

Abstract
Traditional chemotherapy has resulted in only a modest response if any for the three most common cutaneous malignancies of basal cell carcinoma (BCC), squamous cell carcinoma (SCC), and melanoma. Recent advances in understanding of the defects in the pathways driving tumorigenesis have changed the way that we think of these cancers and paved the way to targeted therapy for specific tumors. In this review, we will introduce the novel systemic treatments currently available for these cancers in the context of what is understood about the tumor pathogenesis. We will also introduce ongoing studies that will hopefully broaden our options for highly effective and tolerable treatment.
Keywords: Melanoma, basal cell carcinoma, squamous cell carcinoma, chemotherapy, pathway inhibitor, immunotherapy
INTRODUCTION
The objective of this review is to discuss the novel systemic treatments available for the management of metastatic basal cell carcinoma (BCC), squamous cell carcinoma (SCC), and melanoma. Although surgical excision is the gold standard treatment for all of these cutaneous malignancies, extensive locally destructive or metastatic disease still poses a therapeutic challenge and treatments are rarely curative. Traditional treatment is highly toxic and the non-specificity of the mechanism of action makes it impossible to determine who will respond to treatment. The advent of molecular targeted therapy is changing the therapeutic landscape for these diseases with increased therapeutic index and in many cases with a more tolerable toxicity profile.
Basal Cell Carcinoma
BCC is the most common form of skin cancer with an incidence rate that is 4 to 5 times more likely than SCC. It is typically slow-growing, but if left untreated local invasion may occur, leading to destruction, disfigurement, and rarely metastasis. The options available for treatment of local disease include surgery, destruction, radiation, topical immunomodulation, and topical chemotherapy. Locally advanced BCC may invade underlying muscle, bone, or other contiguous structures. Metastatic disease is rare but can be life threatening. In those cases in which local modalities are insufficient, systemic therapy is warranted. There have been variable successes with cisplatinum-based chemotherapy regimens in the past.1
Recent advances in the understanding of the pathogenesis of BCC have led to the development of therapeutics targeting the biological mechanism driving this malignancy. The Hedgehog (Hh) pathway has been shown to play a key role in the pathogenesis of BCC with the majority of BCC bearing mutations in genes in this developmental pathway. The majority of mutations implicated in BCC pathogenesis involve mutations in the transmembrane proteins with loss of function of patched homologue 1 (Ptch1) or gain of function of smoothened homolog (Smo).2 Mutation in the Ptch1 gene was initially implicated as the cause of the rare autosomal dominant heritable basal cell nevus syndrome (Gorlin syndrome), the hallmark of which is a high susceptibility for the development of BCCs.3-4 It was later found that essentially all BCCs harbored mutation in the Ptch1 gene or other alterations in the Hh signaling pathway.5
Ptch1 is a keratinocyte membrane protein that binds Sonic Hedgehog. In the absence of Sonic Hedgehog, the role of normal Ptch1 is to inhibit Smo. Smo enables the activation of a family of transcription factors called Gli which then enter the nucleus to promote expression of more Gli, Ptch1, as well as other apoptotic factors, and suppression of genes associated with keratinocyte differentiation. This sequence of events leads to cell proliferation and increased survival.
Overexpression of Ptch1, Smo, Gli1, and Gli2 are associated with BCC.6 This makes the Hh pathway both an attractive and logical target for molecular inhibitors for treatment of BCC. A number of hedgehog pathway inhibitors (HPI) are under development in both oral and topical formulations. Currently, all are HPI against Smo.
Vismodegib (Genentech/Roche), previously known as RG3616 or GDC-0449, is the first of the oral small molecule HPI against Smo to be FDA approved for locally advanced or metastatic BCC. In phase I testing, 18/33 patients with locally advanced or metastatic BCC showed a response to the drug and 11 treated patients had stable disease (SD) over a median follow-up of 9.8 months. Measurement of Gli in treated tumors was lower, demonstrating a down-regulation of the Hh pathway thus confirming the molecular mechanism of action.7 In a phase II trial, looking at locally advanced BCC not amenable for surgery or radiation and metastatic BCC, the overall response rate (ORR) was 43% in the locally advanced group and 30% in the metastatic group. The median duration of progression-free survival (PFS) for both groups was 9.5 months.8
LDE225 (Novartis) is another oral HPI targeting Smo. In a phase I dose-escalation study in solid tumors, LDE225 was found to have a dose-dependent inhibition of the Hh pathway, which was measured by downregulation of Gli-1 expression. Although the trial was not designed to test for efficacy, it is notable that only 1 of 7 subjects with BCC progressed while on treatment.9 Phase II trials are currently underway. Other systemic HPIs are currently in development. These include IPI-926 (Infinity Pharmaceuticals), TAK-441 (Millennium Pharmaceuticals), PF-04449913 (Pfizer), LEQ506 (Novartis), and BMS-833923 (Bristol-Myers Squibb).
Topical delivery of HPIs is also under investigation and poses an attractive option in terms of side effect profile. CUR61414 (Curis/Genentech/Roche), was shown to be effective in preclinical models but failed to have clinical activity in superficial or nodular BCCs in a phase I clinical study in humans.10 LDE225 was also formulated as a topical cream. In a randomized, vehicle-controlled, intra-individual trial in subjects with basal cell nevus syndrome, topical LDE225 resulted in clinical responses in 12 of 13 BCCs studied, while tumors treated with vehicle alone showed no efficacy.11 Although these are promising results, further studies will be needed to test if the findings are generalizable for patients with BCC without the syndrome.
Locally advanced and metastatic BCC portends a grave prognosis. The standard of care remains the cisplatin-based chemotherapy. However the landscape of treatment for BCC is on the cusp of changing with the advent of HPIs and targeted molecular approach to treatment and hopefully the prognosis for advanced disease will improve as well.
Melanoma
Melanoma is a devastating disease once metastatic and is the leading cause of skin cancer death.12 Systemic treatment for metastatic melanoma includes chemotherapy, immunotherapy, and more recently targeted therapy. Chemotherapy has been the standard of care for stage IV non-resectable melanoma with only a modest response and no improvement in overall survival.13 With the advent of targeted therapy, we now understand melanoma to be a heterogeneous entity with responses to treatment dependent upon genetic status. Therefore, management of systemic disease now necessitates obtaining genetic analysis prior to a discussion of the options available for an individual patient.
Immunotherapy
Immunotherapy has long been known to play an important role in controlling melanoma and has been used in the adjuvant setting. Interferon alpha 2b and more recently GM-CSF have been shown to improve progression-free and overall survival in high-risk melanoma in the adjuvant setting. For metastatic melanoma, immune therapy options include interleukin-2, ipilimumab, and interleukin-12.
