

The Michael reaction is one of the most general and versatile methods for carbon-carbon bond formation,1 and its Mukaiyama-Michael variant provides an efficient strategy for the addition of silyl enol ethers to α,β-unsaturated carbonyl compounds.2 Catalytic asymmetric reactions with broad variations in α,β-unsaturated carbonyl compounds and chiral catalyst (Lewis acid and Brønsted acid) are well documented,3,4 and the enantioenriched 1,5-dicarbonyl compounds formed from these reactions have proven to be useful building blocks. However, there has been limited variation in the silyl enol ethers used in these reactions, and none of them have incorporated multiple functional groups.
We have recently reported condensation reactions of methyl 3-(trialkylsilanoxy)-2-diazo-3-butenoates (1) in Mukaiyama-aldol,5 Mukaiyama-Michael,6 and Mannich5 processes (Scheme 1) in our efforts to construct functionalized diazo compounds. These reactions are especially facile owing to the stabilization afforded by the diazo functional group to the intermediate formed by electrophilic addition (E+ + 1 → 5). The resulting multifunctional diazoacetoacetates have proven to be valuable building blocks for the efficient synthesis of functionally complex organic compounds.5,7 However, attempts to construct chiral multifunctional diazoacetoacetates have been only moderately successful with the only example being the asymmetric catalytic Mukaiyama-aldol reactions of a limited array of aromatic aldehydes with 1 catalyzed by AgF/(R)-BINAP.8 We now report the first examples of a broadly applicable, highly enantioselective synthesis of chiral γ-functionalized diazoacetoacetates by catalytic Mukaiyama-Michael addition reactions of 3-(tert- butyldimethylsilyloxy)-2-diazo-3- butenoate 1.
Scheme 1.

Diazoacetoacetate synthesis via condensation reactions of 1.
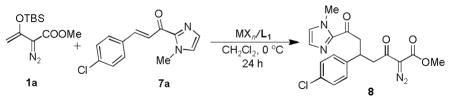
A survey of chiral Lewis acids for the direct Mukaiya–maaldol or Mukaiyama Michael reactions of 1 with α,β-unsaturated carbonyl compounds showed limited reactivity and low enantioselectivity. The success of Evans’ N-oxazolidinone derivatized α,β-unsaturated carbonyl compounds in chiral Lewis acid catalyzed asymmetric reactions9 prompted us to use 6, but even with copper(II) triflate and chiral box and pybox ligands no reaction between 6 and 1 was observed. Since the oxazolidinone basicity of 6 was too strong to effect activation of the α,β-unsaturated carbonyl unit for electrophilic addition, we turned to the less basic α,β-unsaturated 2-acyl imidazole 7,10 and in a reaction with 1 catalyzed by copper(II) triflate ligated with (S,S)-tBu-box L1 (Table 1, entry 5), the Mukaiyama-Michael condensation product 8 was formed in 66% yield but with only 10% ee. In a screening of potential Lewis acids (Table 1), scandium(III) triflate, a preferred catalyst for Mukaiyama-aldol reactions,2c,11 was ineffective for addition to 1a (entry 1). In contrast, the mild Lewis acids, Ni(OTf)2, Zn(OTf)2, and Mg(OTf)2, combined with L1, offered moderate enantioselectivity with moderate to low product yields (entries 2~4), but Cu(SbF6)212 proved to be the most active and effective, giving the desired product in 77% yield with 46% ee (entry 7). By reducing the temperature to −78°C with this copper(II) catalyst, enantioselectvity was improved to 54% ee (entry 8). Because Cu(SbF6)2 in combination with L1 exhibited the highest reactivity in these reactions, this catalytic system was selected for further elaboration.
Table 1.
Selection of Lewis acid for the enantioselective Mukaiyama-Michael addition.[a]
 | |||
|---|---|---|---|
| entry | Lewis acid MXn | yield (%)[b] | ee (%)[c] |
| 1 | Sc(OTf)3 | <5 | 0 |
| 2 | Ni(OTf)2 | 30 | 54 |
| 3 | Zn(OTf)2 | 42 | 39 |
| 4 | Mg(OTf)2 | 26 | 47 |
| 5 | Cu(OTf)2 | 66 | 10 |
| 6 | CuOTf | 31 | 26 |
| 7[d] | Cu(SbF6)2 | 77 | 46 |
| 8[d] | Cu(SbF6)2 | 43 | 54 |
Reactions were performed with 0.25 mmol 7a, Lewis acid (10 mol%) and L1 (12 mol%) in 1.5 mL of DCM, 1.5 equiv of 1a in 0.5 mL of DCM was added over 30 min to the reaction mixture at 0°C (except entry 8 which occurred at −78°C) under a N2 atmosphere. The reaction solution was stirred overnight at 0°C.
