

Abstract
Protection of the blood–brain barrier (BBB) is correlated with improved outcome in stroke. Sphingosine kinase (SphK)-directed production of sphingosine-1-phosphate, which we previously documented as being vital to preconditioning-induced stroke protection, mediates peripheral vascular integrity via junctional protein regulation. We used a hypoxic preconditioning (HPC) model in adult wild-type and SphK2-null mice to examine the isoform-specific role of SphK2 signaling for ischemic tolerance to transient middle cerebral artery occlusion and attendant BBB protection. Reductions in infarct volume and BBB permeability in HPC-treated mice were completely lost in SphK2-null mice. Hypoxic preconditioning-induced attenuation of postischemic BBB disruption in wild types, evidenced by reduced extravascular immunoglobulin G intensity, suggests direct protection of BBB integrity. Measurement of BBB junctional protein status in response to HPC revealed SphK2-dependent increases in triton-insoluble claudin-5 and VE-cadherin, which may serve to strengthen the BBB before stroke. Postischemic loss of VE-cadherin, occludin, and zona occludens-1 in SphK2-null mice with prior HPC suggests that SphK2-dependent protection of these adherens and tight junction proteins is compulsory for HPC to establish a vasculoprotective phenotype. Further elucidation of the mediators of this endogenous, HPC-activated lipid signaling pathway, and their role in protecting the ischemic BBB, may provide new therapeutic targets for cerebrovascular protection in stroke patients.
Keywords: bioactive lipids, focal stroke, neuroprotection, neurovascular unit
Introduction
Blood–brain barrier (BBB) dysfunction and resultant vasogenic edema is a feature of many neurologic disorders, including during Alzheimer's, Parkinson's, multiple sclerosis, bacterial meningitis, and stroke (Weiss et al, 2009). In ischemic stroke, BBB protection is predictive of improved outcome (Latour et al, 2004), but it remains unclear whether preservation of BBB integrity by a given neuroprotective treatment is a cause or consequence of reduced ischemic injury (Obrenovitch, 2008). Moreover, breakdown of the BBB and associated hemorrhagic transformation define the outer therapeutic window for tissue plasminogen activator (tPA) treatment (Aviv et al, 2009). Yet, the molecular basis of BBB dysfunction and protection in stroke still requires considerable elucidation.
Sphingosine-1-phosphate (S1P), a bioactive lipid, exhibits vasculoprotective effects in models of peripheral tissue injury, reducing ischemic damage in the heart (Karliner, 2004) and other tissues (Park et al, 2010). Sphingosine-1-phosphate regulates vascular integrity through several mechanisms, including the modulation of junctional protein expression and localization (Adamson et al, 2010; Lee et al, 2006; Wang and Dudek, 2009). Only recently has this sphingolipid pathway been considered as a potential stroke therapeutic (Czech et al, 2009; Hasegawa et al, 2010; Wacker et al, 2009; Wei et al, 2011). Our earlier results implicate S1P as a proximal transducer of the stress response to hypoxic preconditioning (HPC), ultimately resulting in a protective phenotype against transient focal stroke known as ischemic tolerance (Wacker et al, 2009). Sphingosine-1-phosphate is produced by two sphingosine kinase (SphK) isoforms, SphK1 and SphK2, which often exhibit differing effects. Because only SphK2 protein levels and activity increased in cortical microvessels following HPC, and nonspecific SphK inhibition abrogated HPCs neuroprotective effects (Wacker et al, 2009), an isoform-specific contribution of SphK2 to ischemic tolerance was proposed.
In this study, we used HPC and SphK2-null mice to evaluate if SphK2 establishes ischemic tolerance via promotion of BBB integrity and regulation of cerebroendothelial junctional proteins. Our findings indicate that SphK2 activity is critical to the BBB protection afforded by HPC, secondary to its preischemic and postischemic regulation of the adherens junction protein VE-cadherin, and the tight junction proteins claudin-5, occludin, and zona occludens-1 (ZO-1).
Materials and methods
Mice
Adult, male, C57BL/6 and SphK2 knockout mice were used in these studies. All experimental protocols were approved by the Institutional Animal Care and Use Committee at Washington University and conformed to the National Institutes of Health guidelines for the care and use of animals in research. Homozygous SphK2 knockout mice, congenically backcrossed to C57BL/6 mice for at least seven generations, were generously provided by Dr Richard Proia (NIH). Previous characterization of the SphK2 knockout mice revealed no obvious histological differences, and no compensatory change in SphK1 expression (including after ischemia) (Mizugishi et al, 2005; Pfeilschifter et al, 2011). Animals were housed on a 12-hour light/dark cycle with water and food ad libitum. Efforts were made to reduce the number of mice used and to minimize stress to the animals. All experimental procedures were approved by the Washington University Animal Studies Committee, and surgical procedures were performed using sterile/aseptic techniques in accordance with the institutional guidelines. Experimental conditions were intermixed to prevent batch effects from affecting a single experimental condition; all analyses were blinded.
Hypoxic Preconditioning
Mice were subjected to HPC (4 hours of normobaric 8% oxygen) in modified home cages by flowing hypoxic air through the cage (1.5 L/min). Oxygen content of the outflow air was confirmed through monitoring with an oxygen analyzer (Vascular Technology, Nashua, NH, USA). Food and water were provided ad libitum. Control mice had no exposure to hypoxia.