High dose interleukin-2 can be highly efficacious in a very limited subset of patients with an ORR of 16% and 6-8% complete response rate.14 However, it causes significant toxicities and adverse events requiring intensive monitoring such as hypotension, renal insufficiency, hepatocellular damage, edema, respiratory compromise, myocardial infarction, sepsis, and death. Given the side effect profile, it is often a treatment reserved only for young and fit patients.15 There are currently no biomarkers to determine who could likely benefit from treatment, however it has been found that patients with disease limited to subcutaneous tissue and those who are able to receive more dosages have been more likely to achieve an objective response.16
Ipilimumab (Bristol-Myers Squibb) is a CTLA-4 antibody and has been recently FDA approved for the treatment of metastatic melanoma. CTLA-4 competes for binding of a surface protein B7, thus inhibiting T-cell proliferation and release of immune stimulatory cytokines. Ipilimumab blocks CTLA-4, thus taking the proverbial brakes off the immune system and allowing the immune system to act against melanoma.17 In a phase 3 study comparing ipilimumab, with or without glycoprotein 100 (gp100) peptide vaccine, to gp100 alone in patients with previously treated melanoma, there was a statistically significant improved survival associated with treatment with ipilimumab. Median OS in the ipilimumab groups were not different and was about 10 months compared to 6.1 months for the gp100 alone group. A major drawback of this treatment has been the lag in treatment response.18
IL-12 is a heterodimeric cytokine that regulates both innate and adaptive immune response.19-20 It has been shown to enhance the killing of tumor cells by tumor-infiltrating lymphocytes in patients with melanoma.21-22 Local delivery of IL-12 via direct intratumoral injection of IL-12 plasmid DNA is well-tolerated and has been shown to result in local effects in the treated tumor but no systemic effect.23 Phase I and II trials of systemic IL-12 have been reported with responses in melanoma but is associated with significant toxicity.24-26 More recently, a phase I study of intratumoral electroporation of a DNA plasmid expressing IL-12 into melanoma lesions has been shown to result in regression of untreated metastases in 10 of 19 evaluable patients and is not associated with significant side effects.27 Future studies are proposed.
Although immunotherapy has been shown to make an impact in melanoma beyond just the adjuvant setting, it is still poorly understood why subsets of patients have better response than others. Further investigations will need to be made to determine why responders respond and possibly what can be done to convert a non-responder to a responder. As targeted therapies become the standard of care in melanoma, immunotherapy has become second line treatment reserved for patients who have failed targeted treatment or for those who do not qualify for targeted treatment. Studies are underway combining immunotherapy with targeted therapy such as vemurafenib, a BRAF inhibitor, to potentially boost the efficacy of both.
Therapies targeting molecular signaling
There are 2 main pathways currently recognized to play a role specifically in melanoma pathogenesis. These are the mitogen activated protein kinase (MAPK) pathway and the phosphatidylinositol 3-kinase (PI3K) pathway. Recognizing these pathways and developing drugs that target specific points that are dysregulated in the pathways have several advantages. The specificity allows for targeted treatment with fewer side effects. And because responders are chosen based on their genetic status, it becomes possible to predict clinical response without subjecting non-responders to treatment that will not be effective for them.
MAPK Signaling Pathway
MAPK signaling is initiated by binding of receptor tyrosine kinases, which then lead to activation of Ras, a small G protein on the inner surface of the cell membrane. Once activated, Ras can form complexes with Raf, which then leads to phosphorylation of ERK via activation of MEK. ERK can directly enter the nucleus and effect translation of genes and control cellular proliferation.28
BRAF
Mutation in the BRAF gene occurs in about 66% of melanoma tumors and are commonly found in non-chronically sun-exposed skin.29 BRAF mutation is uncommon in acral-lentiginous melanoma, but we have observed several cases bearing the BRAF mutation at our institution. The BRAF mutation confers increased kinase activity that can lead to increased tumor proliferation.30 Inhibition of the mutated BRAF gene has been shown to be the most effective treatment for melanoma at this time and should be considered first-line treatment for melanoma bearing this mutation. In a recently published phase III trial of oral vemurafenib (Genentech/Roche), a new recently FDA approved BRAF selective inhibitor for metastatic patients bearing the BRAF V600E mutation, showed a 48% response rate for vemurafenib compared to a 5% response rate for dacarbazine. A 6-month interim analysis showed a 63% reduction in risk of death and a 74% reduction in risk of death and disease progression in the vemurafenib versus the dacarbazine group (P<.0001 for both comparisons). Another favorable aspect of treatment with vemurafenib is its relatively benign side effect profile.31 Common side effects of vemurafenib include rash, fatigue, arthralgia, alopecia, photosensitivity, nausea, diarrhea, keratoacanthoma or SCC and to a lesser extent liver function abnormalities and renal insufficiency.
The recent FDA approval of vemurafenib has changed the landscape for management of metastatic melanoma and has caused significant excitement in the melanoma community. Unfortunately, what is not clearly understood is why there is variable response to the medication despite presence of the BRAF mutation, implying that other important factors play a role in melanoma tumorigenesis. Also, among the initial responders, most patients eventually progress on treatment. Some mechanisms of resistance have been proposed but have thus far not been validated.32
MEK
MEK is a downstream target of Raf in the signaling cascade. It has been shown to have mixed results in melanoma. A phase I trial with AZD6244 (AstraZeneca) showed tumor shrinkage in 6 out of 11 patients.33 However a subsequent phase II trial showed PR in some patients, mainly those with BRAF mutations, but there was no benefit in PFS when compared to temozolomide, which is an oral alkylating chemotherapy commonly used in melanoma.34 MEK in combination with temozolomide, docetaxel, or temsirolimus has been shown to be associated with tumor regression in only BRAF mutants and delayed progression in BRAF and NRAS mutants.35 This suggests possibly that MEK plays a bigger role in tumorigenesis in BRAF mutants than NRAS mutants. Studies are currently underway comparing AZD6244 in combination with dacarbazine (in BRAF mutant melanoma only)36 or docetaxel versus chemotherapy alone.37 Two other studies hope to compound the MEK inhibiting effects of AZD6244 by targeting parallel growth pathways with an mTOR and VEGF inhibitor temsirolimus (BRAF mutant melanoma only)38 or a VEGF inhibitor cediranib.39 A newer MEK inhibitor GSK1120212 is currently under investigation in a phase I trial in combination with a BRAF inhibitor GSK2118436 for metastatic BRAF mutant melanoma.40 It is showing great promise, indicating possibly that dual targets in the same pathway are more effective than a single one.
PI3K/AKT Pathway
The PI3 kinase (PI3K) pathway is a prosurvival pathway, antagonizing apoptosis. PI3K is activated by growth factor receptors. It has 2 actions: to regulate cell proliferation via control of entry into the cell cycle and to activate AKT via PDK1. AKT then directly activates transcription factors that cause transcription of prosurvival genes. The PI3K/AKT pathway is constitutively activated in melanoma although mutations in AKT are found in only a small proportion of melanomas.41 Mammalian target of rapamycin (mTOR) is a serine/threonine kinase downstream in this pathway that leads to increased cell growth. Increased activation of mTOR was found in 73% of melanoma cell lines.42 This pathway can be opposed by PTEN.43 Mutation in PTEN has been found in 11% of melanoma tumors44 and 43% of melanoma cell lines.45 A number of inhibitors of this pathway are in the early stages of development, all targeting this pathway from different angles. Thus far, none of the inhibitors of this pathway have demonstrated an objective response.
