

Abstract
Background
Tuberous sclerosis complex (TSC) is associated with tumor development in the brain, retina, kidney, skin, heart, and lung. Seizures, intellectual disability, and characteristic skin lesions commonly manifest in early childhood, but some findings, notably renal angiomyolipomas and pulmonary lymphangioleiomyomatosis (LAM), emerge later, placing adults with undiagnosed TSC at increased risk for morbidity and mortality.
Objective
To describe the clinical presentation and severity of TSC in adult women.
Design
Retrospective cohort study.
Setting
National Institutes of Health Clinical Center, Bethesda, Maryland, 1995 to 2010.
Patients
79 women aged 18 years or older who were enrolled in an observational cohort study of TSC to evaluate disease manifestations.
Measurements
History, physical examination, pulmonary function testing, chest radiography, abdominal computed tomography, high-resolution chest computed tomography, and brain magnetic resonance imaging were used to evaluate patients.
Results
Among the 45 patients who received a diagnosis of TSC in adulthood, 21 presented with symptoms due to LAM, 19 with renal angiomyolipomas, and 10 with seizures. Of the 45 patients, 30 met clinical criteria for TSC in childhood that remained undiagnosed for a median of 21.5 years and 15 were older than 18 years before meeting the clinical criteria for TSC. Patients diagnosed in adulthood and those diagnosed in childhood had similar occurrences of pneumothorax, shortness of breath, hemoptysis, nephrectomy, and death.
Limitation
No men were included in the study, and selection was biased toward patients having pulmonary LAM.
Conclusion
Women who received a TSC diagnosis in adulthood had minimal morbidity during childhood but were still at risk for life-threatening pulmonary and renal manifestations.
Primary Funding Source
Intramural Research Program, National Institutes of Health, National Heart, Lung, and Blood Institute.
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder characterized by the development of hamartomatous tumors in many organs, most notably the brain, retina, kidney, skin, heart, and lung. Tumor predisposition is the result of a mutation in 1 of 2 tumor suppressor genes: TSC1 or TSC2 (1). Affected patients may present early in life with the classic triad of seizures, intellectual disability, and cutaneous angiofibromas, but fewer than 30% have all 3 characteristics and 6% have none (2). The perception of TSC as a universally devastating congenital disease is changing—detailed family studies, often coupled with genetic testing, have revealed mildly affected patients, including seemingly unaffected adults (3–6). As noted in a recent epidemiologic study conducted in Northern Ireland, recognition of TSC in a parent after an affected child receives the diagnosis is not uncommon (7). In the study, nearly 20% of the patients with TSC received their diagnosis after age 15 years, most commonly after TSC was identified in an offspring (7). In a cohort of 243 patients with TSC, 12% received their diagnosis in adulthood (8). This complex is more common than previously reported, with a minimum prevalence of about 1 case in 15 000 to 30 000 persons (7, 9–13).
Although most TSC manifestations appear during early childhood, morbidity and mortality from TSC-related pulmonary and renal disease predominate in adults (14, 15). Lymphangioleiomyomatosis (LAM) is a cystic lung disease that affects approximately 30% to 40% of adult women with TSC (15–19). It may present as pneumothorax, shortness of breath, or hemoptysis and can cause respiratory failure and death. Pulmonary LAM is rare in men, but renal tumors occur in both men and women with TSC. Angiomyolipomas and renal cysts have been observed in 75% and 32%, respectively, of patients with TSC who are older than 22 years (20). Angiomyolipomas can occur in the general population, but patients with TSC are more likely to have multiple or bilateral lesions that present at a younger age than those with sporadic renal tumors (21, 22). Renal disease commonly manifests as hematuria, flank pain, palpable mass, and hemorrhage and is a leading cause of death in this patient population (21, 23).
Patients with TSC should be regularly monitored for new or worsening organ involvement, because early intervention may reduce morbidity and mortality (24). Transcatheter embolization of renal angiomyolipomas preserves kidney function and decreases the risk for hemorrhage or need for subsequent surgery (25). Sirolimus treatment is associated with improved pulmonary function (26, 27), a reduction in the size of renal angiomyolipomas (26), chylous effusions (see accompanying article in this issue by Taveira-DaSilva and colleagues [28]), and LAM (28). Everolimus treatment decreases the size of subependymal giant cell astrocytomas in patients with TSC (29).
Recent advances in the treatment of TSC highlight the need to identify and follow affected patients. Little, however, is known about the clinical characteristics and severity of TSC when it presents in adults. Whether patients diagnosed in adulthood exhibit the same findings as those of patients diagnosed in childhood, or whether they exhibit only those characteristics that commonly emerge later in life, is unclear. The purpose of this study was to determine the clinical presentation and severity of TSC in adult women.
METHODS
Patients
This retrospective cohort study was conducted at the National Institutes of Health Clinical Center, Bethesda, Maryland. Patients were referred by physicians, were self-referred, or were referred through the Tuberous Sclerosis Alliance or the LAM Foundation. They were enrolled in 2 pulmonary LAM protocols: 95-H-0186, which explores factors involved in the pathogenesis of LAM, and 96-H-0100, which examines the role of genetic factors in lung disease. At the initial visit, 21 women with LAM enrolled in protocol 95-H-0186, 31 women with TSC enrolled in protocol 96-H-0100, and 27 women enrolled in both protocols. Patients were known to have either LAM or TSC or both conditions at enrollment. Inclusion criteria were age 18 years or older and being capable of providing written informed consent, which was obtained from all patients at enrollment. Because pulmonary LAM is rarely diagnosed in men, all patients in this study were women. An additional 19 patients with TSC and LAM were excluded from this study because of incomplete data for 3 or more major criteria of TSC.
