal Schwann cell cytoplasmic invaginations are known to be retrogradely transported organelles (Tsukita and Ishikawa 1980). Our observations indicate that Schwann cells (that are not expressing the HSPB1 transgene) are actively participating in the removal of unwanted/accumulated or damaged organelles from axons for selective phagocytosis, a process suggestive of an early axonal disease as previously proposed (Spencer and Schaumburg 1974). In accordance, the increase in the SLI number is likely to be a compensatory mechanism to increase the efficacy of Schwann cell-axon interactions in this model (Sahenk 1999; Quintes, Goebbels et al. 2010).
Understanding the mechanism of increased axonal neurofilament packing density in peripheral nerves of PrP-HSPB1(R136W) mice needs further study. In the hypomyelinated nerves from the peripheral myelin-deficient trembler mice, a similar alteration in the hypophosphorylated neurofilament cytoskeleton is associated with decreased stability of the axonal microtubule cytoskeleton (decreased levels of insoluble tubulin) and changes in the composition and phosphorylation of axonal microtubule-associated proteins, including tau, MAP 1A, and MAP 1B (Kirkpatrick and Brady 1994). As shown previously, an increase in the neurofilament packing density can also be induced secondarily by an agent that directly binds to MAPs (Sahenk and Mendell 1992). In our studies from 12 months old PrP-HSPB1(R136W) mice, we observed no abnormalities in the microtubule morphology such as maloriented fragments suggestive of impaired microtubule stability, nor did we see an apparent decreased in their numbers, although detailed ultrastructural morphometric studies are needed for detecting subtle microtubule density changes in axons.
HSPB1 in an exceptionally multifunctional protein that, in addition to protein chaperone activities, is involved in axonal transport, protein sorting, protein degradation and RNA decay (reviewed in Carra and Landry 2008; Mymrikov, Seit-Nebi et al. 2011). The functional consequences of dHMN/CMT-associated HSPB1 mutations expressed in mammalian cells suggest a defect in neurofilament assembly and dysregulation of microtubules (Evgrafov, Mersiyanova et al. 2004; Ackerley, James et al. 2006; Almeida-Souza, Asselbergh et al. 2011). Mutant HSPB1 forms abnormal, insoluble intracellular aggregates and sequestration of neurofilament protein in the cytoplasm of primary mouse cortical neurons (Ackerley, James et al. 2006). It has been demonstrated biochemically that dHMN/CMT-associated mutations in HSPB1 and HSPB8 form oligomeric complexes with a higher binding affinity, resulting in protein-protein interactions that are “tighter” than normal (Fontaine, Sun et al. 2006; Sun, Fontaine et al. 2010). Interestingly, dHMN/CMT associated HSPB1 mutations that reside in the central α-crystallin domain bind tightly to tubulin while mutations in the C-terminal domain do not, suggesting that the mechanism of neuropathy is not related to this increased binding affinity (Almeida-Souza, Asselbergh et al. 2011). It is likely that, in our model, mutant HSPB1(R136W) binds more tightly to α-tubulin and neurofilaments in axons resulting in the increased neurofilament packing density observed. Ultimately, further in vivo studies to elucidate the molecular mechanism of mutant HSPB1-mediated selective motor neuron injury in these models must account for a pathological mechanism that is common for all HSPB1 mutations.
Conclusions
Expression of mutant HSPB1 in neurons results in axonal neuropathy in mice. The PrP-HSPB1(R136W) mice provide an additional tool to study the molecular basis of mutant HSPB1-mediated motor neuropathy and may provide clues to the relationship between individual HSPB1 mutations and clinical severity in the hereditary neuropathies.
Supplementary Material
Supplementary figure 1. Swollen axons with increased density of axoplasm (asterisk)and myelin ovoids (arrow) as remnants of degenerated axons in the anterior funiculus of the sacral cord (A) and in the anterolateral funiculus of the lumbar cord (B) from a PrP-HSPB1(R136W) mouse at 1.5 year of age. 1μm-thick, toluidine blue stained cross sections. Bar = 10μm.