Perifosine (Aeterna Zentaris) is an AKT inhibitor, inhibiting AKT phosphorylation and translocation to the cell membrane. Unfortunately, a phase II trial using this drug in metastatic melanoma showed only stabilization of disease in 3 out of 14 patients and was associated with side effects requiring missed, delayed, or reduced dose in all patients.46
UCN-01 (Kyowa Hakko Kogyo) is an inhibitor of PDK-1, whose role is activation of AKT thus leading to decreased apoptosis. A phase I trial demonstrated PR in one patient on this medication. A subsequent phase II trial of UCN-01 in metastatic melanoma accrued 16 evaluable patients with 4 patients demonstrating SD and 12 progressive disease. Median PFS was 1.3 months and median OS was 7.3 months. It was relatively well-tolerated.47
Various mTOR inhibitors are also being evaluated in patients with melanoma. Temsirolimus, CCL-779, (Wyeth) is an mTOR inhibitor that has been tested in a melanoma phase II clinical trial with disappointing results with only one PR lasting just 2 months.48 Everolimus (Abbott) is another mTOR inhibitor with dual activity against EGFR. It is currently under investigation in a phase II trial which has thus far demonstrated 7 out of 24 patients with stabilization of disease in an interim analysis.49
Dual pathway inhibition
It is believed that both the MAPK and the PI3K/AKT pathways play key roles in melanoma tumorigenesis. Inhibition of the PI3K/AKT pathway alone has been disappointing without objective response. Raf inhibition, on the other hand, is limited to those bearing the BRAF mutation and is not durable in the majority of cases. There is hope that using a combinatorial approach with inhibitors in both pathways will have an additive positive effect. In cell culture, the combination of sorafenib, a non-selective Raf inhibitor, with sirolimus, an mTOR inhibitor, caused a two-fold increase in apoptosis of melanoma cells relative to sorafenib alone. This was attributable to an upregulation in genes associated with endoplasmic reticulum stress-induced apoptosis.50 BEZ235 (Novartis) is another molecule with dual mTOR and PI3K inhibition that has shown greater activity than temsirolimus in preclinical melanoma models.51 There is also currently a phase Ib study underway combining BEZ235 with the MEK inhibitor MEK162.52
KIT
Kit is a cytokine receptor that belongs to the type III receptor tyrosine kinase family. Kit signaling plays an important role in a number of physiological processes including melanogenesis.53 Overall, this mutation is rare but is most commonly found in melanoma located on chronically sun-damaged skin, mucosa, and acral skin.54 The overall incidence rate of Kit mutant melanoma has been reported to be 8%.55
Imatinib (Novartis) was the earliest Kit inhibitor tested in clinical trials for melanoma. Two previous trials in which imatinib was tested for efficacy against melanoma demonstrated no treatment response.56-57 However, it is important to note that the patients were not tested for Kit mutation and it was determined that most selected patients had tumors that demonstrated little to no Kit expression by immunohistochemistry. A recent phase II open-label, single-arm trial using imatinib only in Kit mutant metastatic melanoma recruited a total of 43 patients and resulted in 23 CR, 13 PR, and 10 patients with SD. The median PFS was 3.5 months.58 Based on this study, it can be concluded that Kit inhibitors can play an important role in the armamentarium against selected melanoma bearing this mutation. Multiple trials are ongoing using newer tyrosine kinase inhibitors such as nilotinib, sunitinib, dasatinib, and masitinib against Kit mutated melanoma.
c-MET
c-Met is a receptor tyrosine kinase that is activated by its ligand hepatocyte growth factor and is essential for normal development, cell migration, growth, survival, differentiation, and angiogenesis.59 In normal skin, c-Met is expressed on epithelial cells and melanocytes, whereas hepatocyte growth factor is produced mainly by mesenchymal cells and interacts with c-Met in a paracrine manner.60 c-Met has been found to be expressed in 88% of melanomas61 with overexpression correlating with the invasive growth of melanoma cells. Many melanomas also secrete hepatocyte growth factor, which can induce sustained activation of c-Met in an autocrine fashion.62 Cabozantinib, XL184, (Exelixis) is a c-Met and VEGFR2 inhibitor found in a phase II randomized discontinuation trial of patients with advanced solid tumors that demonstrated a 5% objective response rate in melanoma. Patients with bony metastases from pancreatic, breast, or melanoma had an objective response of 87%.63 Cabozantinib is currently in a phase II trial among patients with various solid tumors, including melanoma.64 Foretinib, XL880, (GlaxoSmithKline) is another c-Met/VEGFR2 inhibitor that also has shown some objective response activity in patients with melanoma in a phase I trial.65
Epidermal Growth Factor Receptor
The epidermal growth factor receptor (EGFR) is a member of a family of transmembrane protein kinase receptors which consists of the 4 receptors: EGFR (HER1 or ErbB1), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4).66 The EGFR gene resides on chromosome 7. Several different ligands activate these receptors, which then relay signals to the parallel MAPK and PI3K pathways leading to growth effects, angiogenesis, migration, and invasion. EGFR has been shown to play an important role in the growth and survival of many tumors including cutaneous malignancies. Anti-EGFR agents are monoclonal antibodies directed at the extracellular domain of the receptor and low-molecular weight adenosine triphosphate (ATP)-competitive inhibitors of the receptor’s intra-cellular tyrosine kinase (TKI).
ErbB1 has been found to be expressed in up to 96% of primary melanomas and in 90% of metastatic tumors.67 There are gains in chromosome 7, where EGFR resides, in about 50% of melanomas, and increased copy number of chromosome 7 has been associated with poor prognosis in some studies.68-69 ErbB3 is also frequently expressed in melanoma and has been associated with tumor progression and a worse prognosis.70-72 Evidence of the importance of EGFR signaling has been seen in melanoma cell lines70 as well as in animal models.73 A screen for somatic mutations in ErbB4 revealed that 19% of metastatic tumors harbored this mutation.74
The ErbB1 inhibitor erlotinib hydrochloride (Genentech) has been evaluated in a phase II trial of metastatic melanoma and showed no objective responses, but 4 of 14 patients had SD.75 The ErbB1/B2 inhibitor lapatinib is currently under investigation for melanoma bearing the ErbB4 mutation after preclinical data suggests its effectiveness in ErbB4 mutant melanoma. The ErbB1/B2 inhibitor gefitinib (AstraZeneca) was tested, and only 2 of 50 evaluable patients had PR.76 A trial of erlotinib hydrochloride in combination with the vascular endothelial growth factor A inhibitor bevacizumab (Genentech/Roche) showed greater efficacy, with 2 of 23 patients having PR lasting less than 6 months and 5 patients having SD lasting greater than 6 months.77 Toxic effects were greater with this combination, with 1 patient each experiencing myocardial infarction and bowel perforation.
Vascular Endothelial Growth Factor Receptor
Angiogenesis plays a major role in tumor growth. Targeting the vascular endothelial growth factor receptor (VEGF) makes logical sense and has been shown to affect tumor growth by inhibition of angiogenesis. Although this is considered targeted therapy, it does not specifically target melanoma cells. Among the VEGF inhibitors, Bevacizumab is perhaps the most extensively studied anti-VEGF antibody. It is under investigation in combination with immunotherapy, chemotherapy, and other targeted treatments in metastatic melanoma. It appears to have a synergistic effect when used in conjunction with other systemic modalities.