At enrollment, patients were evaluated for TSC diagnostic criteria comprising major and minor features (Table 1) (30). They were admitted for clinical history, physical examination, pulmonary function testing, chest radiography, abdominal computed tomography, high-resolution computed tomography of the chest (28), and brain magnetic resonance imaging (31). Diagnosis of TSC was based on the presence of at least 2 major or 1 major and 2 or more minor diagnostic features (30). Patient-reported history, confirmed by medical records and referral letters when available, was used for age at diagnosis and age at onset of TSC-related symptoms or signs. The age when patients reported manifesting at least 2 major or 1 major and 2 or more minor features was considered the age of TSC penetrance. To evaluate for progression of TSC-related disease, we gave patients the opportunity to return every 6 to 12 months. At return visits, patients had a physical examination, pulmonary function testing, and the other tests described depending on symptoms or disease severity.
Table 1.
Diagnostic Criteria of Tuberous Sclerosis Complex for 79 Adult Women at Enrollment*
| Criterion | Patients, n (%) |
|---|---|
| Major features | |
| Facial angiofibromas or forehead plaque | 73 (92) |
| Nontraumatic ungual or periungual fibroma | 63 (80) |
| Hypomelanotic macules (≥3) | 55 (70) |
| Shagreen patch (connective tissue nevus) | 50 (63) |
| Multiple retinal nodular hamartomas | 9/54 (17)† |
| Cortical tuber | 65 (82) |
| Subependymal nodule | 31 (39) |
| Subependymal giant cell astrocytoma | 3 (4) |
| Cardiac rhabdomyoma, single or multiple | 2/54 (4)† |
| LAM | 64 (81) |
| Renal angiomyolipomas | 71 (90) |
| Minor features | |
| Multiple randomly distributed pits in dental enamel | 66/75 (88)† |
| Hamartomatous rectal polyps | 4/48 (8)† |
| Bone cysts | ND |
| Cerebral white matter migration lines | ND |
| Gingival fibromas | 33/76 (43)† |
| Nonrenal hamartoma | ND |
| Retinal achromic patch | 3/54 (6)† |
| “Confetti” skin lesions | 20 (25) |
| Multiple renal cysts | 24 (30) |
LAM = lymphangioleiomyomatosis; ND = not done.
Tuberous sclerosis complex was confirmed in patients with ≥2 major features or 1 major and ≥2 minor features. The presence of LAM and renal angiomyolipomas in the same patient was counted as 1 major feature for diagnosis, because they occur together in sporadic LAM.
Data were unavailable for some patients (total number is the denominator).
Statistical Analysis
We divided patients into 3 groups based on their age at TSC diagnosis and penetrance: childhood diagnosis (penetrance in childhood; diagnosis at age <18 years), delayed diagnosis (penetrance in childhood; diagnosis at age ≥18 years), or adult penetrance (penetrance and diagnosis at age ≥18 years).
Three patients could not be assigned to a group because of unclear history of penetrance or diagnosis. They were excluded from further analysis, leaving a final sample size of 76 patients. We compared characteristics of the 3 groups, including total numbers of major features and presence or absence of specific findings, by using SPSS Statistics, version 19 (IBM Software, Somers, New York), to calculate analysis of variance, nonparametric analysis of variance (Kruskall–Wallis test), and chi-square statistics as appropriate. Significant differences among the 3 groups (P < 0.05) were followed by comparisons between pairs of groups by using independent samples t, Mann–Whitney, or chi-square tests as appropriate.
Role of Funding Source
This research was funded by the Intramural Research Program, National Institutes of Health, National Heart, Lung, and Blood Institute, and the Congressionally Directed Medical Research Programs. The funding sources had no role in the design, data collection, storage, analysis, drafting of the report, or decision to publish the manuscript.
RESULTS
The 79 patients were between 20 and 69 years of age, and all had multiple diagnostic criteria for TSC at enrollment (Table 1). Most patients had skin and dental lesions characteristic of TSC, including facial angiofibromas or forehead plaques, ungual fibromas, hypomelanotic macules, shagreen patches, and dental pitting (Figure 1, A to F). Angiomyolipomas (Figure 1, G) and LAM (28) were observed in 90% and 81% of patients, respectively. More than 80% of patients had cortical tubers (Figure 1, H), but only 52% (41 of 79 patients) had a history of seizures. Subependymal nodules (Figure 1, I), subependymal giant cell astrocytomas, multiple retinal nodular hamartomas, and cardiac rhabdomyomas were less frequent (Table 1). Twenty of 74 (27%) patients had a positive family history of TSC.
Figure 1. Major and minor diagnostic features of tuberous sclerosis complex in adult women.