Isolated yield of 8 after chromatography.
Determined by chiral HPLC (AD-H, Hexane/i-PrOH = 50:50, flow rate 1.0 mL/min, 254 nm, tr1= 9.0 min, tr2 =11.1 min).
Cu(SbF6)2 was formed in situ from CuCl2 and AgSbF6 under a N2 atmosphere (ref. 12).
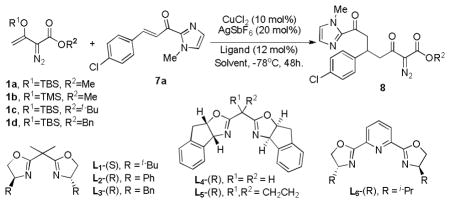
Optimization of this Mukaiyama-Michael transformation was effected on 7a by initially changing the ester alkyl group and silyl ether group of 1(1a–1d). Compared to the TBS group of 1a (Table 2, entry 1), the TMS analog 1b exhibited higher reactivity but had a much lower enantiomeric excess (entry 2). However, although no significant change in %ee was observed with the tert-butyl ester of 1c, a dramatic improvement in enantioselectivity was achieved when the ester alkyl group was changed from methyl to benzyl 1d; % ee improved to 93% (entry 4), and this vinyldiazoester was used in efforts to achieve further optimization.
Table 2.
Optimization of reactant, ligand, and reaction conditions for the enantioselective Mukaiyama-Michael addition.[a]
 | ||||||
|---|---|---|---|---|---|---|
| entry | 1 | ligand | solvent | additive | yield (%)[b] | ee (%)[c] |
| 1 | 1a | L1 | DCM | - | 43 | 54 |
| 2 | 1b | L1 | DCM | - | 64 | 12 |
| 3 | 1c | L1 | DCM | - | 33 | 50 |
| 4 | 1d | L1 | DCM | - | 42 | 93 |
| 5 | 1d | L2 | DCM | - | 30 | 65 |
| 6 | 1d | L3 | DCM | - | 33 | 76 |
| 7 | 1d | L4 | DCM | - | 40 | 87 |
| 8 | 1d | L5 | DCM | - | 34 | 88 |
| 9 | 1d | L6 | DCM | - | <5 | ND |
| 10 | 1d | L1 | DCM | HFIP (1.0 eq) | 67 | 83 |
| 11 | 1d | L1 | DCM | 4Å (0.1g) | 44 | 94 |
| 12 | 1d | L1 | THF | 4Å (0.1g) | <5 | ND |
| 13 | 1d | L1 | Toluene | 4Å (0.1g) | 35 | 90 |
| 14[d] | 1d | L1 | DCM | HFIP (1.0 eq) | 78 | 93 |
| 15[e] | 1d | L1 | DCM | HFIP & 4Å | 81 | 94 |
Reactions were performed as described in Table 1. The chiral catalyst was prepared according to ref 12.
Isolated yield of 8 after chromatography.
Determined by chiral HPLC (See supporting information).
30 mol% Catalyst was used.
Catalyst was prepared in a glove box, and the reaction was run for three days.
A survey of chiral box and pybox ligands showed comparable reactivity with the box ligands L1–L3 as well as L4 and L5 (Table 2), but %ee values were considerably lower with L2 and L3, compared with L1. Although L4 and L5 gave product yields and %ee values that were comparable to those of L1, there was no obvious advantage to their use. Pybox L6 exhibited very low reactivity, and the %ee value from the use of this ligand was not determined.