Bevacizumab with immunotherapy
Phase II trial of bevacizumab and high-dose interferon alpha-2b, which has antiangiogenic properties via down-regulation of basic-fibroblast growth factor, in metastatic melanoma resulted in a median PFS of 4.8 months and OS of 17 months as compared to historical control of bevacizumab alone with PFS of 3 months and OS of 8.5 months. Six patients had PR and 5 patients had SD for greater than 24 weeks.78 In a phase I trial of bevacizumab and ipilimumab for stage III or IV melanoma, of the 21 patients who were evaluable, there were 8 PR, all of whom had durable responses greater than 6 months, and 6 SD. Post-treatment biopsies showed activated vessel endothelium with extensive T-cell trafficking, which were not seen in patients treated with ipilimumab alone. These results suggest a synergistic effect of VEGF and CTLA4 blockade.79
Bevacizumab with chemotherapy
There have been many trials looking at bevacizumab used in conjunction with various chemotherapeutic regimens which include temozolomide, nab-paclitaxel, carboplatin/paclitaxel, and dacarbazine. They have shown a modestly improved PFS and/or OS relative to previously reported survival for bevacizumab alone.80-84
Bevacizumab with other targets
It is still too early to say that bevacizumab has a synergistic effect when used in conjunction with other targeted treatment. In a phase II trial of bevacizumab and everolimus, median PFS was 4 months and OS was 8.6 months.85 A triple combination trial of temozolomide, sorafenib (Raf kinase inhibitor), and bevacizumab of 11 patients with refractory acral advanced melanoma, resulted in 1 CR, 2 PR, and 6 SD.86 However an interim report of another triple combination regimen of bevacizumab, oxaliplatin, and sorafenib in a phase I/II trial accruing 6 patients showed 1 PR, 3 mixed response, and 3 progression of disease.87 Additionally, there are trials ongoing combining dasatinib and bevacizumab.
Ranibizumab
Ranibizumab (Genentech) is a monoclonal antibody fragment derived from the same parent mouse antibody as bevacizumab. It is much smaller than the parent molecule and has been affinity matured to provide stronger binding to VEGF-A. There are currently multiple trials using this molecule for choroidal and uveal melanoma as an adjuvant for tumor control or for control of radiation retinopathy or maculopathy.
Squamous Cell Carcinoma
SCC has been shown to have an increased expression of EGFR, with about 92-100% of SCC demonstrating binding to EGFR antibody.88-89 High EGFR signaling has been associated with aggressive disease, poor response to therapy, increased development of resistance to cytotoxic chemotherapy, poor survival, and poor prognosis.90 Another study found that primary SCC tumors were immunohistochemically focally weakly positive for EGFR while metastatic SCCs were diffusely strongly positive, suggesting that stronger expression of EGFR had a higher potential for metastasis.91
Cetuximab (Merck) is an anti-EGFR monoclonal antibody approved by the FDA for SCC of the head and neck. It binds with higher affinity than natural ligands TGF-α and EGF. Cetuximab inhibits progression in the cell cycle at the G0/G1 phase, increases expression of the cell cycle regulator p27KIP1, and induces apoptosis by increasing expression of pro-apoptotic proteins or by inactivation of anti-apoptotic proteins.92 It can also inhibit angiogenesis via inhibition of VEGF, interleukin-8, and basic fibroblast growth factor.93 Cetuximab has been shown to be effective in case reports for recurrent non-resectable squamous cell carcinoma as well as metastatic disease.94-96 There have been several phase I and II trials using cetuximab in combination with platinum-based chemotherapy documenting safety and efficacy in the combination regimen in metastatic, recurrent, or refractory SCC of the head and neck.97-99 However, larger studies have not been done documenting effectiveness as a monotherapy. Predictive biomarkers for success with cetuximab include presence of EGFR in the tumor and wild-type for K-Ras100 and BRAF101. The rationale is that these mutations constitutively activate the downstream MAPK pathway that is independent of EGFR activity.
Panitumumab, ABX-EGF, (Amgen) is a human IgG2 monoclonal antibody against EGFR that binds to EGFR like cetuximab. Phase I trials have shown it to be well-tolerated and efficacious in colorectal carcinoma and non-small cell lung cancer. An open-label phase II trial is currently underway to study the clinical efficacy in SCC. Matuzumab, EMD 72000, (Merck/Takeda) is a humanized IgG1 monoclonal antibody against EGFR. It has been shown to have tumor response against esophageal SCC, cervical carcinoma, ovarian carcinoma, colorectal carcinoma, and head and neck SCC.102 A phase II trial of matuzumab in patients with platinum-resistant ovarian carcinoma showed that matuzumab was well-tolerated and demonstrated evidence of anti-tumor activity.103 There are currently no studies at this time using this drug in the treatment of non-melanoma skin cancer.
EGFR TKI and Non-melanoma Skin Cancer
TKIs are synthetic low molecular weight molecules that interact with the intracellular tyrosine kinase domain of several receptors including EGFR. They inhibit ligand-induced receptor phosphorylation by competing for intracellular Mg-ATP-binding sites.104 Gefitinib has been shown to inhibit EGFR and MAPK activation and Pak 1 activity in exponentially growing cutaneous squamous carcinoma cells. It has been approved for the treatment of non-squamous cell lung cancer after platinum-based or docetaxel based therapy failure and has been shown to have modest activity in advanced skin SCC.105 Erlotinib is a potent reversible, selective inhibitor of EGFR (ErbB1)106 which is also approved for non-squamous cell lung cancer but has demonstrated effectiveness in other cancers as well.107 There are several studies underway to investigate its utility in cutaneous SCC in combination with radiotherapy, as an adjuvant, or as a neoadjuvant therapy.
Conclusion
Management of cutaneous malignancy has entered a new era. Understanding the molecular basis for tumorigenesis has paved the way towards development of new molecules that inhibit at critical sites necessary for neoplastic growth and survival. Some agents have shown startling efficacy and yet others have equally surprised with their lack of efficacy, making the poignant message that although the frontier of our understanding has advanced significantly, the entire story is yet to be told.
Figure 1.
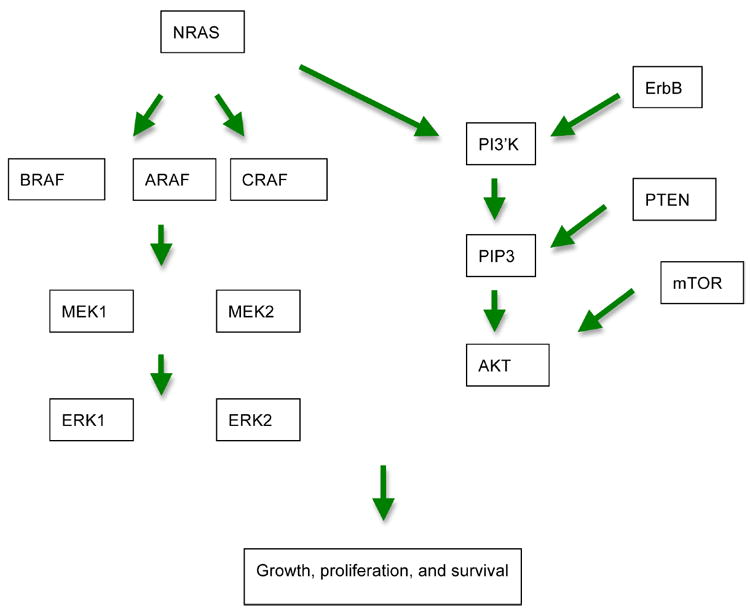
Simplified schematic for pathways involved in melanoma tumorigenesis.
Figure 2.