Supplementary figure 2. Cross section of the lumbrical muscle from a PrP-HSPB1(R136W) mouse at 1.5 year of age showing a swollen terminal axon with increased density of the axoplasm (arrow) and a neuromuscular junction (arrow head) in the immediate vicinity (A) Double arrows point to a small intramuscular nerve bundle with normal size axons for comparison. Panel B shows another area of the same muscle and the arrow heads point to severely atrophic/denervated fibers, which are seen occasionally. 1μm-thick, toluidine blue stained cross sections. Bar = 10μm.
Highlights.
PrP-HSPB1 and PrP-HSPB1(R136W) transgenic mouse lines have been developed.
HSPB1(R136W) expression in neurons results in age-dependent axonal degeneration.
Electrophysiological evidence of distal axonal dysfunction is detected in PrP-HSPB1(R136W) mice
Increased neurofilament density is seen in one year old HSPB1(R136W) mice.
Acknowledgement
We thank Dr. Arthur Burghes and Vicki McGovern for guidance and suggestions. We are also grateful to Dr. Brian Kaspar and his laboratory for immunohistochemistry reagents and expertise. We also thank Dr. Dana McTigue and Todd Lash for the use of the activity monitor equipment. This project was supported by start up funds from The Wexner Medical Center at The Ohio State University, NINDS K08NS067282 and The Fred F. and Herman M. Dreier Fund. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute Of Neurological Disorders And Stroke or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Ackerley S, James PA, et al. A mutation in the small heat-shock protein HSPB1 leading to distal hereditary motor neuronopathy disrupts neurofilament assembly and the axonal transport of specific cellular cargoes. Hum Mol Genet. 2006;15(2):347–354. doi: 10.1093/hmg/ddi452. [DOI] [PubMed] [Google Scholar]
- Almeida-Souza L, Asselbergh B, et al. Small Heat-Shock Protein HSPB1 Mutants Stabilize Microtubules in Charcot-Marie-Tooth Neuropathy. J Neurosci. 2011;31(43):15320–15328. doi: 10.1523/JNEUROSCI.3266-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti S, Previtali SC, et al. Analyzing histopathological features of rare charcot-marie-tooth neuropathies to unravel their pathogenesis. Arch Neurol. 2010;67(12):1498–1505. doi: 10.1001/archneurol.2010.303. [DOI] [PubMed] [Google Scholar]
- Beuche W, Friede RL. A quantitative assessment of myelin sheaths in the peripheral nerves of dystrophic, quaking, and trembler mutants. Acta Neuropathol. 1985;66(1):29–36. doi: 10.1007/BF00698292. [DOI] [PubMed] [Google Scholar]
- Bienfait HM, Baas F, et al. Phenotype of Charcot-Marie-Tooth disease Type 2. Neurology. 2007;68(20):1658–1667. doi: 10.1212/01.wnl.0000263479.97552.94. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, et al. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13(6):159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Brownlees J, Ackerley S, et al. Charcot-Marie-Tooth disease neurofilament mutations disrupt neurofilament assembly and axonal transport. Hum Mol Genet. 2002;11(23):2837–2844. doi: 10.1093/hmg/11.23.2837. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18(2):327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Butchbach ME, Edwards JD, et al. Abnormal motor phenotype in the SMNDelta7 mouse model of spinal muscular atrophy. Neurobiol Dis. 2007;27(2):207–219. doi: 10.1016/j.nbd.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carra S, Landry J. Role of HSPB1 and HSPB8 in hereditary peripheral neuropathies: beyond the chaperone function. In: Asea A, Brown IR, editors. Heat Shock Proteins and the Brain: Implications for Neurodegerative Diseases and Neuroprotection. Springer Science + Business Media B.V.; 2008. pp. 139–155. [Google Scholar]
- Chung KW, Kim SB, et al. Distal hereditary motor neuropathy in Korean patients with a small heat shock protein 27 mutation. Exp Mol Med. 2008;40(3):304–312. doi: 10.3858/emm.2008.40.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2(11):806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- d’Ydewalle C, Krishnan J, et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med. 2011;17(8):968–974. doi: 10.1038/nm.2396. [DOI] [PubMed] [Google Scholar]
