

Abstract
In the mouse, the initial signals that establish left-right (LR) asymmetry are determined in the node by nodal flow. These signals are then transferred to the lateral plate mesoderm (LPM) through cellular and molecular mechanisms that are not well characterized. We hypothesized that endoderm might play a role in this process because it is tightly apposed to the node and covers the outer surface of the embryo, and, just after nodal flow is established, higher Ca2+ flux has been reported on the left side near the node, most likely in the endoderm cells. Here we studied the role of endoderm cells in the transfer of the LR asymmetry signal by analyzing mouse Sox17 null mutant embryos, which possess endoderm-specific defects. Sox17–/– embryos showed no expression or significantly reduced expression of LR asymmetric genes in the left LPM. In Sox17 mutant endoderm, the localization of connexin proteins on the cell membrane was greatly reduced, resulting in defective gap junction formation, which appeared to be caused by incomplete development of organized epithelial structures. Our findings suggest an essential role of endoderm cells in the signal transfer step from the node to the LPM, possibly using gap junction communication to establish the LR axis of the mouse.
Keywords: Left-right asymmetry, Endoderm, Gap junction, Sox17, Nodal
INTRODUCTION
Establishing a three-dimensional axis, with anteroposterior, dorsoventral and left-right (LR) coordinates, is a critical step in embryogenesis. LR axis determination provides positional information for internal organs after the other two axes are established (Beddington and Robertson, 1999). The process of establishing LR asymmetry in the mouse occurs in four steps (Hamada et al., 2002). First, symmetry is broken in the node by a leftward flow of extra-embryonic fluid termed nodal flow (Nonaka et al., 1998). Second, the LR signal established in the node is transferred from the node to the lateral plate mesoderm (LPM), which comprises the mesodermal component of internal organs. Third, Nodal begins to be expressed in the LPM near the node and then extends rostrocaudally, expanding throughout the entire left LPM. This expansion is achieved by positive and negative transcriptional feedback loops between Nodal and Lefty1/2, together with the long-range displacement of the secreted proteins Nodal, Lefty1 and Lefty2 (Marjoram and Wright, 2011; Osada et al., 2000; Saijoh et al., 2000). Nodal also turns on the Pitx2 transcription factor in the left LPM. Finally, the Nodal signal and Pitx2 establish characteristics of the left side in the LPM, and asymmetric morphogenesis takes place in internal organs.
Although the mechanisms of LR determination have been extensively analyzed, it is still unclear how LR signals in the node are transferred to the left LPM. Bilateral expression of Nodal in the peripheral region of the node (in crown cells or perinode) is considered to be essential for this step, as its depletion in this region results in the loss of Nodal expression in the LPM (Brennan et al., 2002; Saijoh et al., 2003). Considering the fact that Nodal is a secreted molecule that can diffuse over long distances, Nodal protein generated in the node might travel to the LPM through the extracellular matrix (ECM), thereby transferring LR information to this region. In support of this contention, Oki et al. demonstrated that sulfated ECM proteoglycans such as chondroitin sulfate and heparan sulfate are necessary to induce Nodal expression in the LPM (Oki et al., 2007). However, there is no direct evidence to support the movement of Nodal from the node to the LPM. Collectively, these data suggest that expression of Nodal protein in the crown cells is essential for Nodal expression in the left LPM; however, it is unclear whether Nodal is itself the signal. Consequently, both the molecular nature of the LR signal and the mechanism by which this signal is transferred to the LPM remain to be determined.
Several studies have reported that after nodal flow is established, higher Ca2+ flux is observed on the left side near the node, most likely in the endoderm cells (Hadjantonakis et al., 2008; McGrath et al., 2003; Tanaka et al., 2005). This observation and the fact that endoderm in the mouse embryo is located adjacent to the node and covers the entire LPM at that stage, led us to hypothesize that endoderm might play a role in the signal transfer from the node to the left LPM.
To address the roles of endoderm in LR determination, we analyzed Sox17 mutant mice. Sox17 is a high-mobility group transcription factor and is specifically expressed in the endoderm and endothelial cells of developing embryos (Kanai-Azuma et al., 2002; Sakamoto et al., 2007). Null mutants die at around embryonic day (E) 10, with an abnormal heart and loss of blood stem cells; however, at E8.0, when LR determination occurs, the Sox17 mutants have defects specifically in endoderm cells (Kanai-Azuma et al., 2002; Kim et al., 2007; Pfister et al., 2011; Sakamoto et al., 2007).
In this study, we found that a majority of Sox17–/– embryos fail to express Nodal, Lefty1/2 and Pitx2 in the LPM, despite the fact that LR determination within the node of Sox17 mutant embryos was essentially normal. We also demonstrated that the LPM has the potential to express these asymmetric genes in Sox17–/– embryos. Our data suggest that the primary LR defect in Sox17–/– embryos is an impaired ability to transfer the LR signal from the node to the LPM. Significantly, membrane localization of gap junction connexin proteins was impaired in Sox17–/– endoderm, resulting in defective intercellular transport between endoderm cells. Collectively, our findings are the first to suggest an essential role of endoderm cells in the signal transfer step from the node to the LPM, possibly using gap junction communication to establish the LR axis of the mouse.
MATERIALS AND METHODS
Mice and embryo collection
Mice carrying the Sox17 null allele (Kanai-Azuma et al., 2002) and the Sox17GFP allele (Kim et al., 2007) were maintained in a mixed genetic background of 129sv, C57BL/6J and CD-1, and 129sv and C57BL/6J, respectively. Prior to harvest, the stages of embryos were monitored and estimated by ultrasound scan of the pregnant mother (Visual Sonics). Embryos were genotyped as described previously (Kanai-Azuma et al., 2002; Kim et al., 2007). All procedures using mice were in accordance of the IACUC protocol at the University of Utah.