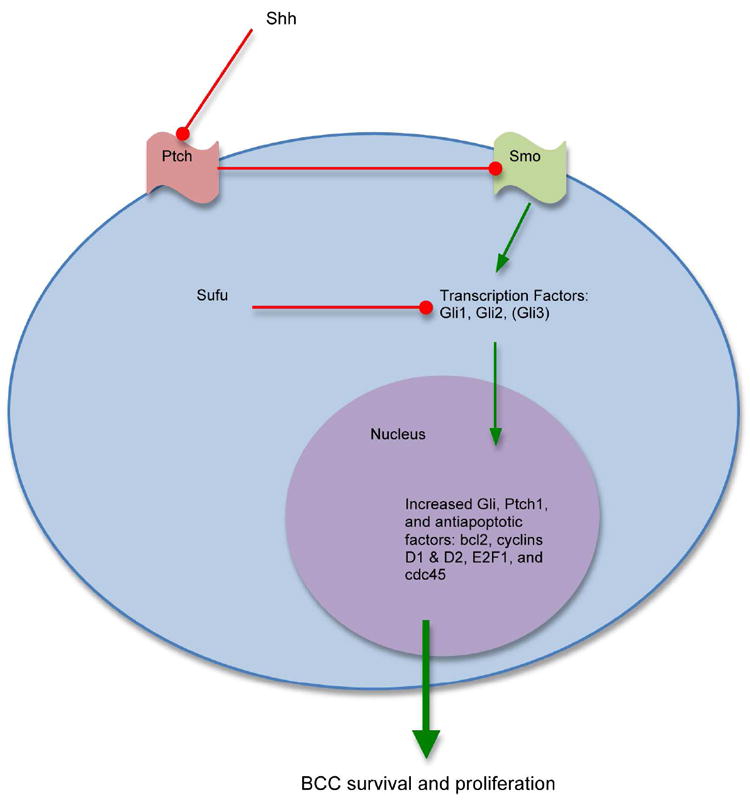
Hedgehog pathway in the pathogenesis of basal cell carcinoma.
Systemic Therapy for Melanoma
| Class | Medication | Mechanism of action |
|---|---|---|
| Immunotherapy | High dose IL-2 | IL-12 mediated killing of tumor via tumor-infiltrating lymphocytes |
| Ipilimumab | CTLA-4 antibody | |
| Intratumoral electroporation of IL-12 | Gene transfer using in vivo DNA electroporation of IL-12 leading to IL-12 mediated killing of tumor | |
| Targeted therapy | Vemurafenib | BRAF inhibitor |
| GSK118436 | BRAF inhibitor | |
| Sorafenib | Non-selective RAF inhibitor | |
| Selumetib (AZD6244) | MEK inhibitor | |
| GSK1120212 | MEK inhibitor | |
| MEK162 | MEK inhibitor | |
| Sirolimus | mTOR inhibitor | |
| Temsirolimus (CCL-779) | mTOR and VEGF inhibitor | |
| Everolimus | mTOR and VEGF inhibitor | |
| BEZ235 | mTOR and PI3K inhibitor | |
| Cediranib | VEGF inhibitor | |
| Perfosine | AKT inhibitor | |
| UCN-01 | PDK-1 inhibitor | |
| Imatinib | KIT inhibitor | |
| Nilotinib | KIT inhibitor | |
| Sunitinib | KIT inhibitor | |
| Dasatinib | KIT inhibitor | |
| Masitinib | KIT inhibitor | |
| Cabozantinib (XL184) | c-MET and VEGFR2 inhibitor | |
| Foretinib (XL880) | c-MET and VEGFR2 inhibitor | |
| Gefitinib | ErbB1/B2 inhibitor | |
| Bevacizumab | EGFR inhibitor | |
| Ranibizumab | Monoclonal antibody fragment VEGF-A inhibitor |
Systemic Therapy for Squamous Cell Carcinoma
| Medication | Mechanism of action |
|---|---|
| Cetuximab | EGFR monoclonal antibody |
| Panitumumab | IgG2 monoclonal antibody against EGFR |
| Matuzumab | Humanized IgG1 monoclonal antibody against EGFR |
| Gefitinib | EGFR TKI |
| Erlotinib | ErbB1 inhibitor |
Acknowledgments
Dr Daud’s institution has received funds for Grants from Merck, Pfizer, Glaxo-Smith Kline, Sherry and OncoSec; consulting fee or honorarium from Merck and BMS; fees for participation in review activities from Scherring, Merck and Bristol-Myers Squibb; has grants/grants pending with Merck, Scherring, Bristol-Myers Squibb, Pfizer and Oncosec; has received funds for services provided to speakers bureaus from Scherring, Bristol-Myers Squibb and Merck. Dr Daud has personally received support for travel to meetings from Scherring.
Footnotes
Conflict of Interest Disclosures: All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. All other authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pfeiffer P, Hansen O, Rose C. Systemic cytotoxic therapy of basal cell carcinoma. A review of the literature. Eur J Cancer. 1990;26:73–7. doi: 10.1016/0277-5379(90)90262-r. [DOI] [PubMed] [Google Scholar]
- 2.Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer. 2008;8:743–754. doi: 10.1038/nrc2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–51. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 4.Johnson RL, Rothman AL, Xie J, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–71. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 5.Xie J, Murone M, Luoh SM, et al. Activating smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–2. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 6.Gupta S, Takebe N, Lorusso P, et al. Targeting the hedgehog pathway in cancer. Ther Adv Med Oncol. 2010;2(4):237–50. doi: 10.1177/1758834010366430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164–72. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 8.Sekulic A, Migden MR, Oro AE, et al. A pivotal study evaluating efficacy and safety of the hedgehog pathway inhibitor (HPI) vismodegib (GDC-0449) in patients with locally advanced or metastatic basal cell carcinoma (BCC). 7th Congress of the European Association of Dermato-Oncology; Nantes, France. 2011. [Google Scholar]
- 9.Tawbi HA, Rodon Ahnert J, Dummer R, et al. Phase I study of LDE225 in advanced solid tumors: Updated analysis of safety, preliminary efficacy, and pharmacokinetic-pharmacodynamic correlation. J Clin Oncol. 2011:29. Abstract 3062. [Google Scholar]
- 10.Tang T, Tang JY, Li D, et al. Targeting superficial or nodular basal cell carcinoma with topically formulated small molecule inhibitor of smoothened. Clin Cancer Res. 2011;17:3378–87. doi: 10.1158/1078-0432.CCR-10-3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skvara H, Kalthoff F, Meingassner JG, et al. Topical treatment of basal cell carcinomas in nevoid basal cell carcinoma syndrome with a smoothened inhibitor. J Invest Dermatol. 2011;131:1735–44. doi: 10.1038/jid.2011.48. [DOI] [PubMed] [Google Scholar]
- 12.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 13.Chapman PB, Einhorn LH, Meyers ML, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999;17:2745–51. doi: 10.1200/JCO.1999.17.9.2745. [DOI] [PubMed] [Google Scholar]
- 14.Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin-2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 15.Algazi AP, Soon CW, Daud AI. Treatment of cutaneous melanoma: current approaches and future prospects. Cancer Manag Res. 2010;2:197–211. doi: 10.2147/CMR.S6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Royal RE, Steinberg SM, Krouse RS, et al. Correlates of response to interleukin-2 therapy in patients treated for metastatic renal cancer and melanoma. Cancer J Sci Am. 1996;2(2):91–98. [PubMed] [Google Scholar]
- 17.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 18.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilumumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trinchieri G. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol. 1995;13:251–276. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- 20.Hendrzak JA, Brunda MJ. Interleukin-12: biologic activity, therapeutic utility, and role in disease. Lab Investig. 1995;72:619–37. [PubMed] [Google Scholar]
- 21.Andrews JVR, Schoof DK, Bertagnolli MM, Peoples GE, Goedegebuure PS, Eberlein TJ. Immunomodulatory effects of interleukin-12 on human tumor-infiltrating lymphocytes. J Immunother. 1993;14:1–10. doi: 10.1097/00002371-199307000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Zeh HJ, III, Hurd S, Storkus WJ, Lotze MT. Interleukin-12 promotes the proliferation and cytolytic activity of immune effectors: implications for the immunotherapy of cancer. J Immunother. 1993;14:155–61. doi: 10.1097/00002371-199308000-00012. [DOI] [PubMed] [Google Scholar]