A. Multiple facial angiofibromas, often scattered symmetrically across the nose and cheeks and concentrated in the nasal groove. B. Forehead plaque, pink to red-brown fibrous plaques that can be located anywhere on the face or scalp, with adjacent white scar from skin biopsy. C. Ungual fibromas, emerging from around or under the nail and more common on the toes than fingers. D. Hypomelanotic macules, with a polygonal or ash-leaf shape, under fluorescent lighting (left) and accentuated with a Wood lamp (right). E. Shagreen patch, a connective tissue nevus usually situated on the lower back. F. Dental pitting, pinpoint to crater-like defects in the enamel. G. Computed tomography scan of multiple renal angiomyolipomas in the left kidney in a patient after right nephrectomy. H. Magnetic resonance image of multiple cortical tubers, including a large cortical tuber in the left parietal lobe (arrow). I. Magnetic resonance image of multiple cortical tubers, calcified subependymal nodules (arrows).
The age at TSC diagnosis ranged from 0 to 67 years. Diagnosis during childhood and adulthood was made in 31 (41%) and 45 (59%) patients, respectively. Of patients with TSC diagnosed in adulthood, 30 (66%) reported having clinical features that fulfilled diagnostic criteria for TSC during childhood (Table 2), but the diagnosis was delayed by a median of 21.5 years. The remaining 15 (33%) had insufficient diagnostic criteria for TSC during childhood, but clinical features sufficient for diagnosis were apparent at a median age of 35 years. Patients with TSC diagnosed in childhood tended to be younger at enrollment but had had TSC features for much longer than those with adult penetrance. The median follow-up was 3 years (Table 2).
Table 2.
Characteristics of 3 Groups of Adult Women With TSC
| Characteristic | Childhood Diagnosis Group (n = 31) |
Delayed Diagnosis Group (n = 30) |
Adult Penetrance Group (n = 15) |
Total (n = 76) |
|---|---|---|---|---|
| Diagnostic and enrollment history | ||||
| Median age at first TSC criterion (IQR), y | 0.1 (0.0 to 1.0) | 0.0 (0.0 to 4.0) | 12.0 (5.0 to 31.0) | 1.0 (0.0 to 5.0) |
| Median age at TSC penetrance (IQR), y* | 3.0 (1.0 to 6.0) | 6.0 (3.0 to 12.0) | 35.0 (27.5 to 42.5) | 6.0 (2.5 to 15.0) |
| Median age at TSC diagnosis (IQR), y | 6.0 (1.1 to 10.0) | 26.5 (23.0 to 36.0) | 44.0 (39.0 to 48.5) | 23.0 (8.0 to 36.0) |
| Median time to TSC diagnosis (IQR), y† | 0.0 (0.0 to 2.5) | 21.5 (15.0 to 28.0) | 1.0 (0.0 to 13.5) | 7.0 (0.0 to 20.5) |
| Median age at enrollment (IQR), y | 32.0 (26.0 to 39.5) | 39.0 (33.0 to 47.0) | 44.0 (41.0 to 49.0) | 37.5 (30.0 to 46.0) |
| Median time from penetrance to enrollment (IQR), y | 26.0 (23.0 to 35.0) | 31.5 (23.0 to 42.0) | 2.0 (0.0 to 11.0) | 25.0 (18.5 to 37.0) |
| Median time from diagnosis to enrollment (IQR), y | 25.3 (23.0 to 32.0) | 11.5 (1.0 to 15.0) | 0.0 (−0.5 to 0.0) | 14.0 (1.0 to 24.0) |
| Median duration of follow-up (IQR), y | 3.0 (1.0 to 7.0) | 3.0 (0.0 to 7.0) | 5.0 (1.0 to 6.0) | 3.0 (1.0 to 6.5) |
| Family history, n (%) | ||||
| Positive family history at TSC diagnosis | 2/30 (6.7)‡ | 3/29 (10)‡ | 1/15 (6.7) | 6 (7.9) |
| Positive family history at enrollment | 7/30 (23)‡ | 10/29 (35)‡ | 3/15 (20) | 20/74 (27) |
| Offspring with TSC | 7/12 (58) | 9/15 (60) | 2/8 (25)§ | 18/35 (51) |
| No offspring | 19 | 15 | 5§ | 39 |
IQR = interquartile range; TSC = tuberous sclerosis complex.
Age when the patient fulfilled the diagnostic criteria for definite TSC by patient recollection of onset of major and minor features.
Age at TSC diagnosis minus age at TSC penetrance.
One patient in each of these groups was adopted, so family history is unavailable.
Information regarding offspring was not available for 2 patients.
Patients in whom diagnosis was delayed were similar to those with TSC diagnosed in childhood. Frequencies of a positive family history (Table 2), major and minor criteria at enrollment (Appendix Table, available at www.annals.org), or morbidity and mortality (Table 3) did not significantly differ. Only 7 (23%) patients in the delayed diagnosis group had seizures at the time of TSC diagnosis, whereas 17 (55%) in the childhood diagnosis group presented with seizures (Table 4). These patients were more likely to present with LAM and renal angiomyolipomas at the time of TSC diagnosis than those with TSC diagnosed in childhood (Table 4).
Table 3.
Tuberous Sclerosis Complex Morbidity and Mortality*
| Variable | Childhood Diagnosis Group (n = 31) |
Delayed Diagnosis Group (n = 30) |
Adult Penetrance Group (n = 15) |
Total (n = 76) |
|---|---|---|---|---|
| Seizures | 21 (68) | 16 (53) | 4 (27)† | 41 (54) |
| Nephrectomy | 9 (29) | 10 (33) | 4 (27) | 23 (30) |
| Renal embolization | 5 (16) | 2 (7) | 0 (0) | 7 (9) |
| Renal transplantation | 2 (6.5) | 1 (3) | 0 (0) | 3 (4) |
| Pneumothorax | 14 (45) | 13 (43) | 4 (27) | 31 (41) |
| Shortness of breath | 7 (23) | 7 (23) | 4 (27) | 18 (24) |
| Hemoptysis | 1 (3) | 2 (7) | 2 (13) | 5 (7) |
| Lung transplantation | 0 (0) | 1 (3) | 1 (7) | 2 (3) |
| Death‡ | 6 (19) | 2 (7) | 1 (7) | 9 (12) |
Morbidity occurred before or during follow-up. Values reported are numbers (percentages).