Transient Focal Cerebral Ischemia
Mice underwent transient middle cerebral artery occlusion (tMCAO) 48 hours following HPC, when applicable. Mice were anesthetized by initial exposure to 5% halothane/70% NO2/30% O2, with continued 2% halothane to maintain anesthesia for the remainder of the procedure. The left MCA was exposed by incision of the temporal muscle and a baseline blood flow was obtained by laser Doppler flowmetry (TSI, Shoreview, MN, USA). The left common carotid artery was exposed via a ventral midline incision on the neck, and a silicon-coated, 6.0-gauge, nylon suture, 12 mm in length, was advanced 9.0 to 10.5 mm to transiently block the origin of the MCA. Body temperature was maintained throughout the surgical procedure by a heating pad, and animals were placed in a 34°C incubator during ischemia. After 45 minutes of ischemia, animals were reanesthetized, and the suture was withdrawn to reestablish perfusion. Mice were allowed to recover from anesthesia for 30 minutes in the 34°C incubator. Blood flow through the MCA was measured by laser Doppler flowmetry at the onset and end of ischemia to confirm blockage of the blood flow throughout the ischemic period, and then immediately following and 24 hours after suture withdrawal to confirm reperfusion from the end of ischemia until kill. Mice retaining >15% of baseline perfusion during ischemia and mice that did not reach at least 50% of baseline by 5 minutes postreperfusion or 70% after 24 hours were excluded. In addition, animals showing evidence of intracerebral bleeding or subarachnoid hemorrhage on brain extraction were excluded from the study. The numbers of excluded mice and reasons for exclusion, including mortality, are presented in Supplementary Table 1, and relative MCA blood flow at the relevant time points for nonexcluded mice in each experimental group is presented as Supplementary Table 2.
Infarct, Swelling, and Neurologic Deficit Quantification
Neurologic deficit was scored 24 hours after reperfusion, according to the following scale:
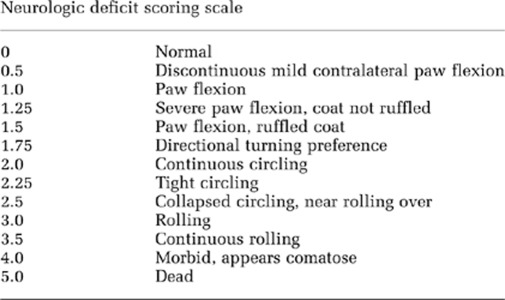
Animals were killed 24 hours following tMCAO by cervical dislocation. The brains were sectioned on a 1-mm thick brain matrix and exposed to a solution of 2,3,5-triphenyl tetrazolium chloride (1% in phosphate-buffered saline (PBS)) to delineate infarct regions. Infarct volume was manually measured in Sigma Scan Pro 4 (Systat Software Inc, San Jose, CA, USA) and total infarct volume was corrected for edema. Ipsilateral swelling was used as an index of cerebral edema, quantified as the increase in ipsilateral hemispheric volume relative to the contralateral hemisphere.
Blood–Brain Barrier Integrity Assessments
At 24 hours post-tMCAO, following halothane overdose, mice were transcardially perfused (20 mL 0.01 M PBS, 40 mL 4% paraformaldehyde/0.01 M PBS). After extraction, the brains were cryoprotected in 30% sucrose, and sliced coronally into 10-μm sections. Postischemic permeability to endogenous, extravasated immunoglobulin G (IgG) was detected using a modified immunohistochemistry protocol with anti-mouse IgG IRDye antibody (LI-COR, Lincoln, NE, USA). In brief, slides were brought to room temperature, rinsed 3 × with deionized water, and hydrated in PBS (0.01 M, pH 7.4) for 5 minutes. Sections (delimited with a PAP hydrophobic barrier pen; Dako, Carpinteria, CA, USA) were coated with LI-COR IRDye 800 anti-mouse secondary antibody (1:200) in 200 μL blocking solution (10% normal goat serum, 0.1% Triton X in PBS) for 2 hours. Following a 10-minute PBS-T (0.1% Tween-20 in PBS) wash, slides were rinsed in deionized water and air dried. Immunoglobulin G intensity was imaged and quantified on an Odyssey Infrared Imaging System (LI-COR). For each mouse, four sections from the rostral (∼1 mm rostral from bregma) and caudal (∼2 mm caudal from bregma) regions of the infarct were quantified for IgG permeability with respect to both crosssectional area of extravasated IgG, and IgG intensity. Intensity and area were measured within the Odyssey Infrared Imaging System software (v3.0, LI-COR).
Immunoblotting
Mice were killed by cervical dislocation 48 hours following HPC or 24 hours after reperfusion from tMCAO. Triton-insoluble fractions were prepared from the entire cortex of the ischemic hemisphere (or from either cortex of HPC-treated or naive animals) to isolate junctional proteins associated with the cytoskeleton (McNeill et al, 1993; Stuart et al, 1996; Wang et al, 2011; Wong, 1997). In brief, tissue was homogenized in 600 μL of cytoskeleton-stabilizing buffer (10 mM HEPES, pH 7.4, 250 mM sucrose, 150 mM KCl, 1 mM EGTA, 3 mM MgCl2, protease inhibitor cocktail (Sigma, St Louis, MO, USA), 1 mM Na3VO4, 1% Triton X-100) and incubated on ice for 20 minutes to lyse the cells. After centrifugation (15 minutes, 15,000 g, 4°C), the pellet was resuspended in 400 μL Laemmli sample buffer (Bio-Rad, Hercules, CA, USA) with 1 M urea. The solution was sonicated for 10 pulses, 20 μL mercaptoethanol was added, and then the solution was incubated 10 minutes at 95°C, allowed to cool to room temperature, and centrifuged (10 minutes, 15,000 g). The supernatant (triton-insoluble fraction) was immunoblotted using primary antibodies for occludin (mouse monoclonal, 1 μg/mL; Invitrogen, Carlsbad, CA, USA), VE-cadherin (rabbit polyclonal, 2.5 μg/mL; Abcam, Cambridge, MA, USA), ZO-1 (rabbit polyclonal, 2.5 μg/mL; Abcam), and claudin-5 (rabbit polyclonal, 0.4 μg/mL; Santa Cruz Biotechnology, Santa Cruz, CA, USA) with the corresponding secondary antibodies from LI-COR for use with the Odyssey Infrared Imaging System. Optical density of the protein bands (occludin=60 kDa; VE-cadherin=130 kDa; ZO-1=220 kDa; claudin-5=22 kDa) were quantified within the Odyssey imaging software and was normalized to total protein content (β-actin). Protein expression in each experimental condition was then normalized to the control condition to determine relative protein expression changes in response to HPC, ischemia, and/or SphK2 gene deletion.