Changing the solvent from dichloromethane to THF completely shut down the reaction, but reaction in toluene afforded a %ee value comparable to that in DCM; however, the reaction rate was slower in toluene. With hexafluoroisopropyl alcohol (HFIP) as an additive3b or using 30 mol% catalyst instead of 10 mol%, a significantly improved yield of 8 was obtained (up to 78% yield with 94% ee, entries 10 and 14 of Table 2), and the result was reproducable by adding 4Å molecular sieve (entry 11).13 Having established optimum conditions with [Cu(II) (S,S)-t-Bu-box](SbF6)2, efforts were undertaken to reduce the amount of catalyst required to obtain high product yields: an 81% yield of 8 with 94% ee was obtained with 10 mol% catalyst, which was prepared in glove box and allowed to undergo reactionover a 3 day period (entry 15).
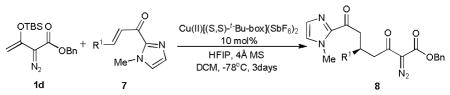
Using these optimized conditions, reactions with a diverse set of α, β-unsaturated 2-acyl imidazoles were examined with Michael donor 1d (Table 3). Aryl and alkyl substitutions all gave high yields and high to excellent enantioselectivity. Those with electron-donating substituents showed higher reactivity and selectivity compared to those with electron-withdrawing substituents (Table 3, entries 1~6 vs 7). α,β-Unsaturated 2-acyl imidazoles with aromatic heterocyclic and naphthyl substituents exhibited comparable reactivities and high ee values (entries 11 and 12). As expected from the electronic effects of aryl substituents, higher enantioselectivity was achieved with the meta-nitro-substituted 7h than with para-nitro-substituted 7g. Surprisingly, the %ee from the reaction with 7m (R = t-Bu, entry 13) was greater than that from 7n (R = cyclohexyl, entry 14)
Table 3.
Catalytic enantioselective Mukaiyama-Michael addition of vinyldiazoacetate 1d with representative Michael acceptors.[a]
 | ||||
|---|---|---|---|---|
| entry | R1 (7) | product 8 | yield (%)[b] | ee (%)[c] |
| 1 | 4-ClC6H4 (7a) | 8a | 81 | 94 |
| 2 | 4-BrC6H4 (7b) | 8b | 83 | 91 |
| 3 | 4-FC6H4 (7c) | 8c | 76 | 93 |
| 4 | 4-MeC6H4 (7d) | 8d | 77 | 91 |
| 5 | C6H5 (7e) | 8e | 75 | 91 |
| 6 | 4-MeOC6H4 (7f) | 8f | 88 | 96 |
| 7 | 4-NO2C6H4 (7g) | 8g | 62 | 86 |
| 8 | 3- NO2C6H4 (7h) | 8h | 68 | 94 |
| 9 | 2-ClC6H4 (7i) | 8i | 75 | 95 |
| 10 | 2,6-2ClC6H3 (7j) | 8j | 65 | 91 |
| 11 | 2-furanyl (7k) | 8k | 80 | 80 |
| 12 | 2-naphthyl (7l) | 8l | 72 | 91 |
| 13 | t-Bu (7m) | 8m | 62 | 95 |
| 14 | cyclohexyl (7n) | 8n | 79 | 81 |
Reactions were carried out on a 0.25 mmol scale in DCM with 1.0 equiv. of HFIP, 4Å molecular sieve (0.1 g) and 10 mol% of catalyst which was prepared in situ according to ref 12. 1.5 equiv of 1d in 0.5 mL of DCM was added over 30 min to the reaction mixture at −78°C under a N2 atmosphere in a dry ice-acetone bath. The reaction solution was stirred for three days at this temperature.
Isolated yield of 8 after chromatography.
Determined by chiral HPLC (See supporting information).
The absolute configuration of the generated stereocenter in 8 was determined by converting the Michael addition product (Scheme 2) to a reported chiral diester having a known absolute configuration that had been formed by desymmetrization of the substituted glutaric anhydride with chiral oxazolidinones.14 Cleavage of the diazoacetoacetate to the diazoacetate and carboxylic acid, a well-known and widely used transformation,15 followed by esterification of the resulting carboxylic acid with chiral (S)-1-(1-naphthyl)ethanol formed the β-substituted esters (9) in high yield without loss of chirality. Methylation of the imidazole functional group according to the reported procedure16 smoothly removed the imidazole and produced chiral diesters 10 in good yield. Comparing the NMR data of compound 10e with reported data for the known (1S,3S)-10e and (1S,3R)-10e14a Confirmed that the product formed from the Mukaiyama-Michael reaction of 1d with 7e is indeed (5S)-8e.