Significant difference vs. the childhood diagnosis group (P < 0.05).
An additional death occurred in a patient with tuberous sclerosis complex and lymphangioleiomyomatosis diagnosed in adulthood who had an uncertain age of penetrance.
Table 4.
Clinical Characteristics at Diagnosis of Tuberous Sclerosis Complex*
| Characteristic | Childhood Diagnosis Group (n = 31) |
Delayed Diagnosis Group (n = 30) |
Adult Penetrance Group (n = 15) |
Total (n = 76) |
|---|---|---|---|---|
| Any skin major feature | 31 (100) | 28 (93) | 10 (66)† | 69 (91) |
| Seizures | 17 (55)‡ | 7 (23) | 3 (20) | 27 (36) |
| Lymphangioleiomyomatosis | 0 (0)§ | 10 (33)§ | 11 (73)§ | 21 (28) |
| Renal angiomyolipomas | 1 (3)‡ | 11 (36) | 8 (53) | 49 (64) |
Values reported are numbers (percentages).
Significant difference vs. the delayed diagnosis and childhood diagnosis groups (P < 0.05).
Significant difference vs. the delayed diagnosis and adult penetrance groups (P < 0.05).
Significant difference among all 3 groups (P < 0.05).
Patients with adult penetrance differed from the other 2 groups. At enrollment, they had statistically significantly fewer major criteria, mostly because of lower frequencies of angiofibromas or forehead plaques, ungual fibromas, hypomelanotic macules, and shagreen patches (Appendix Table). Fewer patients in the adult penetrance group reported skin lesions at the time of TSC diagnosis (Table 4). Skin manifestations appeared later in life in patients with adult penetrance. Six patients had no skin manifestations and 9 had only 1 skin manifestation before age 18 years, whereas all those with TSC diagnosed in childhood had 1 or more skin manifestations before age 18 years. Median ages at onset of angiofibromas in the childhood diagnosis, delayed diagnosis, and adult penetrance groups were 5, 7, and 21.5 years, respectively. Patients with adult penetrance were less likely to present with seizures and more likely to present with LAM or angiomyolipomas at the time of TSC diagnosis than those with TSC diagnosed in childhood (Table 4).
Seizures usually presented during the first 5 years of life, but 8 of 79 (10%) patients reported their first seizures in adulthood. Most patients had complex partial seizures, and 5 had infantile spasms. Twenty-nine percent of the patients were taking anticonvulsants at the time of enrollment, although several more reported having taken them in the past.
Pulmonary complications included shortness of breath, pneumothorax, and hemoptysis, with no significant differences among the 3 groups. In patients with LAM, the mean FEV1 was 82% predicted (SD, 28% predicted) and diffusing capacity of the lung for carbon monoxide was 80% predicted (SD, 26% predicted), with no significant differences among the 3 groups. Two patients had pulmonary transplants (Table 3).
Renal complications included hemorrhage, pain, hematuria, and renal insufficiency, which were treated with partial or total nephrectomy, embolization, and renal transplantation (Table 3). Bilateral angiomyolipomas occurred in 61 of 71 patients, and 1 woman had renal cell carcinoma.
Ten patients died during the study (Table 3). Two deaths were due to respiratory failure (1 after pulmonary hemorrhage and 1 after pneumothorax), and 1 each were from complications after lung transplantation, pulmonary embolus, and complications from obstruction of a ventriculoperitoneal shunt. Five patients died of unknown causes. In the 5 patients with an attributable cause of death, the time between onset of symptoms and death was 2 to 7 years after onset of pulmonary symptoms and 20 years after onset of central nervous system symptoms for the patient with an obstructed shunt.
DISCUSSION
This study shows that the clinical presentation of TSC in adults differs from the typical presentation in children. Women with TSC diagnosed in adulthood frequently presented with pulmonary LAM or renal angiomyolipomas, but they, as shown in other studies of adults with TSC (32), were less likely to have seizures or had seizures later in life (8, 33). Most of the patients in this study had no family history of TSC at the time of diagnosis, which is consistent with the observation that TSC occurs sporadically in about two thirds of patients (34). We propose that TSC should be considered in an adult with any one of the following: bilateral or multiple renal angiomyolipomas, LAM, or TSC-associated skin lesions even in the absence of seizures, intellectual disability, or an affected family member. Patients who present with apparently “mild” TSC during childhood are still at risk for life-threatening manifestations later in life. A delay in diagnosis precludes monitoring for early tumor formation, delays treatment, and hinders patients from making informed decisions regarding childbearing.
All but one of the women who had a delayed diagnosis had skin lesions characteristic of TSC that developed in childhood and, in some cases, were well-documented in their medical records. Imaging studies to reveal other manifestations were typically not done during childhood in this group of women, possibly because they did not have ongoing seizures or obvious intellectual disability. Some women eventually received a diagnosis of forme fruste TSC (35, 36) in the mistaken belief that they had a TSC variant that manifested as only skin lesions.