Statistical Analyses
Comparisons were done with SigmaStat using two-way ANOVA (analysis of variance) for Figures 1, 2, and 3A–3D, ANOVA on Ranks for Figure 1B, and one-way ANOVA for Figures 4A–4D. Statistical comparisons between groups were done with the Holm–Sidak method for multiple pairwise comparisons with P<0.05 accepted as significant.
Figure 1.

Hypoxic preconditioning (HPC) 48 hours before ischemia promoted tolerance in C57BL/6 wild-type mice to transient middle cerebral artery occlusion (tMCAO) (45 minutes), but did not protect SphK2–/– mice with respect to infarct volume (A), neurologic deficits (B), or ipsilateral hemispheric swelling (C). With a shorter tMCAO (35 minutes), HPC was still unable to induce tolerance in SphK2-nulls (A). Data represent mean±s.e.m. N=5 to 6 per group for 35 minutes tMCAO and N=6 to 9 per group for 45 minutes tMCAO. P<0.05 versus (*) C57BL/6; (#) C57BL/6+HPC. SphK, sphingosine kinase.
Figure 2.

Hypoxic preconditioning (HPC) reduced both intensity (B) and area (C) of immunoglobulin G (IgG) permeability following transient middle cerebral artery occlusion (tMCAO) in C57BL/6 wild-type mice, but not in SphK2–/– mice. Intensity of the extravasated IgG was normalized to the nonpreconditioned contralateral hemisphere. Representative coronal sections from rostral and caudal regions (A). Data represent mean±s.e.m. N=6 to 8 per group. P<0.05 versus (*) C57BL/6; (#) C57BL/6+HPC. SphK, sphingosine kinase.
Figure 3.

Triton-insoluble, cytoskeletally associated junctional protein expression was altered 48 hours following hypoxic preconditioning (HPC) in SphK2–/– mice and matched C57BL/6 wild types. Optical density quantification of claudin-5 (A), occludin (B), zona occludens (ZO)-1 (C), and VE-cadherin (D) immunoblots are shown, normalized to β-actin and then naives (C57BL/6 without HPC). Data represent mean±s.e.m. N=6 to 11 per group. P<0.05 versus (*) C57BL/6; (#) C57BL/6+HPC. SphK, sphingosine kinase.
Figure 4.

Triton-insoluble, cytoskeletally associated junctional protein expression was altered 24 hours after transient middle cerebral artery occlusion (tMCAO) in untreated and hypoxic preconditioned (HPC) SphK2–/– mice and matched C57BL/6 wild types. Optical density quantification of claudin-5 (A), occludin (B), zona occludens (ZO)-1 (C), and VE-cadherin (D) immunoblots are shown, normalized to β-actin and then naives (C57BL/6 without HPC or tMCAO). N=6 per group. P<0.05 versus (*) C57BL/6; (#) C57BL/6+HPC; (†) C57BL/6+HPC+tMCAO; (**) all other conditions. SphK, sphingosine kinase.
Results
Hypoxic Preconditioning Does Not Protect Sphingosine Kinase 2-Null Mice
Our previous studies demonstrated that ischemic tolerance to tMCAO was not established when SphK activity was pharmacologically inhibited during HPC (Wacker et al, 2009). Since cortical microvascular SphK2 activity increased following HPC, while SphK1 was unchanged, SphK2 signaling was presumed to be necessary to transduce the gene expression changes underlying the ischemia-tolerant phenotype. To test this, we assessed the efficacy of HPC to induce tolerance in SphK2-null and matched wild-type mice. While HPC reduced infarct volume 38% in wild types, the protective effect of HPC was completely blocked in the SphK2 knockouts (Figure 1A). To allay the concern that baseline infarcts in SphK2-nulls were too severe for HPC to provide any significant degree of ischemic tolerance, we repeated these studies using a shorter-duration ischemic insult (35 minutes). However, even with a less severe ischemic insult, HPC remained ineffective at inducing any protection in the SphK2-null mice. Attendant neurologic deficits and postischemic hemispheric swelling were also not improved by HPC in SphK2 knockouts (Figures 1B and 1C). These findings indicate SphK2-mediated elaboration of S1P is critical to the tissue-wide establishment of tolerance to transient focal stroke by HPC.