Whole-mount in situ hybridization, immunohistochemistry and image processing
Whole-mount in situ hybridization was performed according to standard procedures (Wilkinson, 1998) using the following probes: Sox17, a combination of 0.8 kb and 0.6 kb fragments that cover the Sox17 coding region, and previously described probes (Kanai et al., 1996); Nodal, a combination of three plasmids containing 5′ and 3′ UTR regions and the coding sequence; mouse Cx43 mRNA was detected using a rat 1125 bp cDNA fragment that shares 95% sequence identity with mouse Cx43 (Beahm et al., 2006); Cerl2 (Hashimoto et al., 2004); Cryptic (Shen et al., 1997); Foxf1 (Mahlapuu et al., 2001); Foxh1 (Saijoh et al., 2000); Gdf1 (Rankin et al., 2000); Lefty1/Lefty2 (Meno et al., 1996), which cross-hybridizes with both Lefty1 and Lefty2 mRNA; L-Plunc (Hou et al., 2004); Meox1 (Candia et al., 1992); Pitx2 (Yoshioka et al., 1998); and Raldh2 (Niederreither et al., 1997). Following whole-mount in situ hybridization, embryos were imaged in 80% glycerol and 10 μm sections were prepared by standard paraffin sectioning.
For immunohistochemistry (IHC), embryos at E7.5-8.5 were fixed in 4% paraformaldehyde (PFA) in PBS for 1 hour at room temperature (RT), dehydrated through a methanol series and stored at –20°C until use. Primary antibodies used for IHC: acetylated tubulin (Sigma clone 6-11B-1, 1:200); β-galactosidase (Abcam ab9361, 1:500); chondroitin sulfate (Sigma CS-56, 1:100); Cx40 (Santa Cruz sc-20466, 1:100); Cx43 (Sigma C6219, 1:500); E-cadherin (BD Transduction 610181, 1:100); γ-tubulin (Sigma T3559, 1:100); GFP (Invitrogen A21311, 1:1000); laminin (Sigma L9393, 1:100); and ZO1 (Zymed 33-9100, 1:400). Alexa Fluor dye-labeled secondary antibodies were used (Invitrogen, 1:1000). Nuclei were counterstained with DAPI.
Confocal microscopy images were obtained at 0.3-1 μm intervals using a Leica TCS SP5 system. Images were processed using FluoRender software (http://www.sci.utah.edu/software/13-software/127-fluorender.html). Fluorescent signals are pseudo-colored in the figures to permit viewing by the color-blind.
Scanning electron microscopy
Embryos dissected at E8.0 were fixed in 2.5% glutaraldehyde/1% PFA in 0.1 M Sorensen’s phosphate buffer (pH 7.4) at RT overnight, washed in 0.1 M cacodylate buffer (pH 7.4), and post-fixed in 1% osmium tetroxide in cacodylate buffer for 45 minutes at RT. Embryos were then dehydrated through a graded ethanol series, critical point dried, gold coated, and mounted on a low-vacuum electron microscope (Hitachi S-2460N).
Nodal flow
To assess nodal flow over the node surface, FluorSpheres (0.5 μm, Invitrogen) were added to the medium and the embryo was mounted on a slide-glass chamber under a time-lapse fluorescence microscope kept in a chamber at 37°C. The directional movements and velocities of the beads across the node surface were recorded at 0.2-second intervals and traced using the MTrackJ plug-in in ImageJ (NIH).
Iontophoresis of whole embryos
Embryos were mounted in a fixed position on a silicon-derived embryo-holding chamber containing HEPES-buffered DMEM. Embryos were visualized at 40× using an Olympus BX-WI fixed-stage microscope. A glass micropipette containing a filtered solution of 0.1 M LiCl, 5% Lucifer Yellow and 1% Rhodamine dextran (10 kDa) was positioned within an endodermal cell using a micromanipulator. The iontophoresis was performed using an Axopatch 200A patch clamp and Digidata 1320A (Axon Instruments) at an iontophoretic current of –1.0 nA and a negative holding voltage of 60 mV generating a 25-30 MΩ circuit resistance. After a 10-second iontophoresis of dyes, the micropipette was removed and epifluorescence and bright-field images were captured using a MicroFire M digital camera (Olympus).
RESULTS
Nodal expression is absent in the LPM of Sox17 mutant embryos
We have previously shown that Sox17–/– embryos do not undergo embryonic turning and most mutants have aberrant heart looping (Kanai-Azuma et al., 2002; Pfister et al., 2011; Sakamoto et al., 2007). Further observations (data not shown) suggested that heart looping in Sox17–/– embryos is essentially random. Since both embryonic turning and heart looping are readouts of LR patterning, these data suggested that Sox17–/– embryos have a defect in LR determination. We therefore examined the expression of several genes that are required for LR establishment in Sox17–/– embryos.
We first examined Nodal, which is a master gene for LR determination (Hamada et al., 2002). In Sox17–/– embryos, Nodal expression was absent from the entire LPM (10/12) or was weakly expressed in a small region of the left LPM near the node (2/12). In Sox17+/– (n=8) and wild-type (n=22) embryos, Nodal was expressed throughout the left LPM (Fig. 1A,D,G,J). Despite its absence in the LPM, perinodal expression of Nodal was essentially normal in Sox17–/– embryos (Fig. 1D,G, black arrows).
Fig. 1.

Loss or reduction of LR asymmetric gene expression in Sox17 mutants. (A-I) Expression patterns of Nodal (A,D,G), Lefty1/Lefty2 (B,E,H) and Pitx2 (C,F,I) in Sox17+/– (A-C) and Sox17–/– (D-I) mouse embryos at E8.2. (D,E) Sox17–/– embryos show loss of asymmetric gene expression in the LPM. (G,H) Asymmetric gene expression was limited to a small region near the node in Sox17–/– embryos (red arrowheads in G-I). Ectopic expression domains of Nodal, Lefty2 and Pitx2 were observed in endoderm cells (red arrowheads in D′,E′,F′). Lefty1 (E,H) and Nodal (D,G) expression in the node is indicated by black arrows. (A′-F′) Sections, the positions of which are indicated in A-F. (J) Ratio of LR gene expression in the LPM. Fg, foregut; En, endoderm; NP, neural plate. R, right LPM; L, left LPM. Scale bars: 500 μm in A-I; 200 μm in A′-F′.