Our results indicate that sufficient criteria for diagnosis may not manifest until adulthood in patients with TSC (Figure 2). These women with adult penetrance tended to have fewer major criteria than adults diagnosed in childhood. Throughout childhood, they expressed no diagnostic features or had a single subtle skin manifestation. The onset of angiofibromas was delayed, often until adulthood. Possible explanations for delayed penetrance include mutations associated with a mild phenotype (4), mosaicism (37), and effects of modifier genes (38). Polymorphisms in modifier genes may affect disease severity differently for each organ, and it is possible that modifier genes, which reduced the severity of skin and brain disease during childhood, do not mitigate adult renal and pulmonary manifestations in these adult women.
Figure 2. Clinical characteristics of tuberous sclerosis complex with early or late penetrance.
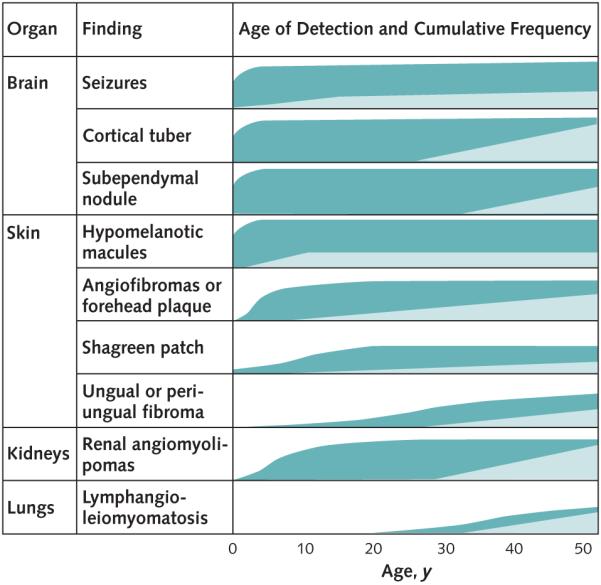
The estimated frequencies of findings (0% to 100%) over time are represented by the filled areas. Patients with tuberous sclerosis complex diagnosed in childhood (dark green) typically experience onset of multiple manifestations of the disease in the first 5 years. Patients with adult penetrance (light green) do not meet clinical criteria for diagnosis in childhood and have lower frequencies of brain and skin involvement in adulthood. The substantial variations in disease severity between patients or the potential for changing disease severity over time for 1 patient are not indicated. Patients at risk for a delay in diagnosis (not shown) have childhood penetrance but minimal illness in childhood.
Our study had several limitations. The patients were referred to a tertiary care research center for studies of pulmonary disease, so they differ from the general population with TSC by being biased toward women with LAM. Among the women with mild TSC in childhood, there may be referral bias toward those who developed more severe disease later in life. Patients were capable of providing informed consent, reducing the numbers of patients with more severe involvement of their central nervous system and probably contributing to the lower frequency of seizures in this study sample than in other TSC studies (32). The data on age at onset were self-reported and retrospective.
Most of our patients had not been tested for mutations in TSC1 or TSC2, but DNA testing is now commercially available and may permit earlier diagnosis, even before patients meet clinical diagnostic criteria (5). A negative genetic test result does not exclude the possibility of TSC, because no mutation is identified in approximately 15% of patients who fulfill diagnostic criteria for TSC (39). Additional support for the diagnosis can be obtained by examining the patient for TSC manifestations that are not part of the current consensus criteria. These include intraoral fibromas at nongingival sites, such as the buccal or labial mucosa and tongue (40); inconspicuous nail findings, such as red comets (partially blanchable, longitudinal red streaks in the nails) (41); and sclerotic bone lesions observed during computed tomography of the chest, abdomen, and pelvis (42).
Once TSC is diagnosed, a management and surveillance plan should be implemented. General monitoring guidelines for patients with TSC include high-resolution computed tomography of the chest in asymptomatic women at age 18 years and again at age 30 to 40 years or more frequently in male or female patients with respiratory symptoms (43); periodic imaging studies of the brain and kidneys in male and female patients; and evaluation of other organ systems as indicated by symptoms (24). In addition to the surgical interventions available for treating TSC tumors, the potential utility of the mammalian target of rapamycin inhibitors for treating TSC-related tumors has been demonstrated in recent trials (26–29).
Although TSC has been considered a disease that causes clinically significant morbidity in childhood, our data revealed that some patients with TSC either remain without the diagnosis or do not present with symptoms until later in life. Patients whose symptoms manifest in adulthood may have a remote or no history of seizures and minimal to no skin lesions but can harbor potentially fatal tumors in their lungs or kidneys. An early and accurate diagnosis of TSC is important to minimize the serious morbidity that often accompanies the disease.
Context
Tuberous sclerosis complex (TSC) is commonly recognized by the occurrence of skin lesions or seizures during childhood. Hamartomatous tumors may damage several other organs later in life, although the clinical picture of adults with TSC is not well-described.
Contribution
This observational study found that most adult women with TSC had diagnostic findings that remained unrecognized during childhood. In other patients, disease manifestations sufficient for diagnosis were not present until adulthood. Despite the absence of seizures or skin lesions in many adults with TSC, life-threatening pulmonary and renal complications were often present.
Caution
The cohort was derived from a study evaluating lymphangioleiomyomatosis; therefore, pulmonary manifestations may have been more common than in other TSC populations.