- Di Giorgio FP, Carrasco MA, et al. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10(5):608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierick I, Baets J, et al. Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: a genotype-phenotype correlation study. Brain. 2008;131(Pt 5):1217–1227. doi: 10.1093/brain/awn029. [DOI] [PubMed] [Google Scholar]
- Dimos JT, Rodolfa KT, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321(5893):1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- Evgrafov OV, Mersiyanova I, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004;36(6):602–606. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- Fontaine JM, Sun X, et al. Abnormal small heat shock protein interactions involving neuropathy-associated HSP22 (HSPB8) mutants. Faseb J. 2006;20(12):2168–2170. doi: 10.1096/fj.06-5911fje. [DOI] [PubMed] [Google Scholar]
- Gould RM, Byrd AL, et al. The number of Schmidt-Lanterman incisures is more than doubled in shiverer PNS myelin sheaths. J Neurocytol. 1995;24(2):85–98. doi: 10.1007/BF01181552. [DOI] [PubMed] [Google Scholar]
- Hindson BJ, Ness KD, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlden H, Laura M, et al. Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type 2. Neurology. 2008;71(21):1660–1668. doi: 10.1212/01.wnl.0000319696.14225.67. [DOI] [PubMed] [Google Scholar]
- Huang L, Min JN, et al. Insights into function and regulation of small heat shock protein 25 (HSPB1) in a mouse model with targeted gene disruption. Genesis. 2007;45(8):487–501. doi: 10.1002/dvg.20319. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Abe A, et al. A clinical phenotype of distal hereditary motor neuronopathy type II with a novel HSPB1 mutation. J Neurol Sci. 2009;277(1-2):9–12. doi: 10.1016/j.jns.2008.09.031. [DOI] [PubMed] [Google Scholar]
- Irobi J, De Jonghe P, et al. Molecular genetics of distal hereditary motor neuropathies. Hum Mol Genet. 2004;13(Spec No 2):R195–202. doi: 10.1093/hmg/ddh226. [DOI] [PubMed] [Google Scholar]
- Irobi J, Van K, et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 2004;36(6):597–601. doi: 10.1038/ng1328. [DOI] [PubMed] [Google Scholar]
- James PA, Rankin J, et al. Asymmetrical late onset motor neuropathy associated with a novel mutation in the small heat shock protein HSPB1 (HSP27) J Neurol Neurosurg Psychiatry. 2008;79(4):461–463. doi: 10.1136/jnnp.2007.125179. [DOI] [PubMed] [Google Scholar]
- Jordanova A, Irobi J, et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet. 2006;38(2):197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- Kijima K, Numakura C, et al. Small heat shock protein 27 mutation in a Japanese patient with distal hereditary motor neuropathy. J Hum Genet. 2005;50(9):473–476. doi: 10.1007/s10038-005-0280-6. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick LL, Brady ST. Modulation of the axonal microtubule cytoskeleton by myelinating Schwann cells. J Neurosci. 1994;14(12):7440–7450. doi: 10.1523/JNEUROSCI.14-12-07440.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K, de Almeida DF, et al. Sensory nerve conduction of the plantar nerve compared with other nerve conduction tests in rats. Clin Neurophysiol. 2004;115(7):1677–1682. doi: 10.1016/j.clinph.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Luigetti M, Fabrizi GM, et al. A novel HSPB1 mutation in an Italian patient with CMT2/dHMN phenotype. J Neurol Sci. 2010;298(1-2):114–117. doi: 10.1016/j.jns.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Maatkamp A, Vlug A, et al. Decrease of Hsp25 protein expression precedes degeneration of motoneurons in ALS-SOD1 mice. Eur J Neurosci. 2004;20(1):14–28. doi: 10.1111/j.1460-9568.2004.03430.x. [DOI] [PubMed] [Google Scholar]
- Miller CC, Ackerley S, et al. Axonal transport of neurofilaments in normal and disease states. Cell Mol Life Sci. 2002;59(2):323–330. doi: 10.1007/s00018-002-8425-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mymrikov EV, Seit-Nebi AS, et al. Large potentials of small heat shock proteins. Physiol Rev. 2011;91(4):1123–1159. doi: 10.1152/physrev.00023.2010. [DOI] [PubMed] [Google Scholar]