We next examined two other left-specific genes, Lefty2 and Pitx2, that are downstream targets of Nodal (Osada et al., 2000; Saijoh et al., 2000). As expected, both Lefty2 and Pitx2 were either completely absent (6/11, 7/13, respectively) or were expressed in a restricted region of the left LPM near the node (5/11, 6/13, respectively) in Sox17–/– embryos at a stage when they are expressed throughout the entire left LPM in both Sox17+/– and wild-type embryos (Fig. 1B,C,E,F,H-J). We did not observe right side or bilateral expression of these genes in Sox17–/– embryos, suggesting defective but not randomized LR determination in Sox17–/– embryos. In addition, we unexpectedly observed that Nodal, Lefty2 and Pitx2 were ectopically expressed in the foregut endoderm of Sox17–/– embryos at the early somite stage (Fig. 1A′-F′, red arrowheads).
Sox17 is expressed at the correct place and time to affect LR determination
To explore the cause of the impaired expression of laterality genes in Sox17–/– embryos, we performed a detailed in situ hybridization investigation of the spatial and temporal aspects of Sox17 expression during the period in which LR determination occurs. Sox17 expression was initiated in definitive endoderm cells migrating anteriorly from the primitive streak; however, no expression was observed within the primitive streak, including the anterior primitive streak that contains the node and endoderm precursors (Fig. 2A,A′). At the late streak stage, the distal part of the embryo, including the prospective node region (red arrowheads), was covered with endoderm cells that express Sox17 (Fig. 2B,B′). These observations suggest that Sox17-expressing cells could affect node formation and/or function, including the initial LR determination in the node.
Fig. 2.

Dynamic Sox17 expression in definitive endoderm cells at stages when LR determination occurs. (A-D) Sox17 expression was initiated in migrating lateral endoderm cells at mid-streak stage (A) and became expanded at the late-streak stage (B) and bud stage (C), then the expression was refined to the anterior foregut region at the headfold stage (D). Black arrowheads indicate Sox17-expressing cells. Red lines indicate the primitive streak region. (E-G) A new domain of Sox17 expression is observed in the endoderm cells near the node from 0 ss (E) to 2-4 ss (F,G, arrows). (H) At E8.5, Sox17 is prominently expressed in the foregut and hindgut pockets. (A′,B′,D′,D″,G′-G″′) Sections of A, B, D and G as indicated, with G″′ as a magnification of G″. Red arrowheads and red circles indicate the position of the node precursors (B-D) and the node (E-H); green arrowheads indicate the LPM. Fg, foregut; Hg, hindgut; PS, primitive streak. Scale bars: 500 μm.
During the bud stages, Sox17 expression expanded from anterior to posterior; however, newly generated endoderm cells migrating from the primitive streak near the node did not express Sox17 (Fig. 2C, red arrowhead). Sox17 expression was restricted to the foregut region of the embryo at the headfold stage (Fig. 2D, black arrowheads). Interestingly, a new domain of Sox17 expression was initiated in endoderm cells adjacent to the node at the 0 somite stage (ss) and expanded posteriorly (Fig. 2E-G, arrows). This endodermal Sox17 expression neighboring the node corresponds to a population of cells that show asymmetric Ca2+ flux just after nodal flow is established (Tanaka et al., 2005). Thus, we predict that Sox17-expressing endoderm could play a role in transferring LR signals from the node to the LPM.
After 2 ss, the endoderm overlying the LPM expressed Sox17 (Fig. 2F,G). Hence, it is also possible that Sox17-expressing endoderm is involved in LPM development. At this stage, Sox17 is again expressed only in endoderm cells and not in the node, including the perinode cells (supplementary material Fig. S1).
This expression pattern of Sox17 suggests three possible explanations for the LR determination defects observed in Sox17–/– embryos: (1) the initial LR determination in the node is defective; (2) the transfer of LR signals from the node to the left LPM is impaired; or (3) the defective LPM cannot express or amplify Nodal after the LR signal from the node is received.
Sox17 mutants can respond to Nodal signaling in the LPM
First, we examined the possibility that Sox17–/– LPM has an impaired ability to respond to Nodal signaling. We examined LPM-expressed genes including Nodal signal components in Sox17–/– embryos, including Foxf1, a key gene for LPM differentiation (Fig. 3A,E) (Tsiairis and McMahon, 2009), the co-receptor Cryptic (Cfc1 – Mouse Genome Informatics) (Fig. 3B,F) (Yan et al., 1999), the transcription factor Foxh1 (Fig. 3C,G) (Saijoh et al., 2000), and the co-ligand of Nodal Gdf1 (Fig. 3D,H) (Rankin et al., 2000; Tanaka et al., 2007). All of these genes were expressed in Sox17–/– embryos with expression patterns similar to those found in Sox17+/– control embryos (Fig. 3A-H).
Fig. 3.

The LPM is properly specified in Sox17 mutants. (A-H) Foxf1 (A,E), Cryptic (B,F), Foxh1 (C,G) and Gdf1 (D,H) were expressed normally in Sox17+/– and Sox17–/– mouse embryos at 5-6 ss. (I-O) Induction of endogenous Nodal expression by ectopic Nodal activity in the right LPM. A lipofection mixture of the Nodal and GFP expression vectors was injected into the right LPM (I, arrow) at the headfold stage. GFP-expressing cells represent cells that expressed exogenous Nodal (arrows in J,K). Induced endogenous Nodal was expressed throughout the entire right LPM in Sox17+/– (L,M, red arrows) and Sox17–/– (N,O, red arrows) embryos. Black arrows in L-O indicate the sites of exogenous Nodal. Note that the left side expression was reduced (L) or completely repressed (M) by the right side expression of Nodal in Sox17+/– embryos. Scale bars: 500 μm.