Implication
The presentation or diagnosis of TSC may be delayed until adulthood.
—The Editors
Acknowledgment
The authors thank the LAM Foundation and the Tuberous Sclerosis Alliance for assistance with patient recruitment. They also thank Martha Vaughan for critical review of the manuscript and Angelo M. Taveira-DaSilva and Mary Haughey for assistance in data collection. The authors have listed everyone who contributed significantly to the work.
Grant Support: From the Intramural Research Program, National Institutes of Health, National Heart, Lung, and Blood Institute, and grants from the National Cancer Institute (R01 CA100907; Dr. Darling) and the Congressionally Directed Medical Research Programs (TS080064; Dr. Darling).
Appendix
Appendix Table.
Features That Differed in Frequency Between Groups at the Time of Enrollment
| Feature | Childhood Diagnosis Group (n = 31) |
Delayed Diagnosis Group (n = 30) |
Adult Penetrance Group (n = 15) |
Total (n = 76) |
|---|---|---|---|---|
| Mean number of major features at enrollment (IQR) | 7.0 (6.0–8.0) | 6.5 (5.0–7.2) | 4.0 (4.0–6.0)* | 7.0 (5.0–7.0) |
| Major skin features, n (%) | ||||
| Facial angiofibromas or forehead plaque | 30 (97) | 30 (100) | 11 (71)* | 71 (93) |
| Nontraumatic ungual or periungual fibroma | 29 (94) | 25 (83) | 8 (53)† | 62 (82) |
| Hypomelanotic macules (≥3) | 27 (87) | 21 (70) | 5 (33)* | 53 (70) |
| Shagreen patch | 25 (81) | 20 (67) | 4 (27)* | 49 (64) |
IQR = interquartile range.
Significant difference vs. the delayed diagnosis and childhood diagnosis groups (P < 0.05).
Significant difference vs. the childhood diagnosis group (P < 0.005).
Footnotes
Publisher's Disclaimer: Disclaimer: The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or reflecting those of the Department of Defense or the Uniformed Services University of the Health Sciences.
Potential Conflicts of Interest: Disclosures can be viewed at www.acponline.org/authors/icmje/ConflictOfInterestForms.do?msNum[H11005]M10-2008.
Reproducible Research Statement: Study protocol: Available from Cardiovascular and Pulmonary Branch/National Heart, Lung, and Blood Institute (contact Dr. Moss: mossj@nhlbi.nih.gov). Statistical code: Not available. Data set: Available from Cardiovascular and Pulmonary Branch/National Heart, Lung, and Blood Institute after approval by the institutional review boards of the National Heart, Lung, and Blood Institute and the requesting investigator and completion of a human subjects Material Transfer Agreement (contact Dr. Moss: mossj@nhlbi.nih.gov).
Author Contributions: Conception and design: D. Seibert, J. Moss, T.N. Darling.
Analysis and interpretation of the data: D. Seibert, C.H. Hong, F. Takeuchi, C. Olsen, J. Moss, T.N. Darling.
Drafting of the article: D. Seibert, T.N. Darling.
Critical revision of the article for important intellectual content: J. Moss, T.N. Darling.
Final approval of the article: C. Olsen, J. Moss, T.N. Darling.
Provision of study materials or patients: J. Moss.
Statistical expertise: C. Olsen.
Obtaining of funding: J. Moss, T.N. Darling.
Administrative, technical, or logistic support: C.H. Hong, F. Takeuchi, J. Moss.
Collection and assembly of data: C.H. Hong, C. Olsen, O. Hathaway, T.N. Darling.
References
- 1.Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372:657–68. doi: 10.1016/S0140-6736(08)61279-9. [PMID: 18722871] [DOI] [PubMed] [Google Scholar]
- 2.Gomez MR. Tuberous sclerosis. 2nd ed Raven Pr; New York: 1988. [Google Scholar]
- 3.O’Callaghan FJ, Osborne JP. Advances in the understanding of tuberous sclerosis. Arch Dis Child. 2000;83:140–2. doi: 10.1136/adc.83.2.140. [PMID: 10906022] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jansen AC, Sancak O, D’Agostino MD, Badhwar A, Roberts P, Gobbi G, et al. Unusually mild tuberous sclerosis phenotype is associated with TSC2 R905Q mutation. Ann Neurol. 2006;60:528–39. doi: 10.1002/ana.21037. [PMID: 17120248] [DOI] [PubMed] [Google Scholar]
- 5.Vail EA, Rakowski SK, Numis AL, Thiele EA. Role of mutational analysis in diagnosis of tuberous sclerosis complex. Clin Genet. 2009;75:282–5. doi: 10.1111/j.1399-0004.2008.01129.x. [PMID: 19250385] [DOI] [PubMed] [Google Scholar]
- 6.Quist SR, Franke I, Sutter C, Bartram CR, Gollnick HP, Leverkus M. Periungual fibroma (Koenen tumors) as isolated sign of tuberous sclerosis complex with tuberous sclerosis complex 1 germline mutation [Letter] J Am Acad Dermatol. 2010;62:159–61. doi: 10.1016/j.jaad.2009.01.021. [PMID: 20082901] [DOI] [PubMed] [Google Scholar]