- 23.Mahvi DM, Henry MB, Albertini MR, et al. Intratumoral injection of IL-12 plasmid DNA—results of a phase I/IB clinical trial. Cancer Gene Ther. 2007;14(8):717–23. doi: 10.1038/sj.cgt.7701064. [DOI] [PubMed] [Google Scholar]
- 24.Gollob JA, Mier JW, Veenstra K, et al. Phase I trial of twice-weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: Ability to maintain IFN-gamma induction is associated with clinical response. Clin Cancer Res. 2000;6:1678–1692. [PubMed] [Google Scholar]
- 25.Alatrash G, Hutson TE, Molto L, et al. Clinical and immunologic effects of subcutaneously administered interleukin-12 and interferon alfa-2b: Phase I trial of patients with metastatic renal cell carcinoma or malignant melanoma. J Clin Oncol. 1004;22:2891–2900. doi: 10.1200/JCO.2004.10.045. [DOI] [PubMed] [Google Scholar]
- 26.Younes A, Pro B, Robertson MJ, et al. Phase II clinical trial of interleukin-12 in patients with relapsed and refractory non-Hodgkin’s lymphoma and Hodgkin’s disease. Clin Cancer Res. 2004;10:5432–5438. doi: 10.1158/1078-0432.CCR-04-0540. [DOI] [PubMed] [Google Scholar]
- 27.Daud AI, DeConti RC, Andrews S, et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol. 2008;26(36):5896–903. doi: 10.1200/JCO.2007.15.6794. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2645111/?tool=pubmed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fecher L, Amaravadi RK, Flaherty KT. The MAPK pathway in melanoma. Curr Opin Oncol. 2008;20:183–189. doi: 10.1097/CCO.0b013e3282f5271c. [DOI] [PubMed] [Google Scholar]
- 29.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. et al. PubMed. [DOI] [PubMed] [Google Scholar]
- 30.Taube JM, Begum S, Shi C, Eshleman JR, Westra WH. Benign nodal nevi frequently harbor the activating V600E BRAF mutation. Am J Surg Pathol. 2009;33(4):568–571. doi: 10.1097/PAS.0b013e31818a64fb. [DOI] [PubMed] [Google Scholar]
- 31.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMahon M. Parsing out the complexity of RAF inhibitor resistance. Pigment Cell Melanoma Res. 2011;24(2):361–5. doi: 10.1111/j.1755-148X.2010.00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adjei AA, Cohen RB, Franklin W. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26(13):2139–2146. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dummer R, Robert C, Chapman PB, et al. AZD6244 (ARRY-142886) vs temozolomide (TMZ) in patients (pts) with advanced melanoma: an open-label, randomized, multicenter, phase II study. J Clin Oncol. 2008;26(15 suppl) et al. Abstract 9033. [Google Scholar]
- 35.Patel SP, Lazar AJ, Mahoney S, et al. Clinical responses to AZD6244 (ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma. J Clin Oncol. 2010 doi: 10.1002/cncr.27790. Abstract 58. [DOI] [PubMed] [Google Scholar]
- 36.Comparison of AZD6244 in Combination with Dacarbazine Versus Dacarbazine Alone in BRAF Mutation Positive Melanoma Patients – Full Text View – ClinicalTrials.gov. [December 12, 2011]; Available at: http://clinicaltrials.gov/ct2/show/NCT00936221.
- 37.Docetaxel With or Without AZD6244 in Melanoma (DOC-MEK) – Full Text View – ClinicalTrials.gov. [December 12, 2011]; Available at: http://clinicaltrials.gov/ct2/show/NCT01256359.
- 38.Temsirolimus/AZD 6244 for Treatment-naïve With BRAF Mutant Unresectable Stage IV – Full Text View – ClinicalTrials.gov. [December 12, 2011]; Available at: http://clinicaltrials.gov/ct2/show/NCT01166126.
- 39.Cediranib Maleate and Selumetinib in Treating Patients with Solid Malignancies – Full Text View – ClinicalTrials.gov. [December 12, 2011]; Available at: http://clinicaltrials.gov/ct2/show/NCT01364051.
- 40.Investigate Saftey, Pharmacokinetics and Pharmacodynamics of GSK2118436 & GSK1120212 – Full Text View – ClinicalTrials.gov. [December 12, 2011]; Available at: http://clinicaltrials.gov/ct2/show/NCT01072175.
- 41.Stahl JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S, Robertson GP. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003;63:2881–2890. [PubMed] [Google Scholar]
- 42.Karbowniczek M, Spittle CS, Morrison T, Wu H, Henske EP. mTOR is activated in the majority of malignant melanomas. J Invest Dermatol. 2008;128(4):980–987. doi: 10.1038/sj.jid.5701074. [DOI] [PubMed] [Google Scholar]
- 43.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reifenberger J, Wolter M, Boström J, et al. Allelic losses on chromosome arm 10q and mutation of the PTEN (MMAC1) tumour suppressor gene in primary and metastatic malignant melanomas. Virchows Arch. 2000;436(5):487–493. doi: 10.1007/s004280050477. et al. [DOI] [PubMed] [Google Scholar]
- 45.Guldberg P, thorStraten P, Birck A, Ahrenkiel V, Kirkin AF, Zeuthen J. Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer Res. 1997;57(17):3660–3663. [PubMed] [Google Scholar]
- 46.Ernst DS, Eisenhauer E, Wainman N, et al. Phase II study of perifosine in previously untreated patients with metastatic melanoma. Invest New Drugs. 2005;23(6):569–75. doi: 10.1007/s10637-005-1157-4. [DOI] [PubMed] [Google Scholar]
- 47.Li T, Christensen SD, Frankel PH, et al. A phase II study of cell cycle inhibitor UCN-01 in patients with metastatic melanoma: a California Cancer Consortium trial. Invest New Drugs. 2010 doi: 10.1007/s10637-010-9562-8. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Margolin K, Longmate J, Baratta T, et al. CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer. 2005;104(5):1045–1048. doi: 10.1002/cncr.21265. [DOI] [PubMed] [Google Scholar]
- 49.Rao RD, Windschitl HE, Allred JB, et al. Phase II trial of the mTOR inhibitor everolimus (RAD-001) in metastatic melanoma. J Clin Oncol. 2006;24(18 suppl) et al. Abstract 8043. [Google Scholar]
- 50.Meier FE, Beck D, Niessner K, et al. Effect of mTOR inhibitors on sorafenib-induced endoplasmic reticulum stress and apoptosis in melanoma cells. J Clin Oncol. 2010;28(suppl) et al. Abstract e19027. [Google Scholar]
- 51.Maira SM, Stauffer F, Brueggen J, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 52.Safety, Pharmacokinetics and Pharmacodynamics of BEZ235 Plus MEK162 in Selected Advanced Solid Tumor Patients – Full Text View – ClinicalTrials.gov. [December 12, 2011]; Available at: http://clinicaltrials.gov/ct2/show/NCT01337765.