Scheme 2.

Synthesis of chiral 3-substituted pentanedioic acid esters.
In summary, we have developed a catalytic, highly enantioselective Mukaiyama-Michael addition of 3-(trialkylsilanoxy)-2-diazo-3-butenoate to α,β-unsaturated 2-acyl imidazoles with a chiral copper(II) Lewis acid. This methodology offers access to a broad selection of highly functionalized chiral diazoacetoacetates that can be conveniently transformed to chiral diester compounds whose asymmetric center is chemically differentiated solely by different alkyl ester groups. The further utility of these Michael addition products is under investigation.
Experimental Section
The copper catalyst was prepared in glove box according to the Evans’ procedure:12 CuCl2 (0.025 mmol), and chiral ligand (0.030 mmol) in DCM (0.5 mL) were stirred for 2 hours in an oven-dried flask, then AgSbF6 (0.050 mmol) in DCM (0.5 mL) was added dropwise, and this solution was stirred for another 3 hours in the absence of light. The resulting green catalyst suspension was filtered with cotton, and the solution was added to the oven-dried reaction flask, which contained 4Å molecular sieves (100 mg) and the Michael acceptor (0.25 mmol). The reaction flask was then sealed with a rubber stopper before being removed from the glove box. The temperature of the reaction was lowed to −78°C with dry ice-acetone bath, the additive (0.25 mmol) was introduced at this moment followed by dropwise addition of the diazo compound (0.38 mmol) in DCM (0.5 mL) by syringe. The reaction was carried out at this temperature for three days, then quenched with saturated NH4Cl and purified by flash chromatography on silica gel (eluent: hexanes: EtOAc = 5:1 to 2:1) to give the pure products.
Footnotes
Support for this research to MPD from the National Institutes of Health (GM 46503) and National Science Foundation (CHE-0748121) is gratefully acknowledged. WH thanks the National Science Foundation of China (20932003), and the MOST of China (2011CB808600).
Contributor Information
Dr. Xinfang Xu, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742
Prof. Wen-Hao Hu, Email: whu@chem.ecnu.edu.cn, Institute of Drug Discovery and Development, East China Normal University, 3663 Zhongshan Bei Road, Shanghai 200062, China
Prof. Michael P. Doyle, Email: mdoyle3@umd.edu, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, Fax: (+11) 301-314-2779
References
- 1.For comprehensive reviews: Little R, Masjedizadeh M, Wallquist O, Mcloughlin J. Org React. 1995;47:315.Tomioka K, Nagaoka Y. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 3. Springer; Heidelberg: 1999. Chapter 29.1.Kanai M, Shibasaki M. In: Catalytic Asymmetric Synthesis. 2. Ojima I, editor. Wiley; New York: 2000. p. 569.Feringa BL. Acc Chem Res. 2000;33:346. doi: 10.1021/ar990084k.Christoffers J, Baro A. Angew Chem Int Ed. 2003;42:1688. doi: 10.1002/anie.200201614.Wang E, Wang J, Ji H. Angew Chem Int Ed. 2005;44:1369.Gridnev ID, Watanabe M, Wang H, Ikariya T. J Am Chem Soc. 2010;132:16637. doi: 10.1021/ja107597w.Evans DA, Mito S, Seidel D. J Am Chem Soc. 2007;129:11583. doi: 10.1021/ja0735913.Jautze S, Peters R. Synthesis. 2010;3:365.Boncel S, Gondela A, Walczak K. Synthesis. 2010;10:1573.Soloshonok VA, Ueki H, Ellis TK. ACS Symposium Series; 2009. p. 72.Yang H, Kim S. Synlett. 2008;4:555.Kazmaier U. Angew Chem Int Ed. 2009;48:5790. doi: 10.1002/anie.200901261.Enders D, Wang C, Liebich JX. Chem Eur J. 2009;15:11058. doi: 10.1002/chem.200902236.Yoo W, Miyamura H, Kobayashi S. J Am Chem Soc. 2011;133:3095. doi: 10.1021/ja110142y.Krishna PR, Sreeshailam A, Srinivas R. Tetrahedron. 2009;65:9657.Santanu M, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471. doi: 10.1021/cr0684016.Melchiorre P, Jørgensen KA. J Org Chem. 2003;68:4151. doi: 10.1021/jo026837p.