To directly test whether the LPM in Sox17 mutants can express Nodal in response to Nodal signaling, exogenous introductions of Nodal expression were performed (Fig. 3I-O). Nodal expression is regulated by a positive-feedback mechanism (Osada et al., 2000; Saijoh et al., 2000); thus, if the LPM in Sox17–/– embryos has an ability to respond to Nodal signaling, exogenous Nodal should be able to induce endogenous Nodal expression in the LPM of Sox17–/– embryos. Nodal expression vectors were introduced into a small area of the right LPM together with GFP expression vectors as a reporter of transfected cells, using a lipofection technique (Fig. 3I-K) (Nakamura et al., 2006). Both the control LPM (6/6; Fig. 3L,M) and the Sox17–/– LPM (5/5; Fig. 3N,O) expressed Nodal throughout the right LPM in response to ectopic Nodal activity in this small region. These data suggested that the LPM in Sox17–/– embryos is capable of initiating and expanding Nodal expression when supplied with a source of Nodal.
Node formation is partially affected but LR determination at the node appears to be normal in Sox17 mutants
Next, we examined the possibility that the initial LR determination in the node is affected in Sox17–/– embryos. The node precursors are formed at the most anterior part of the primitive streak, which is covered by endoderm cells (Fig. 2B,B′). Hence, these two tissues could interact substantially with each other during development, and endoderm may affect the differentiation of the node precursors (Sulik et al., 1994). In Sox17–/– embryos, Nodal was expressed in the perinodal region (Fig. 1D,G, black arrows), as observed in Sox17+/– embryos; however, expression at the headfold stage was extended more posteriorly in Sox17–/– embryos (Fig. 4H, arrow), whereas expression in control embryos was horseshoe shaped (Fig. 4D).
Fig. 4.

Node morphogenesis is partially defective but the node is able to direct LR asymmetry in Sox17 mutants. (A-H) Sox17+/– (A-D) and Sox17–/– (E-H) embryos. (A-C,E-G) Immunohistochemistry (IHC) of γ-tubulin for basal bodies (magenta in A,B,E,F), acetylated tubulin (Actu) for cilia (green in B,F; magenta in C,G) and ZO1 for tight junctions (green in A,C,E,G) at 1-2 ss. (D,H) Nodal mRNA in the node region at the headfold stage. Note that in Sox17–/– embryos, some endoderm cells remained in the node region (indicated by asterisks), resulting in the posterior elongation of the node. The shape of nodes is highlighted in red (C″,G″). An arrow indicates isolated node cells posteriorly (G″). Nodal expression also showed posterior elongation of the node in the Sox17–/– embryo (H, arrow). The boxed regions in A-C,E-G are magnified in A′-C′,E′-G′. (I-J′) Node structures and cilia formation were examined by scanning electron microscopy. The boxed regions in I and J are magnified in I′ and J′. Note that two large endoderm cells remained in the node (arrowheads), and ectopic ciliated cells (arrow) were observed in the separate region posterior to the Sox17–/– node (J). (K,L) Nodal flow in Sox17+/– and Sox17–/– nodes. Each line represents the trajectory of bead movement over the node surface, as taken from movies (supplementary material Movies 1, 2). The black lines outline the boundary of the node. (M-R) Both Sox17+/– and Sox17–/– perinode cells express Nodal (M,P), L-Plunc (N,Q) and Cerl2 (O,R) asymmetrically. Ectopic Cerl2 expression was observed in the posterior of the Sox17–/– node (R, arrow).
Acetylated tubulin (cilia) and γ-tubulin (basal bodies) immunohistochemistry (IHC) showed that cilia formed normally in the Sox17–/– node, although cilia formation was also observed in segregated node-like cells found posterior to the node (Fig. 4A-G, arrow in 4G″). We found some endoderm cells in the node (Fig. 4E,G, asterisks) and the node did not form the typical teardrop shape in Sox17–/– embryos (Fig. 4E-G″). Scanning electron microscopy also confirmed normal cilia formation as well as the presence of isolated endoderm cells (arrowheads) in the node and segregated groups of ciliated cells (arrow) posterior to the node in Sox17–/– embryos (Fig. 4I,J). Since ectopic endoderm cells in the node of Sox17–/– embryos might prevent the differentiation of node precursors into mature ciliated cells, the number of node cells was examined by γ-tubulin and ZO1 (Tjp1 – Mouse Genome Informatics) staining (Fig. 4A,E). No statistically significant difference was seen between Sox17–/– and control nodes [299±5.5 (n=5) and 302±5.9 (n=5), respectively; P=0.76, Student’s t-test].
As cell number and cilia formation in the node appeared normal in Sox17–/– embryos, we next determined whether the abnormal shape of the node might affect the direction and strength of the nodal flow that provides a bias to determine the initial LR asymmetry. When the flow was visualized with fluorescent beads, the flow rate and direction were similar in Sox17–/– and control embryos; however, the smooth pattern of the flow was slightly affected by the presence of the remaining endoderm cells in the Sox17–/– nodes (Fig. 4K,L; supplementary material Movies 1, 2; n=9 and n=7 for control and mutant embryos, respectively). Importantly, a significant leftward flow developed in all Sox17–/– embryos examined.
To evaluate the outcome of nodal flow on the generation of LR polarity across the node, we examined the asymmetric expression of the perinodal genes Nodal, L-Plunc (Bpifb1 – Mouse Genome Informatics) and Cerl2 (Dand5 – Mouse Genome Informatics) (Hou et al., 2004; Pearce et al., 1999). All three genes were asymmetrically expressed in the node in most Sox17–/– embryos [Nodal, 7/11 (64%); L-Plunc, 9/11 (82%); Cerl2, 10/11 (91%); Fig. 4P-R], similar to control embryos [Nodal, 15/22 (68%); L-Plunc, 11/11 (100%); Cerl2, 9/9 (100%); Fig. 4M-O]. These data suggested that in Sox17–/– embryos the nodal flow is strong enough to generate LR asymmetry in the node despite some abnormal features of the node.