- 53.Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature. 1988;335(6185):88–89. doi: 10.1038/335088a0. [DOI] [PubMed] [Google Scholar]
- 54.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 55.Terada T. Low incidence of KIT gene mutations and no PDGFRA gene mutations in primary cutaneous melanoma: an immunohistochemical and molecular genetic study of Japanese cases. Int J Clin Oncol. 2010;15(5):453–6. doi: 10.1007/s10147-010-0087-0. [DOI] [PubMed] [Google Scholar]
- 56.Ugurel S, Hildenbrand R, Zimpfer A, et al. Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer. 2005;92(8):1398–1405. doi: 10.1038/sj.bjc.6602529. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wyman K, Atkins MB, Prieto V, et al. Multicenter phase II trial of high-dose imatinib mesylate in metastatic melanoma: significant toxicity with no clinical efficacy. Cancer. 2006;106(9):2005–2011. doi: 10.1002/cncr.21834. [DOI] [PubMed] [Google Scholar]
- 58.Carvajal RD, Anonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305(22):2327–34. doi: 10.1001/jama.2011.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gentile A, Trusolino L, Comoglio PM. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 2008;27(1):85–94. doi: 10.1007/s10555-007-9107-6. [DOI] [PubMed] [Google Scholar]
- 60.Hsu MY, Meier F, Herlyn M. Melanoma development and progression: a conspiracy between tumor and host. Differentiation. 2002;70(9–10):522–536. doi: 10.1046/j.1432-0436.2002.700906.x. [DOI] [PubMed] [Google Scholar]
- 61.Puri N, Ahmed S, Janamanchi V, et al. c-Met is a potentially therapeutic target for treatment of human melanoma. Clin Cancer Res. 2007;13(7):2246–53. doi: 10.1158/1078-0432.CCR-06-0776. [DOI] [PubMed] [Google Scholar]
- 62.Economou MA, All-Ericsson C, Bykov V, et al. Receptors for the liver synthesized growth factors IGF-1 and HGF/SF in uveal melanoma: intercorrelation and prognostic implications. Acta Ophthalmol. 2008;86(thesis 4):20–25. doi: 10.1111/j.1755-3768.2008.01182.x. [DOI] [PubMed] [Google Scholar]
- 63.Gordon MS, Vogelzang NJ, Schoffski P, et al. Activity of cabozantinib (XL184) in soft tissue and bone: Results of a phase II randomized discontinuation trial (RDT) in patients (pts) with advanced solid tumors. J Clin Oncol. 2011;29(suppl) et al. Abstract 3010. [Google Scholar]
- 64.Study of XL184 in adults with advanced malignancies. ClinicalTrials.gov. [December 12, 2011]; Available at: http://www.clinicaltrials.gov/ct2/show/study/NCT00940225.
- 65.Eder JP, Appleman L, Heath E, et al. A phase I study of a novel spectrum selective kinase inhibitor (SSKI), XL880, administered orally in patients (pts) with advanced solid tumors (STs) J Clin Oncol. 2006;24(18 suppl) et al. Abstract 3041. [Google Scholar]
- 66.Yarden Y. The EGFR family and its ligands in human cancer: signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37(suppl 4):S3–S8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 67.Sparrow LE, Heenan PJ. Differential expression of epidermal growth factor receptor in melanocytic tumours demonstrated by immunohistochemistry and mRNA in situ hybridization. Australas J Dermatol. 1999;40(1):19–24. doi: 10.1046/j.1440-0960.1999.00310.x. [DOI] [PubMed] [Google Scholar]
- 68.Trent JM, Meyskens FL, Salmon SE, et al. Relation of cytogenetic abnormalities and clinical outcome in metastatic melanoma. N Engl J Med. 1990;322(21):1508–1511. doi: 10.1056/NEJM199005243222107. published correction appears in N Engl J Med. 1990;323(18) 1283 et al. [DOI] [PubMed] [Google Scholar]
- 69.Rákosy Z, Vízkeleti L, Ecsedi S, et al. EGFR gene copy number alterations in primary cutaneous malignant melanomas are associated with poor prognosis. Int J Cancer. 2007;121(8):1729–1737. doi: 10.1002/ijc.22928. et al. [DOI] [PubMed] [Google Scholar]
- 70.Ueno Y, Sakurai H, Tsunoda S, et al. Heregulin-induced activation of ErbB3 by EGFR tyrosine kinase activity promotes tumor growth and metastasis in melanoma cells. Int J Cancer. 2008;123(2):340–347. doi: 10.1002/ijc.23465. et al. [DOI] [PubMed] [Google Scholar]
- 71.Djerf EA, Trinks C, Abdiu A, Thunell LK, Hallbeck EL, Waltz TM. ErbB receptor tyrosine kinases contribute to proliferation of malignant melanoma cells: inhibition by gefitinib (ZD1839) Melanoma Res. 2009;19(3):156–166. doi: 10.1097/CMR.0b013e32832c6339. [DOI] [PubMed] [Google Scholar]
- 72.Buac K, Xu M, Cronin J, Weeraratna AT, Hewitt SM, Pavan WJ. NRG1/ERBB3 signaling in melanocyte development and melanoma: inhibition of differentiation and promotion of proliferation. Pigment Cell Melanoma Res. 2009;22(6):773–784. doi: 10.1111/j.1755-148X.2009.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wellbrock C, Gómez A, Schartl M. Signal transduction by the oncogenic receptor tyrosine kinase Xmrk in melanoma formation of Xiphophorus. Pigment Cell Res. 1997;10(1–2):34–40. doi: 10.1111/j.1600-0749.1997.tb00463.x. [DOI] [PubMed] [Google Scholar]
- 74.Prickett TD, Agrawal NS, Wei X, et al. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet. 2009;41(10):1127–1132. doi: 10.1038/ng.438. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wyman K, Kelley M, Puzanov I, et al. Phase II study of erlotinib given daily for patients with metastatic melanoma (MM) J Clin Oncol. 2006;24(18 suppl) et al. Abstract 18002. [Google Scholar]
- 76.Patel S, Bedikian A, Kim K, et al. A phase II study of gefitinib in patients with metastatic melanoma. J Clin Oncol. 2009;27(15 suppl) et al. Abstract 9057. [Google Scholar]
- 77.Wyman K, Spigel D, Puzanov I, et al. A multicenter phase II study of erlotinib and bevacizumab in patients with metastatic melanoma. J Clin Oncol. 2007;25(18 suppl) et al. Abstract 8539. [Google Scholar]
- 78.Grignol VP, Olencki T, Relekar K, et al. A phase 2 trial of bevacizumab and high-dose interferon alpha 2B in metastatic melanoma. J Immunother. 2011;34(6):509–15. doi: 10.1097/CJI.0b013e31821dcefd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hodi FS, Friedlander PA, Atkins MB, et al. A phase I trial of ipilimumab plus bevacizumab in patients with unresectable stage III or stage IV melanoma. J Clin Oncol. 2011;29(suppl) et al. Abstract 8511. [Google Scholar]
- 80.von Moos R, Seifert B, Simcock M, et al. First-line temozolomide combined with bevacizumab in metastatic melanoma: a multicenter phase II trial (SAKK 50/07) Ann Oncol. 2011 doi: 10.1093/annonc/mdr126. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 81.Boasberg PD, Weber RW, Cruickshank S, et al. Phase II trial of nab-paclitaxel and bevacizumab as first-line therapy in patients with unresectable melanoma. J Clin Oncol. 2011;29(suppl) doi: 10.1097/COC.0b013e318287bbae. et al. Abstract 8543. [DOI] [PubMed] [Google Scholar]