Protection Against Blood–Brain Barrier Disruption Requires Sphingosine Kinase 2
The SphK2 product S1P decreases vascular permeability in many cell and tissue types, and reducing BBB compromise after stroke may improve outcome (Latour et al, 2004; Obrenovitch, 2008; Weiss et al, 2009). To test if SphK2 signaling preserves postischemic BBB integrity in HPC-treated animals, we measured both the intensity and crosssectional area of extravasated IgG (transcardial perfusion eliminated vascular IgG) as indices of BBB breakdown 24 hours after tMCAO in WT and SphK2-nulls, with and without HPC. In nonpreconditioned wild types, ipsilateral disruption of the BBB was clearly evident (Figure 2A); however, HPC reduced the intensity of ipsilateral IgG leak by 51% (P<0.05; Figure 2B). In contrast, SphK2-null mice treated with HPC exhibited slightly greater IgG intensity ipsilaterally compared with untreated wild-type or SphK2-null mice, reflecting a complete loss (P<0.05) of HPC-induced BBB protection in the absence of SphK2 signaling. This is consistent with the results seen using water content as a marker of BBB disruption and dimethylsphingosine to inhibit SphK (Supplementary Figure S1). Interestingly, a significant increase in the intensity of IgG extravasation was also observed contralaterally following HPC in SphK2-null mice relative to wild types (Figure 2B). The crosssectional area of IgG leak in the ipsilateral hemisphere was reduced by 20% in HPC-treated wild types (P<0.05; Figure 2C), but, as with IgG intensity, reductions in this permeability metric were significantly (P<0.05) abrogated in SphK2-null mice. Collectively, these results indicate that SphK2 signaling is critical for the endogenous, HPC-mediated protection of the BBB in focal stroke with reperfusion.
Further examination of BBB permeability in relation to infarct volume was revealing in several ways. First, in nonpreconditioned wild types, the mean area of IgG extravasation was 45% greater than the mean infarct area from rostrally and caudally matched brain regions. This correlative finding is consistent with the notion that BBB leak is not solely the secondary result of brain infarction. Second, HPC had a more profound effect on infarct area than area of increased BBB permeability in wild types; specifically, infarct area decreased 37%, whereas the area of IgG extravasation only decreased 20% with HPC. This resulted in an even greater difference between the areas of infarct and IgG leakage in HPC-treated relative to untreated mice, a finding in line with the contention that BBB protection by HPC is not simply secondary to a smaller infarct. Immunoglobulin G intensity, however, correlates highly (r=0.851; P=0.007) with infarction. Together, these findings suggest that preservation of BBB integrity by HPC may play a role in the resultant reduction in infarction, and represents a primary vasculoprotective phenotype of the ischemia-tolerant brain.
Sphingosine Kinase 2 Signaling Regulates Triton-Insoluble Junctional Protein Expression Following Hypoxic Preconditioning
Studies in the peripheral vasculature indicate a role for S1P in altering the expression and intracellular localization of junctional proteins (Adamson et al, 2010; Lee et al, 2006; Wang and Dudek, 2009), suggesting this as a possible mechanism for the HPC-induced, SphK2-dependent reduction of poststroke BBB dysfunction that we documented herein. Thus, we sought to examine the cortical expression of junctional proteins in triton-insoluble fractions, which yield proteins in direct or indirect association with the cytoskeleton, presumably through linkages at cell junction sites (Stuart et al, 1996; Wang et al, 2011; Wong, 1997). First, we compared baseline expression in wild types and SphK2 knockouts, and then measured expression changes in response to HPC at a time (48 hours post-HPC) coinciding with ischemic onset in our model (Figures 3A–3D). Representative blots are shown in Supplementary Figure S2. We found that baseline occludin expression was significantly lower in SphK2-nulls. Following HPC, VE-cadherin expression increased by 29% (P<0.05) in wild types, but was unchanged in SphK2-nulls. Claudin-5 expression increased 106% (P<0.05) in HPC-treated wild types, but, as with VE-cadherin, HPC in SphK2-nulls did not alter its expression. Hypoxic preconditioning had no effect on expression of occludin in either wild types or knockouts. Neither SphK2 gene deletion nor HPC had any significant effect on the ZO-1 expression. Together, these findings indicate that baseline SphK2 signaling helps localize occludin to the BBB, and that the HPC-induced increase in SphK2 activity may strengthen BBB integrity before stroke by upregulating VE-cadherin and claudin-5 proteins at junctional sites.
Hypoxic Preconditioning and Sphingosine Kinase 2 Signaling Regulate Triton-Insoluble Junctional Protein Expression Following Ischemia
Having demonstrated that HPC protected against postischemic BBB breakdown in a SphK2-dependent manner, we also assessed expression of these four tight junction and adherens junction proteins in triton-insoluble fractions at a postischemic time point coinciding with our determination of IgG permeability (Figures 4A–4D). Representative blots are shown in Supplementary Figure S3. The 44% loss of ZO-1 and 33% loss of VE-cadherin in response to ischemia (P<0.05) in untreated wild-type mice was prevented in mice with prior HPC. A similar pattern was noted for occludin, but did not reach statistical significance. Claudin-5 expression was not changed by ischemia in either untreated or HPC-treated wild types. In the nonpreconditioned SphK2-nulls, the ischemia-induced reductions in the expression of ZO-1, VE-cadherin, and occludin were no different from wild types, but in HPC-treated SphK2 knockouts, preservation of the expression of these proteins was lost. Collectively, these findings indicate that the HPC-induced, SphK2-mediated prevention of the ischemia-induced loss of ZO-1, VE-cadherin, and occludin at junctional sites contributes to maintaining BBB integrity following transient focal cerebral ischemia.
Discussion
Our studies show that, while HPC reduces both infarct volume and extent of BBB breakdown in wild-type mice subjected to focal stroke, HPC-induced neurovascular tolerance is completely lost in SphK2 knockout mice. Moreover, the changes in cytoskeletally associated junctional protein expression we observed in preconditioned and ischemic, wild-type and SphK2-null mice support our hypothesis that both preischemic and postischemic changes in SphK2-mediated junctional protein expression contribute to the BBB protection characterizing HPC-induced ischemic tolerance.