Partial defects in proteoglycan distribution in Sox17–/– embryos
Finally, we examined the possibility that disrupted transfer of the LR signals from the node to the left LPM accounts for the LR defects observed in Sox17–/– embryos. Previously, we reported that proteoglycans with glucosaminoglycan moieties such as chondroitin sulfate and heparan sulfate play important roles in transferring the LR signals from the node to the LPM (Oki et al., 2007). Chondroitin sulfate is distributed at the interface between endoderm and mesoderm in control embryos, and was shown to directly interact with Nodal proteins (Oki et al., 2007). We performed IHC of chondroitin sulfate and laminin at 2-4 ss to study the effect of Sox17 mutation on proteoglycan distribution and ECM development (Fig. 5A-C). We found that 7 of 17 (41%) Sox17–/– embryos showed regional defects in chondroitin sulfate distribution concomitant with laminin matrix disorganization (Fig. 5B-B″), whereas the remaining 10 of 17 (59%) Sox17–/– embryos showed an almost normal distribution of ECM (Fig. 5C-C″; control embryos, n=6). These defects in chondroitin sulfate distribution in the ECM might cause reduced LR signal transfer from the node; however, the loss of Nodal expression in the LPM in 90% of Sox17–/– embryos cannot be accounted for by the reduced chondroitin sulfate distribution and defective ECM composition seen in only 41% of these embryos.
Fig. 5.

Defects in proteoglycan and ECM distribution are not the primary cause of LR abnormalities in Sox17 mutants. Co-IHC of chondroitin sulfate, laminin and GFP in Sox17GFP/+ and Sox17GFP/GFP mouse embryos at 3-4 ss. The Sox17GFP allele is a null mutation. Chondroitin sulfate (proteoglycan) and laminin (ECM) are distributed uniformly in Sox17GFP/+ embryos (A-A″), whereas Sox17GFP/GFP embryos showed either regional defects (B-B″, stars in B′; n=7/17) or almost normal phenotypes (C-C″; n=10/17) in the organization of chondroitin sulfate and laminin. Ect, ectoderm; Mes, mesoderm.
Since paraxial mesoderm lies between the node and the LPM, it could conceivably play a role in mediating transfer of the LR signal from the node to the LPM. To test this, we determined whether the paraxial mesoderm develops normally in Sox17 mutants. In Sox17–/– embryos, both Meox1, which regulates somitogenesis (supplementary material Fig. S2A,C), and Raldh2 (Aldh1a2 – Mouse Genome Informatics), a key gene for retinoic acid synthesis that coordinates symmetric somitogenesis and LR asymmetry (Vermot and Pourquie, 2005) (supplementary material Fig. S2B,D), were expressed normally, suggesting that paraxial mesoderm is normal in Sox17–/– embryos.
Reduced gap junction function in Sox17 mutant epithelial cells
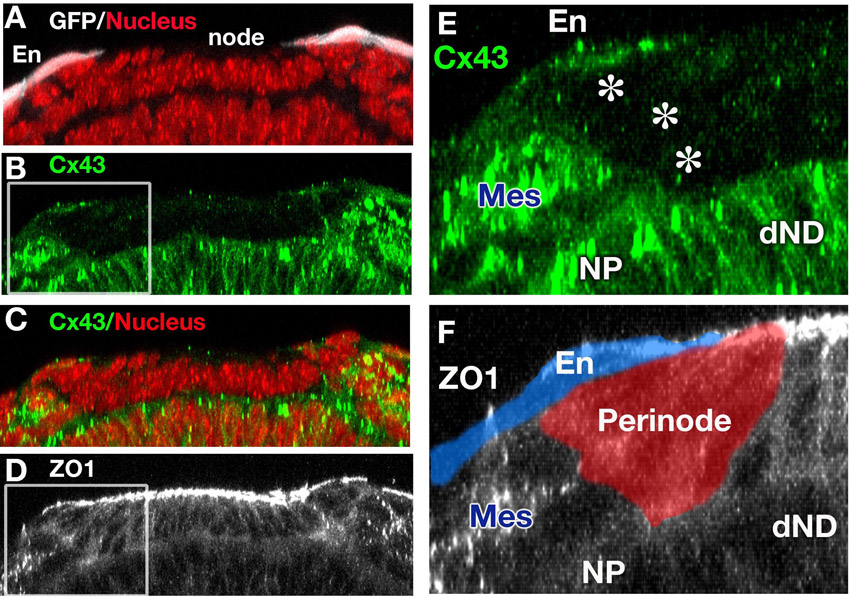
After nodal flow is established, intracellular upregulation of Ca2+ flux is observed in endoderm cells immediately left of the node (Hadjantonakis et al., 2008; McGrath et al., 2003; Tanaka et al., 2005). This suggests that Ca2+ flux might have an important role in signal transfer of LR determination from the node to the LPM (Mercola, 2003). Since the relay of Ca2+ flux is mediated by gap junctional transport of small molecules such as IP3 or Ca2+ itself, we examined the expression of a gap junction protein, connexin 43 (Cx43; Gja1 – Mouse Genome Informatics), by whole-mount IHC, which had not been described previously for the time when LR asymmetry is established. In control embryos, we found that Cx43 was localized in the cell membrane in both extra-embryonic and embryonic endoderm, but not in the node including perinode cells (Fig. 6A,E; supplementary material Fig. S3). In Sox17–/– embryos, Cx43 was localized in the cell membrane of extra-embryonic endoderm but was greatly reduced in the endoderm cells in the embryonic region (Fig. 6B,F). We also observed that the localization of another connexin, Cx40 (Gja5 – Mouse Genome Informatics), on the cell membrane was attenuated in the endoderm of Sox17–/– versus control embryos (data not shown).
Fig. 6.