- 2.Reviews of the Mukaiyama-Michael reaction: Kobayashi S, Sugiura M, Kitagawa H, Lam WWL. Chem Rev. 2002;102:2227. doi: 10.1021/cr010289i.Takahashi A, Yanai H, Zhang M, Sonoda T, Mishima M, Taguchi T. J Org Chem. 2010;75:1259. doi: 10.1021/jo902641g.Borths CJ, Carrera DE, MacMillan DWC. Tetrahedron. 2009;65:6746. doi: 10.1016/j.tet.2009.06.066.Tamagaki H, Nawate Y, Nagase R, Tanabe Y. Chem Commun. 2010;46:5930. doi: 10.1039/c0cc01110j.Tumanov MORVV, Smit WA. Angew Chem Int Ed. 2008;47:9739. doi: 10.1002/anie.200803927.
- 3.Examples of Lewis acid catalyzed asymmetric reactions, see: Evans DA, Rovis T, Kozlowski MC, Tedrow JS. J Am Chem Soc. 1999;121:1994.Evans DA, Willis MC, Johnston JN. Org Lett. 1999;1:865. doi: 10.1021/ol9901570.Evans DA, Johnson JS, Olhava EJ. J Am Chem Soc. 2000;122:1635.Evans DA, Rovis T, Kozlowski MC, Downey CW, Tedrow JS. J Am Chem Soc. 2000;122:9134.Evans DA, Scheidt KA, Johnston JN, Willis MC. J Am Chem Soc. 2001;123:4480. doi: 10.1021/ja010302g.Harada T, Iwai H, Takatsuki H, Fujita K, Kubo M, Oku A. Org Lett. 2001;3:2101. doi: 10.1021/ol016062r.Suga H, Kitamura T, Kakehi A, Baba T. Chem Commun. 2004:1414. doi: 10.1039/b402826k.Desimoni G, Faita G, Guala M, Laurenti A, Mella M. Chem Eur J. 2005;11:3816. doi: 10.1002/chem.200401213.Ishihara K, Fushimi M. Org Lett. 2006;8:1921. doi: 10.1021/ol060651l.Takenaka N, Abell J, Yamamoto H. J Am Chem Soc. 2007;129:742. doi: 10.1021/ja0668320.Zheng W, Zhang Z, Kaplan MJ, Antilla JC. J Am Chem Soc. 2011;133:3339. doi: 10.1021/ja109824x.Trost BM, Hitce J. J Am Chem Soc. 2009;131:4572. doi: 10.1021/ja809723u.Gendrineau T, Chuzel O, Eijsberg H, Genet JP, Darses S. Angew Chem, Int Ed. 2008;47:7669. doi: 10.1002/anie.200803230.Evans DA, Thomson RJ, Franco F. J Am Chem Soc. 2005;127:10816. doi: 10.1021/ja053820q.Sammis GM, Jacobsen EN. J Am Chem Soc. 2003;125:4442. doi: 10.1021/ja034635k.Mazet C, Jacobsen EN. Angew Chem Int Ed. 2008;47:1762. doi: 10.1002/anie.200704461.Madhavana N, Weck M. Adv Synth Catal. 2008;350:419.
- 4.Examples of organocatalytic asymmetric reactions, see: Zhang F, Corey EJ. Org Lett. 2001;3:639. doi: 10.1021/ol015592k.Brown SP, Goodwin NC, MacMillan DWC. J Am Chem Soc. 2003;125:1192. doi: 10.1021/ja029095q.Zhu Y, Malerich JP, Rawal VH. Angew Chem Int Ed. 2010;49:153. doi: 10.1002/anie.200904779.Zhu SL, Yu SY, Ma DW. Angew Chem Int Ed. 2008;47:545. doi: 10.1002/anie.200704161.Huang HC, Jin ZC, Zhu KL, Liang XM, Ye JX. Angew Chem Int Ed. 2011;50:3232. doi: 10.1002/anie.201008255.Belmessieri D, Morrill LC, Simal C, Slawin AMZ, Smith AD. J Am Chem Soc. 2011;133:2714. doi: 10.1021/ja109975c.Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471. doi: 10.1021/cr0684016.