- 82.Perez DG, Suman V, Amatruda T, et al. Phase II trial of carboplatin, weekly paclitaxel, and biweekly bevacizumab in patients with unresectable stage IV melanoma. J Clin Oncol. 2007;25(18 supple) doi: 10.1002/cncr.23987. et al. Abstract 8560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kottschade LA, Suman V, Perez DG, et al. A randomized phase II trial of temozolomide (TMZ) and bevacizumab (BEV) or nab-paclitaxel (nab-P)/carboplatin (CBDCA) and bevacizumab (BEV) in patients with unresectable stage IV metastatic melanoma: A North Central Cancer Treatment Group Study (N0775) J Clin Oncol. 2011;29(suppl) et al. Abstract 8532. [Google Scholar]
- 84.di Pietro A, Ferrucci P, Munzone E, et al. Dacarbazine (DTIC) plus bevacizumab (B) combination therapy in chemotherapy (CTh)-naïve advanced melanoma (MM) patients (pts): A phase II study. J Clin Oncol. 2010;28(15 suppl) et al. Abstract 8536. [Google Scholar]
- 85.Hainsworth JD, Infante JR, Spigel DR, et al. Bevacizumab and everolimus in the treatment of patients with metastatic melanoma: a phase 2 trial of the Sarah Cannon Oncology Research Consortium. Cancer. 2010;116(17):4122–9. doi: 10.1002/cncr.25320. [DOI] [PubMed] [Google Scholar]
- 86.Si L, Chi Z, Yuan X, et al. Durable response of the triple combination of temozolomide, sorafenib, and bevacizumab to treat refractory stage IV acral melanoma. J Clin Oncol. 2010;28(15 suppl) et al. Abstract 8564. [Google Scholar]
- 87.McClay EF, Bessudo A, Frakes L, et al. A phase I/II trial of the combination of bevacizumab, oxaliplatin, and sorafenib in patients with metastatic melanoma. J Clin Oncol. 2008:26. Abstract 20020. [Google Scholar]
- 88.Bauknecht T, Gross G, Hagedorn M. Epidermal growth factor receptors in different skin tumors. Dermatologica. 1985;171:16–20. doi: 10.1159/000249380. [DOI] [PubMed] [Google Scholar]
- 89.Liu B, Zhang H, Li S, et al. The expression of c-erbB-1 and c-erbB-2 oncogenes in basal cell carcinoma and squamous cell carcinoma of skin. Chin Med Sci J. 1996;11:106–9. [PubMed] [Google Scholar]
- 90.Brabender J, Danenberg KD, Metzger R, et al. Epidermal growth factor receptor and HER2-neu mRNA expression in non-small cell lung cancer is correlated with survival. Clin Cancer Res. 2001;7:1850–5. [PubMed] [Google Scholar]
- 91.Shimizu T, Izumi H, Oga A, et al. Epidermal growth factor receptor overexpression and genetic aberrations in metastatic squamous cell carcinoma of the skin. Dermatology. 2001;202:203–6. doi: 10.1159/000051637. [DOI] [PubMed] [Google Scholar]
- 92.Gingras AC, Kennedy SG, O’Leary MA, et al. 4E-B1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt (PKB) signaling pathway. Genes Dev. 1998;12:502–13. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Perrotte P, Matsumoto T, Inoue K, et al. Anti-epidermal growth factor receptor antibody C225 inhibts angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clin Cancer Res. 1999;5:257–65. [PubMed] [Google Scholar]
- 94.Suen JK, Bressler L, Shord SS, et al. Cutaneous squamous cell carcinoma responding serially to single-agent cetuximab. Anti-Cancer Drugs. 2007;18:827–9. doi: 10.1097/CAD.0b013e32809ef9e0. [DOI] [PubMed] [Google Scholar]
- 95.Van-Galvan S, Buceta RL, Ma D-L, et al. Cetuximab induced hypertrichosis of the scalp and eyelashes. J Am Acad Dermatol. 2010;62:531–2. doi: 10.1016/j.jaad.2009.02.023. [DOI] [PubMed] [Google Scholar]
- 96.Arnold AW, Bruckner-Tuderman L, Zuger C, Itin HP. Cetuximab therapy of metastasizing cutaneous squamous cell carcinoma in a patient with severe recessive dystrophic epidermolysis bullosa. Dermatology. 2009;219:80–3. doi: 10.1159/000218714. [DOI] [PubMed] [Google Scholar]
- 97.Thienelt CD, Bunn PA, Jr, Hanna N, et al. A multi-centered pahse I/II study of cetuximab in combination with paclitaxel and carboplatin in untreated patients with stage IV non-small cell lung cancer. J Clin Oncol. 2005;23:8786–93. doi: 10.1200/JCO.2005.03.1997. [DOI] [PubMed] [Google Scholar]
- 98.Vega-Villegas E, Awada R, Mesia L, et al. A phase I study of cetuximab in combination with cisplatin or carboplatin and 5-FU in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Proc Am Assoc Cancer Res. 2003;22:2020. doi: 10.1200/JCO.2005.04.3547. [DOI] [PubMed] [Google Scholar]
- 99.Herbst RS, Arquette M, Shin DM, et al. Phase II multicenter study of the epidermal growth factor receptor antibody cetuximab and cisplatin for recurrent and refractory squamous cell carcinoma of the head and neck. J Clin Oncol. 23:5578–87. doi: 10.1200/JCO.2005.07.120. [DOI] [PubMed] [Google Scholar]
- 100.Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the respons of metastaic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67:2643–8. doi: 10.1158/0008-5472.CAN-06-4158. [DOI] [PubMed] [Google Scholar]
- 101.Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–12. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 102.Vanhoefer U, Tewes M, Rojo F, et al. Phase I study of the humanized antiepidermal growth factor receptor monoclonal antibody in patients with advanced solid tumors that express the EGFR. J Clin Oncol. 2004;22:175–84. doi: 10.1200/JCO.2004.05.114. [DOI] [PubMed] [Google Scholar]
- 103.Seiden MV, Burris HA, Matulonis U, et al. A phase II trial of EMD 72000 (matuxumab), a humanized anti-EGFR monoclonal antibody in patients with platinum-resistance ovarian and primary peritoneal malignancies. Gynecol Oncol. 2007;104:727–31. doi: 10.1016/j.ygyno.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 104.Ciardiello F, Caputo R, Bianco R, et al. Antitumor effect of potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa) an epidermal growth factor receptor selective tyrosine kinase inhibitor. Clini Cancer Res. 2000;6:2053–63. [PubMed] [Google Scholar]
- 105.Glisson BS, Kim ES, Kies MS, et al. Phase II study of gefitinib in patients with metastatic/recurrent squamous cell carcinoma of the skin. J Clin Oncol. 2006;24(18 suppl) et al. Abstract 5531. [Google Scholar]
- 106.Moyer JD, Barbacci EG, Iwata KK, et al. Induction of apoptosis and cell cycle arrest by an epidermal growth receptor inhibitor tyrosine kinase. Cancer Res. 1997;57:4838–48. [PubMed] [Google Scholar]
- 107.Akita RW, Sliwkowski MX. Preclinical studies with Erlotinib (Tarceva) Semin Oncol. 2003;30:15–24. [PubMed] [Google Scholar]