Defective Cx43 localization and gap junctional transport in Sox17–/– endoderm cells. (A-F) Staining for Cx43 (green in A-F), nuclei (magenta in A,B; blue in C,D), ZO1 (magenta in C-F) and GFP (white in E,F). Asterisks indicate the node (A,B). In control embryos, Cx43 protein is normally localized in cell membranes of extra-embryonic (ExE) and embryonic (Emb) endoderm cells (A,A′,E,F). In Sox17–/– embryos at 3 ss, localization to cell membranes is disrupted only in embryonic endoderm cells and not in the extra-embryonic endoderm cells (B,B′,E,F). By contrast, ZO1 was localized on the cell boundaries in both Sox17–/– and Sox17+/– endoderm cells in the embryonic (C-F) and extra-embryonic (E,F) regions. (A′,B′,C,D) Embryonic endoderm cells. The boxed regions in A and B are magnified in A′ and B′. (G-J) Gap junctional transport was examined by iontophoresis of fluorescent dyes. A mixture of Lucifer Yellow and Rhodamine dextran (10 kDa) was injected into endoderm cells of the embryonic region (G,H) or into extra-embryonic endoderm cells (I,J). Three minutes after the injection, Lucifer Yellow dye was transferred to the surrounding cells in the embryonic region in Sox17+/– embryos (G) as well as to extra-embryonic endoderm cells in both Sox17+/– and Sox17–/– embryos (I,J), but only weakly transferred in the embryonic endoderm of Sox17–/– embryos (H). (K-L″) Cx43 mRNA expression in Sox17+/– (K-K″) and Sox17–/– (L-L″) embryos at 2 ss. Position of sections (K′,K″,L′,L″) are indicated in K and L. Red arrows indicate Cx43 expression in endoderm cells near the node (K″,L″). Note that no Cx43 expression is observed in the ventral side of the node. FgEn, foregut endoderm; HF, headfold; n, node. Scale bars: 50 μm in A,B,G; 5 μm in C,D; 10 μm in E,F; 100 μm in K-L″.
We next assessed the level of Cx43 mRNA and found that Cx43 expression was reduced in endoderm cells near the node in Sox17–/– embryos (Fig. 6L″, red arrows) as compared with control embryos (Fig. 6K″), whereas Cx43 mRNA was expressed normally in both the foregut and extra-embryonic endoderm of Sox17–/– embryos (Fig. 6K-L″).
Next, gap junction function was tested by iontophoresis of a gap junction permeable dye, Lucifer Yellow. Three minutes after dye injection, the Lucifer Yellow spread from the injected cell into neighboring cells of the embryonic endoderm (n=10, average 33.1 cells and 4.5 cell diameters; Fig. 6G) or extra-embryonic endoderm in control embryos (Fig. 6I), whereas the impermeable dye Rhodamine dextran was retained in the injected cell. In Sox17–/– embryos, the Lucifer Yellow dye, but not Rhodamine dextran, spread into neighboring cells of the extra-embryonic endoderm (Fig. 6J); however, in embryonic endoderm, both injected dyes stayed in the injected cell (n=6/23 injections) or the Lucifer Yellow expanded into significantly fewer cells (n=17/23, average 6.6 cells and 1.4 cell diameters; P<0.001, Student’s t-test) (Fig. 6H) than in the control embryo.
In conjunction with the reduction of Cx43 mRNA and in the membrane localization of connexin proteins, these data clearly indicate that Sox17–/– endoderm has defective gap junction communication, which is likely to be the major cause of defective LR determination in Sox17–/– embryos.
Sox17 mutants exhibit defects in the formation of epithelial structures in endoderm cells
Why was Cx43 not distributed in the cell membrane in Sox17 mutants? We first tested the possibility that the mislocalization of connexin proteins might be due to a lack of cell-cell contacts. The tight junction component ZO1 was localized normally on the cell membrane of Sox17–/– endoderm cells, similar to control endoderm (Fig. 6C,D), suggesting that Sox17 mutant endoderm develops an epithelial structure with tight junctions.
We further evaluated the epithelial organization of Sox17 mutant endoderm that lies in close proximity to the node, at stages corresponding to LR signal transfer from the node to the LPM (1-3 ss). We made use of Sox17GFP knock-in mice, in which a GFP reporter cassette replaces the coding region of the Sox17 locus, resulting in null mutation and placing GFP expression under the control of the endogenous transcriptional regulation of Sox17 (Kim et al., 2007).
In Sox17GFP/+ embryos at 2 ss, whole-mount IHC of GFP and the tight junction component ZO1 showed GFP expression in the tightly packed endoderm cells as a contiguous epithelial layer overlying mesoderm cells, encompassing the entire embryonic region except the midline and the node (Fig. 7A). Endoderm cells were attached to each other over a relatively large area of contact (Fig. 7A′,A″). In Sox17GFP/GFP embryos, although the endoderm layer covered the entire embryo in a manner similar to that of control endoderm, the endoderm cells constituting the endoderm layer were thinner and larger than those of the control embryo, with a greatly reduced area of contact between cells (Fig. 7B-B″). Unexpectedly, some GFP-positive cells in Sox17GFP/GFP embryos were found interspersed among the underlying mesoderm cells on either side of the midline (Fig. 7B′,B″). We hypothesized that these misplaced cells might be delaminated from the outer endoderm layer, possibly owing to the loss of epithelial cell polarity, or that these cells might be derived from endodermal cells that have failed to epithelialize after migrating out from the primitive streak. From these observations, we hypothesized that Sox17 is required for uniform structural and functional epithelial integrity in endoderm cells.
Fig. 7.

Loss of Sox17 results in defective epithelial polarity and disorganized cellular adhesion. (A-B″) GFP and ZO1 IHC in Sox17GFP/+ and Sox17GFP/GFP mouse embryos at 3 ss (A,B). Optical transverse sections from the reconstructed confocal images showed a compact epithelial layer of definitive endoderm cells in Sox17GFP/+ embryos (A′,A″), whereas in Sox17GFP/GFP embryos the epithelial layer was uneven and a portion of the GFP-positive cell population was found interspersed among the mesoderm cells (B′,B″). (C-D′) Co-IHC of α-tubulin and Cx43 together with nuclear staining in Sox17+/– and Sox17–/– embryos at 3 ss. Microtubules labeled with α-tubulin showed a parallel arrangement in Sox17–/– endoderm (star, D), in contrast to the radial arrangement in Sox17+/– embryos, accompanied by a reduction in the membrane localization of Cx43 (optical transverse sections in C′,D′). (E,F) E-cadherin, GFP and actin distribution in endoderm near the node. Note the disorganized localization of E-cadherin in the Sox17GFP/GFP embryos (F) as compared with the control embryos (E), suggesting weak adherens junctions. Stars indicate individual cells; the blue stars indicate cells with much reduced E-cadherin distribution in the cell boundary. Mes, mesoderm; N, node.