- 7.Devlin LA, Shepherd CH, Crawford H, Morrison PJ. Tuberous sclerosis complex: clinical features, diagnosis, and prevalence within Northern Ireland. Dev Med Child Neurol. 2006;48:495–9. doi: 10.1017/S0012162206001058. [PMID: 16700943] [DOI] [PubMed] [Google Scholar]
- 8.Staley BA, Vail EA, Thiele EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics. 2011;127:e117–25. doi: 10.1542/peds.2010-0192. [PMID: 21173003] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunt A, Lindenbaum RH. Tuberous sclerosis: a new estimate of prevalence within the Oxford region. J Med Genet. 1984;21:272–7. doi: 10.1136/jmg.21.4.272. [PMID: 6492092] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sampson JR, Scahill SJ, Stephenson JB, Mann L, Connor JM. Genetic aspects of tuberous sclerosis in the west of Scotland. J Med Genet. 1989;26:28–31. doi: 10.1136/jmg.26.1.28. [PMID: 2918523] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shepherd CW, Beard CM, Gomez MR, Kurland LT, Whisnant JP. Tuberous sclerosis complex in Olmsted County, Minnesota, 1950-1989. Arch Neurol. 1991;48:400–1. doi: 10.1001/archneur.1991.00530160068015. [PMID: 2012513] [DOI] [PubMed] [Google Scholar]
- 12.Webb DW, Fryer AE, Osborne JP. Morbidity associated with tuberous sclerosis: a population study. Dev Med Child Neurol. 1996;38:146–55. doi: 10.1111/j.1469-8749.1996.tb12086.x. [PMID: 8603782] [DOI] [PubMed] [Google Scholar]
- 13.O’Callaghan FJ, Shiell AW, Osborne JP, Martyn CN. Prevalence of tuberous sclerosis estimated by capture-recapture analysis [Letter] Lancet. 1998;351:1490. doi: 10.1016/S0140-6736(05)78872-3. [PMID: 9605811] [DOI] [PubMed] [Google Scholar]
- 14.Neumann HP, Schwarzkopf G, Henske EP. Renal angiomyolipomas, cysts, and cancer in tuberous sclerosis complex. Semin Pediatr Neurol. 1998;5:269–75. doi: 10.1016/s1071-9091(98)80005-3. [PMID: 9874854] [DOI] [PubMed] [Google Scholar]
- 15.Hancock E, Tomkins S, Sampson J, Osborne J. Lymphangioleiomyomatosis and tuberous sclerosis. Respir Med. 2002;96:7–13. doi: 10.1053/rmed.2001.1206. [PMID: 11863212] [DOI] [PubMed] [Google Scholar]
- 16.Costello LC, Hartman TE, Ryu JH. High frequency of pulmonary lymphangioleiomyomatosis in women with tuberous sclerosis complex. Mayo Clin Proc. 2000;75:591–4. doi: 10.4065/75.6.591. [PMID: 10852420] [DOI] [PubMed] [Google Scholar]
- 17.Franz DN, Brody A, Meyer C, Leonard J, Chuck G, Dabora S, et al. Mutational and radiographic analysis of pulmonary disease consistent with lymphangioleiomyomatosis and micronodular pneumocyte hyperplasia in women with tuberous sclerosis. Am J Respir Crit Care Med. 2001;164:661–8. doi: 10.1164/ajrccm.164.4.2011025. [PMID: 11520734] [DOI] [PubMed] [Google Scholar]
- 18.Moss J, Avila NA, Barnes PM, Litzenberger RA, Bechtle J, Brooks PG, et al. Prevalence and clinical characteristics of lymphangioleiomyomatosis (LAM) in patients with tuberous sclerosis complex. Am J Respir Crit Care Med. 2001;164:669–71. doi: 10.1164/ajrccm.164.4.2101154. [PMID: 11520735] [DOI] [PubMed] [Google Scholar]
- 19.Muzykewicz DA, Sharma A, Muse V, Numis AL, Rajagopal J, Thiele EA. TSC1 and TSC2 mutations in patients with lymphangioleiomyomatosis and tuberous sclerosis complex [Letter] J Med Genet. 2009;46:465–8. doi: 10.1136/jmg.2008.065342. [PMID: 19419980] [DOI] [PubMed] [Google Scholar]
- 20.O’Callaghan FJ, Noakes MJ, Martyn CN, Osborne JP. An epidemiological study of renal pathology in tuberous sclerosis complex. BJU Int. 2004;94:853–7. doi: 10.1111/j.1464-410X.2004.05046.x. [PMID: 15476522] [DOI] [PubMed] [Google Scholar]
- 21.Lendvay TS, Marshall FF. The tuberous sclerosis complex and its highly variable manifestations. J Urol. 2003;169:1635–42. doi: 10.1097/01.ju.0000058253.40352.60. [PMID: 12686801] [DOI] [PubMed] [Google Scholar]
- 22.Seyam RM, Bissada NK, Kattan SA, Mokhtar AA, Aslam M, Fahmy WE, et al. Changing trends in presentation, diagnosis and management of renal angiomyolipoma: comparison of sporadic and tuberous sclerosis complex-associated forms. Urology. 2008;72:1077–82. doi: 10.1016/j.urology.2008.07.049. [PMID: 18805573] [DOI] [PubMed] [Google Scholar]
- 23.Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc. 1991;66:792–6. doi: 10.1016/s0025-6196(12)61196-3. [PMID: 1861550] [DOI] [PubMed] [Google Scholar]
- 24.Roach ES, DiMario FJ, Kandt RS, Northrup H. Tuberous Sclerosis Consensus Conference: recommendations for diagnostic evaluation. National Tuberous Sclerosis Association. J Child Neurol. 1999;14:401–7. doi: 10.1177/088307389901400610. [PMID: 10385849] [DOI] [PubMed] [Google Scholar]