While S1P signaling has long been known to mediate protection in peripheral ischemia (Karliner, 2004), only recently has this bioactive lipid pathway drawn attention in cerebral ischemia. Increased SphK2 was observed in response to cerebral ischemia both in vitro and in vivo, but whether this signaling was protective or deleterious in nature was not determined (Blondeau et al, 2007). Early reports from non-central nervous system tissues on the promotion of cell survival suggested S1P produced by SphK1 in the cytosol was antiapoptotic, while SphK2, due to targeting S1P to the endoplasmic reticulum, promoted apoptosis (Maceyka et al, 2005). However, evidence has since accumulated for a prosurvival role for SphK2 in some cell types (Schnitzer et al, 2009; Vessey et al, 2011), in agreement with both our previous pharmacologic data and our current genetic evidence for SphK2-mediated cytoprotection in the ischemic brain. With respect to elucidating the molecular basis of preconditioning-induced tolerance, our group documented increased cerebromicrovascular SphK2 protein expression and activity following HPC, attenuated HPC-induced ischemic tolerance with inhibition of both SphK isoforms, and efficacious pharmacological preconditioning by the S1P receptor agonist FTY720 (Wacker et al, 2009). Since then, infarct volume measurements in SphK2-null mice were used to demonstrate that this enzyme is an endogenous mediator of protection against cerebral ischemia (Pfeilschifter et al, 2011), and that it mediates cerebral preconditioning to either isoflurane or hypoxia (Yung et al, 2011). Also, the neuroprotective efficacy of postischemic treatment with FTY720 was demonstrated, acting through antiapoptotic (Hasegawa et al, 2010) and antiinflammatory (Czech et al, 2009; Wei et al, 2011) pathways. Herein, we not only confirm the necessity and isoform specificity of SphK2 activity in the ability of HPC to promote ischemic tolerance, but we also implicate a functional role for this enzyme in initiating the cerebroprotective phenotype induced by preconditioning, as well as identify, in part, the molecular basis of BBB protection by SphK2.
The extent of postischemic BBB breakdown strongly correlates with lesion size (Latour et al, 2004); however, whether BBB-specific protection always causes a corresponding reduction in lesion volume remains controversial (Obrenovitch, 2008; Weiss et al, 2009). If reduced BBB damage resulted secondarily from a smaller lesion instead of causing the smaller lesion, one would expect the area of infarct and the area of IgG permeability to be approximately equivalent in both untreated and preconditioned brains. However, in untreated mice, postischemic vasogenic edema occurred over an area/volume that was greater than the ultimate infarction area/volume, consistent with BBB breakdown in some brain regions in the absence of frank infarction. That this mismatch still characterized the brain of HPC-treated mice lends further support to the idea that BBB breakdown is not the result of tissue infarction. Alternatively, the area/volume mismatch in BBB breakdown and infarction could be explained by differing sensitivities of the respective methodologies used for their measurement. However, the characterization of preconditioning-induced vascular protection as a widespread reduction of BBB permeability across the hemisphere is in agreement with previous reports (Hua et al, 2008; Masada et al, 2001). Additionally, the infarction volume that we measured is on the high end of those reported in the literature, so a gross underestimation of infarct volume is unlikely. Together, these findings suggest that the mismatch in the area/volumes of BBB breakdown/infarction is genuine, and are consistent with the notion that HPC-induced reductions in BBB permeability were not solely an accompaniment to reductions in infarction volume. Our examinations of junctional protein expression reveal, for the first time, a preconditioning-induced, SphK2-based mechanism for the observed BBB protection.
Tight junction proteins are vital to the regulation of the BBB (Sandoval and Witt, 2008). Following hypoxia, claudin-5 expression increases (Willis et al, 2010), consistent with our finding following HPC. We also found HPC to increase the adherens junction protein VE-cadherin, which contains several known hypoxia response elements in its promoter and is upregulated via hypoxia-inducible factor-2 (Le Bras et al, 2007). VE-cadherin upregulates claudin-5 expression by suppressing the nuclear localization of the FoxO1-β-catenin complex that inhibits claudin-5 expression (Taddei et al, 2008); their SphK2-dependent, co-upregulation by HPC before stroke that we observed may help establish the BBB-protective phenotype we documented in HPC-treated mice.
Generally speaking, cerebral ischemia is associated with the loss of tight junction proteins, such as occludin and ZO-1 (Jiao et al, 2011; Sandoval and Witt, 2008). Preconditioning with a toll-like receptor 2 ligand (Hua et al, 2008) or brief ischemia (An and Xue, 2009; Gesuete et al, 2011) attenuates the extent of this loss, consistent with our present finding that HPC preserves ZO-1 and occludin levels poststroke. Surprisingly, we found no change in postischemic claudin-5 with or without HPC, in wild types or knockouts. As a regulator of BBB resistance to small molecule (<800 D) permeability (Nitta et al, 2003), perhaps claudin-5 is not a prominent participant in postischemic BBB breakdown when the leakage of even large molecules, as we quantified with IgG, is extensive. In fact, some previous studies even report claudin-5 levels increasing concurrent with elevated BBB permeability (Willis et al, 2010). Regardless of these paradoxical findings regarding claudin-5, our present results clearly demonstrate preconditioning-induced protection against the loss of the postischemic junctional proteins VE-cadherin and ZO-1 that is SphK2-dependent, suggesting that S1P production by SphK2 may be a common mechanism for tight junction protection independent of the preconditioning stimulus.