To determine whether the abnormal endoderm development in Sox17 mutants affects the membrane localization of gap junctions, we compared microtubule organization and Cx43 localization by co-IHC in control and Sox17 mutant endoderm cells (Shaw et al., 2007; Thomas et al., 2005). In control embryos, microtubules labeled by α-tubulin were distributed in a radial fashion and along the cell membrane (Fig. 7C,C′), whereas in Sox17 mutant embryos, which showed severely reduced Cx43 membrane distribution, α-tubulin was primarily present in a fibroblast-like parallel arrangement in the cytoplasmic region and did not show clear accumulation at the cell boundaries (Fig. 7D,D′) (Bre et al., 1990).
Adherens junctional complexes comprising E-cadherin and β-catenin constitute the main source of mechanical adhesion between adjacent epithelial cells (Baum and Georgiou, 2011). In endoderm cells neighboring the node of control embryos at 3 ss, E-cadherin was evenly localized in the basolateral membrane (Fig. 7E), whereas in Sox17–/– embryos E-cadherin distribution was reduced in most cells and was absent from some endoderm cells (Fig. 7F, blue stars). Labeling of actin filaments with phalloidin also showed an uneven localization of actin fibers at the cell boundary in endoderm cells of Sox17 mutants (Fig. 7F). Similar patterns were observed for β-catenin, which was not localized at cell boundaries in some endoderm cells (data not shown). These defects suggest weak intercellular adhesion between endoderm cells, even though ZO1 is localized normally on the cell membrane of Sox17 mutant endoderm cells (Fig. 6D).
Abnormal cytoskeletal assembly together with the incomplete formation of adherens junctions demonstrates defects in the epithelial features of Sox17 mutant endoderm. These defects could result in the loss of gap junction communication, either by lowering the levels of Cx43 transcription or by reducing the localization of Cx43 to the cell membrane, which might cause defects in transferring LR signals from the node to the LPM.
DISCUSSION
All tissues that have previously been reported to play a role in LR determination are mesodermal derivatives, such as the node, LPM, notochord and somites. Here, we report a novel role for endoderm cells in transferring LR signal information from the node to the LPM.
Sox17 mutants develop endoderm-specific defects and lose signal transfer
Sox17–/– endoderm cells exhibit epithelial disorganization. Despite this, the tight junction component ZO1 is still expressed normally, suggesting that the endoderm in the mutant maintains partial epithelium formation. Endoderm constitutes the outermost layer of the early embryo and plays an important role in homeostasis of the embryo by forming a tight junction barrier to the external environment. It should be noted that mesodermal and ectodermal, but not endodermal, derivatives form normally in Sox17 mutants (Fig. 3; data not shown).
The overall shape of the node in Sox17–/– embryos is affected by the presence of isolated endoderm cells that fail to clear from the node region; however, LR determination at the node seems to develop normally, as evidenced by the relatively normal nodal flow and the asymmetric expression of perinodal genes. Similarly, the LPM in Sox17 mutants also develops normally, with the potential to express and propagate Nodal, as shown when triggering Nodal proteins were exogenously supplied directly into the LPM. Therefore, the most likely cause of the absent or reduced LR gene expression observed in Sox17 mutants is a failure to transfer the LR signal from the node to the LPM.
Conserved gap junction function in establishing LR asymmetry in mammals
In this study we found that the localization of the gap junction molecules Cx43 and Cx40 to the cell membrane of the embryonic endoderm was greatly impaired in Sox17 mutants. Physiological assays of gap junction communication indicated that it was dysfunctional in mutant endoderm. These findings suggest the importance of gap junctions in endoderm for LR signal transfer. In addition to our data, it was recently reported that a pharmacological inhibitor of gap junction communication, 18-α-glycyrrhetinic acid, could inhibit Nodal expression in the left LPM of wild-type embryos (Viotti et al., 2012). Although these inhibitor experiments did not specifically identify the endoderm as the tissue responsible for the gap junction communication in LR determination, when combined with our findings from genetic analyses of Sox17 mutants, the data support the idea that gap junctions in the endoderm are necessary for signal transfer from the node to the LPM in the mouse embryo.
Gap junctions have been reported to function in LR determination in several vertebrate models (Levin and Mercola, 1998; Levin and Mercola, 1999). In rabbits, gap junctions are used to prevent signal transfer from the node to the right LPM in cooperation with Fgf8 signaling (Feistel and Blum, 2008). Although, in this case, the tissues responsible for the gap junction communication were not clearly identified, Cx43 is expressed in the endoderm as well as in the mesoderm and ectoderm of the rabbit embryo, similar to the mouse (Fig. 6; supplementary material Fig. S3). These data suggest that a role for endoderm cell gap junctions in mediating signals from the node to the LPM is conserved in mammals.
Impaired epithelial integrity in the Sox17 mutant endoderm and possible roles of endoderm in signal transfer from the node to the LPM
This is the first report that demonstrates the role of Sox17 in the development of the epithelial characteristics of endoderm at the cellular level. We examined the distribution of cell adhesion and cytoskeleton components in Sox17–/– and control embryos and found that endoderm in Sox17 mutants exhibits compromised epithelial integrity, in which the localization of E-cadherin/β-catenin on the cell membrane is disturbed and microtubules and actin filaments show aberrant arrangement. These findings strongly suggest that the severe impairment of gap junction communication in mutant endoderm is a consequence of the failure to properly localize connexin proteins on cell boundaries due to impaired epithelial polarity. Thus, epithelial maturity of endoderm cells near the node is essential for functional gap junction communication among endoderm cells and is involved in transferring signals from the node to the LPM in LR determination.