- 25.Ewalt DH, Diamond N, Rees C, Sparagana SP, Delgado M, Batchelor L, et al. Long-term outcome of transcatheter embolization of renal angiomyolipomas due to tuberous sclerosis complex. J Urol. 2005;174:1764–6. doi: 10.1097/01.ju.0000177497.31986.64. [PMID: 16217279] [DOI] [PubMed] [Google Scholar]
- 26.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–51. doi: 10.1056/NEJMoa063564. [PMID: 18184959] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, et al. the National Institutes of Health Rare Lung Diseases Consortium and the MILES Trial Group Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364:1595–606. doi: 10.1056/NEJMoa1100391. [PMID: 21410393] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taveira-DaSilva AM, Hathaway O, Stylianou M, Moss J. Changes in lung function and chylous effusions in patients with lymphangioleiomyomatosis treated with sirolimus. Ann Intern Med. 2011;154:797–805. doi: 10.1059/0003-4819-154-12-201106210-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–11. doi: 10.1056/NEJMoa1001671. [PMID: 21047224] [DOI] [PubMed] [Google Scholar]
- 30.Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. 1998;13:624–8. doi: 10.1177/088307389801301206. [PMID: 9881533] [DOI] [PubMed] [Google Scholar]
- 31.Moss J, DeCastro R, Patronas NJ, Taveira-DaSilva A. Meningiomas in lymphangioleiomyomatosis. JAMA. 2001;286:1879–81. doi: 10.1001/jama.286.15.1879. [PMID: 11597290] [DOI] [PubMed] [Google Scholar]
- 32.Pulsifer MB, Winterkorn EB, Thiele EA. Psychological profile of adults with tuberous sclerosis complex. Epilepsy Behav. 2007;10:402–6. doi: 10.1016/j.yebeh.2007.02.004. [PMID: 17392032] [DOI] [PubMed] [Google Scholar]
- 33.Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51:1236–41. doi: 10.1111/j.1528-1167.2009.02474.x. [PMID: 20041940] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Au KS, Williams AT, Gambello MJ, Northrup H. Molecular genetic basis of tuberous sclerosis complex: from bench to bedside. J Child Neurol. 2004;19:699–709. doi: 10.1177/08830738040190091101. [PMID: 15563017] [DOI] [PubMed] [Google Scholar]
- 35.Kerr LA, Blute ML, Ryu JH, Swensen SJ, Malek RS. Renal angiomyolipoma in association with pulmonary lymphangioleiomyomatosis: forme fruste of tuberous sclerosis? Urology. 1993;41:440–4. doi: 10.1016/0090-4295(93)90504-4. [PMID: 8488612] [DOI] [PubMed] [Google Scholar]
- 36.Siegel JM. Proceedings: tuberous sclerosis (forme fruste) vs. Cowden syndrome. Arch Dermatol. 1974;110:476–7. doi: 10.1001/archderm.110.3.476b. [PMID: 4451408] [DOI] [PubMed] [Google Scholar]
- 37.Qin W, Kozlowski P, Taillon BE, Bouffard P, Holmes AJ, Janne P, et al. Ultra deep sequencing detects a low rate of mosaic mutations in tuberous sclerosis complex. Hum Genet. 2010;127:573–82. doi: 10.1007/s00439-010-0801-z. [PMID: 20165957] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dabora SL, Roberts P, Nieto A, Perez R, Jozwiak S, Franz D, et al. Association between a high-expressing interferon-gamma allele and a lower frequency of kidney angiomyolipomas in TSC2 patients. Am J Hum Genet. 2002;71:750–8. doi: 10.1086/342718. [PMID: 12192641] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Camposano SE, Greenberg E, Kwiatkowski DJ, Thiele EA. Distinct clinical characteristics of tuberous sclerosis complex patients with no mutation identified. Ann Hum Genet. 2009;73:141–6. doi: 10.1111/j.1469-1809.2008.00496.x. [PMID: 19133941] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sparling JD, Hong CH, Brahim JS, Moss J, Darling TN. Oral findings in 58 adults with tuberous sclerosis complex. J Am Acad Dermatol. 2007;56:786–90. doi: 10.1016/j.jaad.2006.11.019. [PMID: 17239986] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aldrich CS, Hong CH, Groves L, Olsen C, Moss J, Darling TN. Acral lesions in tuberous sclerosis complex: insights into pathogenesis. J Am Acad Dermatol. 2010;63:244–51. doi: 10.1016/j.jaad.2009.08.042. [PMID: 20462663] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avila NA, Dwyer AJ, Rabel A, Darling T, Hong CH, Moss J. CT of sclerotic bone lesions: imaging features differentiating tuberous sclerosis complex with lymphangioleiomyomatosis from sporadic lymphangioleiomymatosis. Radiology. 2010;254:851–7. doi: 10.1148/radiol.09090227. [PMID: 20177097] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson SR. The ERS guidelines for LAM: trying a rationale approach to a rare disease. Respir Med. 2010;104(Suppl 1):S33–41. doi: 10.1016/j.rmed.2010.03.015. [PMID: 20451364] [DOI] [PubMed] [Google Scholar]