- Nagai M, Re DB, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10(5):615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 2009;8(7):654–667. doi: 10.1016/S1474-4422(09)70110-3. [DOI] [PubMed] [Google Scholar]
- Pinheiro LB, Coleman VA, et al. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal Chem. 2012;84(2):1003–1011. doi: 10.1021/ac202578x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintes S, Goebbels S, et al. Neuron-glia signaling and the protection of axon function by Schwann cells. J Peripher Nerv Syst. 2010;15(1):10–16. doi: 10.1111/j.1529-8027.2010.00247.x. [DOI] [PubMed] [Google Scholar]
- Reilly MM, Shy ME. Diagnosis and new treatments in genetic neuropathies. J Neurol Neurosurg Psychiatry. 2009;80(12):1304–1314. doi: 10.1136/jnnp.2008.158295. [DOI] [PubMed] [Google Scholar]
- Sahenk Z. Abnormal Schwann Cell-Axon Interactions In Cmt Neuropathies. The effects of mutant Schwann cells on the axonal cytoskeleton and regeneration-associated myelination. Ann N Y Acad Sci. 1999;883:415–426. [PubMed] [Google Scholar]
- Sahenk Z, Galloway G, et al. TrkB and TrkC agonist antibodies improve function, electrophysiologic and pathologic features in Trembler J mice. Exp Neurol. 2010;224(2):495–506. doi: 10.1016/j.expneurol.2010.05.013. [DOI] [PubMed] [Google Scholar]
- Sahenk Z, Mendell JR. Ultrastructural study of zinc pyridinethione-induced peripheral neuropathy. J Neuropathol Exp Neurol. 1979;38(5):532–550. doi: 10.1097/00005072-197909000-00007. [DOI] [PubMed] [Google Scholar]
- Sahenk Z, Mendell JR. Alterations in slow transport kinetics induced by estramustine phosphate, an agent binding to microtubule-associated proteins. J Neurosci Res. 1992;32(4):481–493. doi: 10.1002/jnr.490320404. [DOI] [PubMed] [Google Scholar]
- Solla P, Vannelli A, et al. Heat shock protein 27 R127W mutation: evidence of a continuum between axonal Charcot-Marie-Tooth and distal hereditary motor neuropathy. J Neurol Neurosurg Psychiatry. 2010;81(9):958–962. doi: 10.1136/jnnp.2009.181636. [DOI] [PubMed] [Google Scholar]
- Spencer PS, Schaumburg HH. A review of acrylamide neurotoxicity. Part II. Experimental animal neurotoxicity and pathologic mechanisms. Can J Neurol Sci. 1974;1(3):152–169. doi: 10.1017/s0317167100119201. [DOI] [PubMed] [Google Scholar]
- Sun X, Fontaine JM, et al. Abnormal interaction of motor neuropathy-associated mutant HspB8 (Hsp22) forms with the RNA helicase Ddx20 (gemin3) Cell Stress Chaperones. 2010 doi: 10.1007/s12192-010-0169-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Liu X, et al. 2005 [Google Scholar]
- Tsukita S, Ishikawa H. The movement of membranous organelles in axons. Electron microscopic identification of anterogradely and retrogradely transported organelles. J Cell Biol. 1980;84(3):513–530. doi: 10.1083/jcb.84.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia RH, Yosef N, et al. Dorsal caudal tail and sciatic motor nerve conduction studies in adult mice: Technical aspects and normative data. Muscle Nerve. 2010 doi: 10.1002/mus.21588. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Shen H, et al. The protective effects of Achyranthes bidentata polypeptides in an experimental model of mouse sciatic nerve crush injury. Brain Res Bull. 2010;81(1):25–32. doi: 10.1016/j.brainresbull.2009.07.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1. Swollen axons with increased density of axoplasm (asterisk)and myelin ovoids (arrow) as remnants of degenerated axons in the anterior funiculus of the sacral cord (A) and in the anterolateral funiculus of the lumbar cord (B) from a PrP-HSPB1(R136W) mouse at 1.5 year of age. 1μm-thick, toluidine blue stained cross sections. Bar = 10μm.
Supplementary figure 2. Cross section of the lumbrical muscle from a PrP-HSPB1(R136W) mouse at 1.5 year of age showing a swollen terminal axon with increased density of the axoplasm (arrow) and a neuromuscular junction (arrow head) in the immediate vicinity (A) Double arrows point to a small intramuscular nerve bundle with normal size axons for comparison. Panel B shows another area of the same muscle and the arrow heads point to severely atrophic/denervated fibers, which are seen occasionally. 1μm-thick, toluidine blue stained cross sections. Bar = 10μm.