Indeed, in the peripheral vasculature, S1P is an established regulator of vascular integrity, mediating cytoskeleton rearrangements, redistributing focal adhesions, and localizing tight junction and adherens junction proteins (Adamson et al, 2010; Lee et al, 2006; Wang and Dudek, 2009). The S1P-based regulation of VE-cadherin, occludin, ZO-1, and claudin-5 expression and localization in rat mesenteric microvessels, human dermal microvascular or umbilical vein endothelial cells, and Chinese hamster ovary cells (Adamson et al, 2010; Lee et al, 2006) is consistent with our results, but we show for the first time localization of these proteins to cell–cell contacts in the BBB. Although the S1P-R1 receptor was implicated in anticoagulant Protein S-mediated protection of the ischemic BBB (Zhu et al, 2010), and the S1P receptor agonist FTY720 reduced BBB permeability in experimental autoimmune encephalomyelitis (Foster et al, 2009), knowledge to date regarding S1P-based regulation of the BBB is relatively scant. Herein, we identified a novel role for SphK2 in affecting basal, hypoxic, and postischemic BBB integrity secondary to its regulation of the expression of key cytoskeletally associated tight junction and adherens junction proteins.
While our current findings have identified multiple novel mechanistic aspects of HPC-induced protection of the BBB—the necessity of increased SphK2 activity after HPC, the SphK2-dependent protection of BBB integrity following ischemia, the SphK2-mediated upregulation of VE-cadherin and claudin-5 following HPC, and the SphK2-mediated protection against occludin, claudin-5, and VE-cadherin loss at endothelial cell junctions following ischemia—additional molecular events proximal and distal to these steps remain to be elucidated. The intermediary pathways that allow SphK2 to regulate the expression and localization of the junctional proteins at the BBB have yet to be clarified, but it would not be unexpected if SphK2-derived S1P signaling proceeded via mediators and pathways already known to regulate BBB tight junctions and/or BBB permeability, such as nitric oxide, vascular endothelial growth factor, proteases, and reactive oxygen species, as well as the actin cytoskeleton, caveolae, and pericytes (Sandoval and Witt, 2008). Indeed, there is evidence from studies in the peripheral vasculature that S1P activates endothelial nitric oxide synthase via PI3K/Akt, counteracts the deleterious effects of reactive oxygen species on vascular endothelium via ERK1/2 and endothelial nitric oxide synthase (Igarashi and Michel, 2008), and regulates pericyte recruitment via N-cadherin signaling (Paik et al, 2004). Sphingosine-1-phosphate R1 receptor activation also induces intracellular calcium release from the endoplasmic reticulum and Rac-1 activation, promoting adherens junction formation at the endothelial cell periphery that can block vascular endothelial growth factor-induced vascular permeability (Hoang et al, 2011; Mehta et al, 2005). Finally, vascular barrier enhancement by S1P may require lipid rafts where activated tyrosine kinases phosphorylate signaling molecules that lead to reduced vascular permeability (Zhao et al, 2009). One or more of these regulatory schemes may also transduce SphK2/S1P signaling into junctional protein localization/expression changes in the endothelium of the BBB. Taken together with our results, S1Ps role as a central regulator of vascular integrity continues to be validated; our present work now suggests the S1P pathway as a potential therapeutic target for BBB protection, giving impetus for the further elucidation of the remaining mechanisms that underlie this defining characteristic of the ischemia-tolerant brain.
While we were able to identify changes in cytoskeletally linked junctional protein expression in the cerebral cortex, the brain region that is most protected by preconditioning in our model and others, some caveats of our study remain. One is that our findings do not allow us to address regional variations in junctional protein expression in response to HPC and/or SphK2 gene deletion, (i.e., in the ischemic core, penumbra, and noninjured tissue). That being said, while previous studies have employed immunohistochemistry to show ischemia-induced regional differences in the expression pattern for tight junction proteins, as well as vessel staining pattern discontinuities (Li et al, 2007; McColl et al, 2008), because the primary aim of our work here was to identify SphK2-mediated protection of the BBB, and because its product S1P is known to specifically regulate the intracellular localization of junctional proteins but not their overall expression changes, immunohistochemistry would not have allowed us to reveal the regulatory changes we report herein using a cell fractionation/immunoblotting approach. Furthermore, this method enabled us to measure intracellular protein localization to junctional sites, a quantitative end point that encompasses many posttranslational modifications—such as phosphorylation state—that regulate the localization of these proteins to the cell junctions, where they are ultimately responsible for functional effects on vascular permeability.
In summary, our findings reveal that HPC-induced ischemic tolerance and the concomitant protection of the BBB depend on SphK2 signaling. Moreover, SphK2-generated S1P participates in both the normal maintenance of occludin at cytoskeletally linked cell junctions, as well as the mediation of HPC-induced increases in the expression of claudin-5 and VE-cadherin at these junctions, which may be compulsory for induction of the vasculoprotective phenotype by HPC. This contention is underscored by our finding that, in the absence of SphK2, not only were postischemic losses of VE-cadherin, occludin, and ZO-1 not attenuated by HPC, they were exacerbated. Further elucidation of the regulation of this endogenous, HPC-activated lipid signaling pathway, the specific cells elaborating and responding to S1P, and its role in BBB protection after stroke, may provide new therapeutic targets that can be modulated for cerebrovascular protection in stroke patients.