- 5.Doyle MP, Kundu K, Russell AE. Org Lett. 2005;7:5171. doi: 10.1021/ol052003s. [DOI] [PubMed] [Google Scholar]
- 6.Yu L, Zhang Y, Jee N, Doyle MP. Org Lett. 2008;10:1605. doi: 10.1021/ol800298n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Yu L, Bakshi K, Zavalij P, Doyle MP. Org Lett. 2010;12:4304. doi: 10.1021/ol101744h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou L, Doyle MP. Org Lett. 2010;12:796. doi: 10.1021/ol902872y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kundu K, Doyle MP. Tetrahedron: Asymmetry. 2006;17:574. [Google Scholar]
- 9.a) Johnson JS, Evans DA. Acc Chem Res. 2000;33:325. doi: 10.1021/ar960062n. [DOI] [PubMed] [Google Scholar]; b) Evans DA, Chapman KT, Bisaha J. J Am Chem Soc. 1988;110:1238. [Google Scholar]
- 10.a) Evans DA, Fandrick KR, Song HJ, Scheidt KA, Xu RS. J Am Chem Soc. 2007;129:10029. doi: 10.1021/ja072976i. [DOI] [PubMed] [Google Scholar]; b) Evans DA, Song HJ, Fandrick KR. Org Lett. 2006;8:3351. doi: 10.1021/ol061223i. [DOI] [PubMed] [Google Scholar]; c) Guan XY, Yang LP, Hu WH. Angew Chem Int Ed. 2010;49:2190. doi: 10.1002/anie.200904905. [DOI] [PubMed] [Google Scholar]; d) Trost BM, Lehr K, Michaelis DJ, Xu J, Buckl AK. J Am Chem Soc. 2010;132:8915. doi: 10.1021/ja103771w. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Myers MC, Bharadwaj AR, Milgram BC, Scheidt KA. J Am Chem Soc. 2005;127:14675. doi: 10.1021/ja0520161. [DOI] [PubMed] [Google Scholar]; f) Boersma AJ, Feringa BL, Roelfes G. Org Lett. 2007;9:3647. doi: 10.1021/ol7015274. [DOI] [PubMed] [Google Scholar]
- 11.a) Evans DA, Masse CE, Wu J. Org Lett. 2002;4:3375. doi: 10.1021/ol026488l. [DOI] [PubMed] [Google Scholar]; b) Ishikawa S, Hamada T, Manabe K, Kobayashi S. J Am Chem Soc. 2004;126:12236. doi: 10.1021/ja047896i. [DOI] [PubMed] [Google Scholar]
- 12.Ligated Cu(SbF6)2 was formed from CuCl2 and ligand, which were thoroughly mixed over 2 h in DCM, by treatment with AgSbF6 (2.0 equiv) according to reference: Evans DA, Kozlowski MC, Murry JA, Burgey CS, Campos KR, Connell BT, Staples RJ. J Am Chem Soc. 1999;121:669.; and Ref. 3e.
- 13.a) Hasegawa A, Ono F, Kanemasa S. Tetrahedron Lett. 2008;49:5220. [Google Scholar]; b) Ono F, Hasegawa M, Kanemasa S, Tanaka J. Tetrahedron Lett. 2008;49:5105. [Google Scholar]
- 14.For the (1S,3S) and (1S,3R) isomers see: Theisen PD, Heathcock CH. J Org Chem. 1993;58:142.Verma R, Mithran S, Ghosh SK. J Chem Soc, Perkin Trans. 1999;1:257.
- 15.a) Regitz M, Maas G. In: Diazo Compounds 2 Properties and Sythesis. Regitz M, Maas G, editors. Academic Press, Inc; Orlando: 1986. [Google Scholar]; b) Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides. Wiley; New York: 1998. [Google Scholar]; c) Doyle MP, Catino AJ. Tetrahedron: Asymmetry. 2003;14:925. [Google Scholar]; d) Doyle MP, Wang YH, Ghorbani P, Bappert E. Org Lett. 2005;7:5035. doi: 10.1021/ol0520121. [DOI] [PubMed] [Google Scholar]
- 16.Evans DA, Song HJ, Fandrick KR. Org Lett. 2006;8:3351. doi: 10.1021/ol061223i.; and ref. 10c.