Acknowledgments
The authors thank Enrico Cristante for his advice on immunoblotting of junctional proteins.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Journal of Cerebral Blood Flow & Metabolism website (http://www.nature.com/jcbfm)
This study was supported by NIH NHLBI F32 Grant HL101202 (BKW), NIH NHLBI R01 HL79278 (JMG), NIH P01 NS32636 (JMG), the Spastic Paralysis Research Foundation of the Illinois–Eastern Iowa District of Kiwanis International, and an NIH Neuroscience Blueprint Core Grant P30 NS057105 to Washington University.
Supplementary Material
References
- Adamson RH, Sarai RK, Altangerel A, Thirkill TL, Clark JF, Curry FR. Sphingosine-1-phosphate modulation of basal permeability and acute inflammatory responses in rat venular microvessels. Cardiovasc Res. 2010;88:344–351. doi: 10.1093/cvr/cvq184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An P, Xue YX. Effects of preconditioning on tight junction and cell adhesion of cerebral endothelial cells. Brain Res. 2009;1272:81–88. doi: 10.1016/j.brainres.2009.03.031. [DOI] [PubMed] [Google Scholar]
- Aviv RI, d'Esterre CD, Murphy BD, Hopyan JJ, Buck B, Mallia G, Li V, Zhang L, Symons SP, Lee TY. Hemorrhagic transformation of ischemic stroke: prediction with CT perfusion. Radiology. 2009;250:867–877. doi: 10.1148/radiol.2503080257. [DOI] [PubMed] [Google Scholar]
- Blondeau N, Lai Y, Tyndall S, Popolo M, Topalkara K, Pru JK, Zhang L, Kim H, Liao JK, Ding K, Waeber C. Distribution of sphingosine kinase activity and mRNA in rodent brain. J Neurochem. 2007;103:509–517. doi: 10.1111/j.1471-4159.2007.04755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Pfeilschifter W, Mazaheri-Omrani N, Strobel MA, Kahles T, Neumann-Haefelin T, Rami A, Huwiler A, Pfeilschifter J. The immunomodulatory sphingosine 1-phosphate analog FTY720 reduces lesion size and improves neurological outcome in a mouse model of cerebral ischemia. Biochem Biophys Res Comm. 2009;389:251–256. doi: 10.1016/j.bbrc.2009.08.142. [DOI] [PubMed] [Google Scholar]
- Foster CA, Mechtcheriakova D, Storch MK, Balatoni B, Howard LM, Bornancin F, Wlachos A, Sobanov J, Kinnunen A, Baumruker T. FTY720 rescue therapy in the dark agouti rat model of experimental autoimmune encephalomyelitis: expression of central nervous system genes and reversal of blood-brain-barrier damage. Brain Pathol. 2009;19:254–266. doi: 10.1111/j.1750-3639.2008.00182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesuete R, Orsini F, Zanier ER, Albani D, Deli MA, Bazzoni G, De Simoni MG. Glial cells drive preconditioning-induced blood-brain barrier protection. Stroke. 2011;42:1445–1453. doi: 10.1161/STROKEAHA.110.603266. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Suzuki H, Sozen T, Rolland W, Zhang JH. Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke. 2010;41:368–374. doi: 10.1161/STROKEAHA.109.568899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang MV, Nagy JA, Senger DR. Active Rac1 improves pathologic VEGF neovessel architecture and reduces vascular leak: mechanistic similarities with angiopoietin-1. Blood. 2011;117:1751–1760. doi: 10.1182/blood-2010-05-286831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua F, Ma J, Ha T, Kelley J, Williams DL, Kao RL, Kalbfleisch JH, Browder IW, Li C. Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J Neuroimmunol. 2008;199:75–82. doi: 10.1016/j.jneuroim.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi J, Michel T. S1P and eNOS regulation. Biochim Biophys Acta. 2008;1781:489–495. doi: 10.1016/j.bbalip.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Jiao H, Wang Z, Liu Y, Wang P, Xue Y. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J Mol Neurosci. 2011;44:130–139. doi: 10.1007/s12031-011-9496-4. [DOI] [PubMed] [Google Scholar]
- Karliner JS. Mechanisms of cardioprotection by lysophospholipids. J Cell Biochem. 2004;92:1095–1103. doi: 10.1002/jcb.20129. [DOI] [PubMed] [Google Scholar]
- Latour LL, Kang DW, Ezzeddine MA, Chalela JA, Warach S. Early blood-brain barrier disruption in human focal brain ischemia. Ann Neurol. 2004;56:468–477. doi: 10.1002/ana.20199. [DOI] [PubMed] [Google Scholar]
- Le Bras A, Lionneton F, Mattot V, Lelievre E, Caetano B, Spruyt N, Soncin F. HIF-2alpha specifically activates the VE-cadherin promoter independently of hypoxia and in synergy with Ets-1 through two essential ETS-binding sites. Oncogene. 2007;26:7480–7489. doi: 10.1038/sj.onc.1210566. [DOI] [PubMed] [Google Scholar]
- Lee JF, Zeng Q, Ozaki H, Wang L, Hand AR, Hla T, Wang E, Lee MJ. Dual roles of tight junction-associated protein, zonula occludens-1, in sphingosine 1-phosphate-mediated endothelial chemotaxis and barrier integrity. J Biol Chem. 2006;281:29190–29200. doi: 10.1074/jbc.M604310200. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu ZY, Ogle M, Wei L. Erythropoietin prevents blood brain barrier damage induced by focal cerebral ischemia in mice. Neurochem Res. 2007;32:2132–2141. doi: 10.1007/s11064-007-9387-9. [DOI] [PubMed] [Google Scholar]
- Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH, Jr, Milstien S, Spiegel S. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005;280:37118–37129. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- Masada T, Hua Y, Xi G, Ennis SR, Keep RF. Attenuation of ischemic brain edema and cerebrovascular injury after ischemic preconditioning in the rat. J Cereb Blood Flow Metab. 2001;21:22–33. doi: 10.1097/00004647-200101000-00004. [DOI] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. 2008;28:9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill H, Ryan TA, Smith SJ, Nelson WJ. Spatial and temporal dissection of immediate and early events following cadherin-mediated epithelial cell adhesion. J Cell Biol. 1993;120:1217–1226. doi: 10.1083/jcb.120.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Konstantoulaki M, Ahmmed GU, Malik AB. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem. 2005;280:17320–17328. doi: 10.1074/jbc.M411674200. [DOI] [PubMed] [Google Scholar]
- Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol. 2005;25:11113–11121. doi: 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrenovitch TP. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol Rev. 2008;88:211–247. doi: 10.1152/physrev.00039.2006. [DOI] [PubMed] [Google Scholar]
- Paik JH, Skoura A, Chae SS, Cowan AE, Han DK, Proia RL, Hla T. Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev. 2004;18:2392–2403. doi: 10.1101/gad.1227804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SW, Kim M, Chen SW, Brown KM, D'Agati VD, Lee HT. Sphinganine-1-phosphate protects kidney and liver after hepatic ischemia and reperfusion in mice through S1P1 receptor activation. Lab Invest. 2010;90:1209–1224. doi: 10.1038/labinvest.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeilschifter W, Czech-Zechmeister B, Sujak M, Mirceska A, Koch A, Rami A, Steinmetz H, Foerch C, Huwiler A, Pfeilschifter J. Activation of sphingosine kinase 2 is an endogenous protective mechanism in cerebral ischemia. Biochem Biophys Res Comm. 2011;413:212–217. doi: 10.1016/j.bbrc.2011.08.070. [DOI] [PubMed] [Google Scholar]
- Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32:200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Schnitzer SE, Weigert A, Zhou J, Brune B. Hypoxia enhances sphingosine kinase 2 activity and provokes sphingosine-1-phosphate-mediated chemoresistance in A549 lung cancer cells. Mol Cancer Res. 2009;7:393–401. doi: 10.1158/1541-7786.MCR-08-0156. [DOI] [PubMed] [Google Scholar]
- Stuart RO, Sun A, Bush KT, Nigam SK. Dependence of epithelial intercellular junction biogenesis on thapsigargin-sensitive intracellular calcium stores. J Biol Chem. 1996;271:13636–13641. doi: 10.1074/jbc.271.23.13636. [DOI] [PubMed] [Google Scholar]
- Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923–934. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]
- Vessey DA, Li L, Jin Z-Q, Kelley M, Honbo N, Zhang J, Karliner JS.2011A sphingosine kinase form 2 knockout sensitizes mouse myocardium to ischemia/reoxygenation injury and diminishes responsiveness to ischemic preconditioning Oxid Med Cell Longevity2011: 8; doi: 10.1155/2011/961059 [DOI] [PMC free article] [PubMed]
- Wacker BK, Park TS, Gidday JM. Hypoxic preconditioning-induced cerebral ischemic tolerance: role of microvascular sphingosine kinase 2. Stroke. 2009;40:3342–3348. doi: 10.1161/STROKEAHA.109.560714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Chiang ET, Simmons JT, Garcia JG, Dudek SM. FTY720-induced human pulmonary endothelial barrier enhancement is mediated by c-Abl. Eur Respir J. 2011;38:78–88. doi: 10.1183/09031936.00047810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Dudek SM. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc Res. 2009;77:39–45. doi: 10.1016/j.mvr.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Yemisci M, Kim HH, Yung LM, Shin HK, Hwang SK, Guo S, Qin T, Alsharif N, Brinkmann V, Liao JK, Lo EH, Waeber C. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann Neurol. 2011;69:119–129. doi: 10.1002/ana.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss N, Miller F, Cazaubon S, Couraud PO. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta. 2009;1788:842–857. doi: 10.1016/j.bbamem.2008.10.022. [DOI] [PubMed] [Google Scholar]
- Willis CL, Meske DS, Davis TP. Protein kinase C activation modulates reversible increase in cortical blood-brain barrier permeability and tight junction protein expression during hypoxia and posthypoxic reoxygenation. J Cereb Blood Flow Metab. 2010;30:1847–1859. doi: 10.1038/jcbfm.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong V. Phosphorylation of occludin correlates with occludin localization and function at the tight junction. Am J Physiol. 1997;273:C1859–C1867. doi: 10.1152/ajpcell.1997.273.6.C1859. [DOI] [PubMed] [Google Scholar]
- Yung LM, Wei Y, Qin T, Wang Y, Smith CD, Waeber C. Sphingosine kinase 2 mediates cerebral preconditioning and protects the mouse brain against ischemic injury. Stroke. 2011;43:199–204. doi: 10.1161/STROKEAHA.111.626911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Singleton PA, Brown ME, Dudek SM, Garcia JG. Phosphotyrosine protein dynamics in cell membrane rafts of sphingosine-1-phosphate-stimulated human endothelium: role in barrier enhancement. Cell Signal. 2009;21:1945–1960. doi: 10.1016/j.cellsig.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Wang Y, Singh I, Bell RD, Deane R, Zhong Z, Sagare A, Winkler EA, Zlokovic BV. Protein S controls hypoxic/ischemic blood-brain barrier disruption through the TAM receptor Tyro3 and sphingosine 1-phosphate receptor. Blood. 2010;115:4963–4972. doi: 10.1182/blood-2010-01-262386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.