How do endoderm cells near the node mediate signal transfer from the node to the LPM? In accordance with the evidence of higher Ca2+ flux in the left endoderm near the node and the fact that the inhibition of gap junction communication results in defects in LR determination (Viotti et al., 2012), one explanation is that endoderm directly transfers second messengers such as Ca2+ through gap junctions from the node to the LPM to induce Nodal expression in the LPM. However, the higher Ca2+ flux in the left endoderm is observed only near the node, suggesting short-range signal transduction. Therefore, it is unlikely that Ca2+ itself signals over a long distance to transmit the LR information from the node to the LPM.
In addition to several studies showing that Nodal proteins act over a long range by diffusion (Chen and Schier, 2001; Constam, 2009; Le Good et al., 2005), a recent report strongly suggests that Nodal proteins produced in the perinode travel a long distance to induce Nodal expression in the left LPM (Kawasumi et al., 2011). Given these data, endoderm might play a role in supporting long-range Nodal movement. The long-range travel of Nodal proteins appears to require ECM components such as chondroitin sulfate (Oki et al., 2007). In Sox17 mutants, the ECM components chondroitin sulfate and laminin were formed relatively normally; however, it remains possible that there are other, as yet unidentified, ECM components that are required for Nodal movement and are affected in the mutant endoderm because of the incomplete epithelial polarity. An alternative possibility is that endoderm plays a role in the modification of Nodal proteins. The range of travel and activities of Nodal are regulated by processing of prepro-Nodal proteins (Ben-Haim et al., 2006), N-glycosylation (Le Good et al., 2005) and dimer formation with Nodal itself or another TGFβ member, Gdf1 (Tanaka et al., 2007). Since Sox17-expressing endoderm is tightly attached to the perinode where Nodal is produced, endoderm might assist in these modifications through cell-cell communication with perinode cells.
Ectopic expression of Nodal in endoderm cells of Sox17–/– embryos
We found that the foregut endoderm of Sox17–/– embryos expresses ectopic Nodal at early somite stages (Fig. 1D,D′,G). Nodal is an upstream regulatory factor that induces endoderm formation during gastrulation (Tremblay et al., 2000) but the expression is immediately shut down in differentiated endoderm (Collignon et al., 1996). These observations suggest that Sox17 might be directly or indirectly required to turn off Nodal expression in differentiating endoderm. If this were the case, we would predict that continued expression of Nodal might also contribute to the failure of endoderm formation in Sox17–/– embryos. Further analysis will be required to test this directly.
In summary, we have identified a novel and important role of endoderm cells in signal transfer from the node to the LPM during the establishment of LR asymmetry in mice. The detailed molecular mechanisms of how endoderm participates in the signal transfer remain to be examined in future studies. However, our present studies detailing Sox17 mutants will help further investigations and will significantly extend the understanding of the complex mechanisms involved in the determination of LR asymmetry in early development.
Supplementary Material
Acknowledgments
We gratefully acknowledge the technical assistance of Christin Schaaf. We also thank Masakazu Hashimoto for technical advice on visualizing nodal flow; Ann Foley, Hinako Kidokoro and Gary Schoenwolf for critical reading of the manuscript; Anna-Katerina Hadjantonakis for sharing unpublished data; Sean Morrison for providing Sox17GFP mice; and Hideo Otsuna for advice on imaging analysis using the FluoRender software.
Footnotes
Funding
The project was supported by grants from the National Institute of Child Health and Human Development [R01HD066121] and the March of Dimes Birth Defects Foundation [FY08-427] to Y.S.; the National Institute on Deafness and Other Communication Disorders [R01DC002994 to M.T.L.]; and the Ministry of Education, Science, Sports and Culture of Japan to M.K.-A.; R.S.S. was supported by an American Heart Association Predoctoral Fellowship [10PRE4450044]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.079921/-/DC1
References
- Adachi H., Saijoh Y., Mochida K., Ohishi S., Hashiguchi H., Hirao A., Hamada H. (1999). Determination of left/right asymmetric expression of nodal by a left side-specific enhancer with sequence similarity to a lefty-2 enhancer. Genes Dev. 13, 1589–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum B., Georgiou M. (2011). Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J. Cell Biol. 192, 907–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beahm D. L., Oshima A., Gaietta G. M., Hand G. M., Smock A. E., Zucker S. N., Toloue M. M., Chandrasekhar A., Nicholson B. J., Sosinsky G. E. (2006). Mutation of a conserved threonine in the third transmembrane helix of alpha- and beta-connexins creates a dominant-negative closed gap junction channel. J. Biol. Chem. 281, 7994–8009 [DOI] [PubMed] [Google Scholar]
- Beddington R. S., Robertson E. J. (1999). Axis development and early asymmetry in mammals. Cell 96, 195–209 [DOI] [PubMed] [Google Scholar]
- Ben-Haim N., Lu C., Guzman-Ayala M., Pescatore L., Mesnard D., Bischofberger M., Naef F., Robertson E. J., Constam D. B. (2006). The nodal precursor acting via activin receptors induces mesoderm by maintaining a source of its convertases and BMP4. Dev. Cell 11, 313–323 [DOI] [PubMed] [Google Scholar]
- Bre M. H., Pepperkok R., Hill A. M., Levilliers N., Ansorge W., Stelzer E. H., Karsenti E. (1990). Regulation of microtubule dynamics and nucleation during polarization in MDCK II cells. J. Cell Biol. 111, 3013–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan J., Norris D. P., Robertson E. J. (2002). Nodal activity in the node governs left-right asymmetry. Genes Dev. 16, 2339–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candia A. F., Hu J., Crosby J., Lalley P. A., Noden D., Nadeau J. H., Wright C. V. (1992). Mox-1 and Mox-2 define a novel homeobox gene subfamily and are differentially expressed during early mesodermal patterning in mouse embryos. Development 116, 1123–1136 [DOI] [PubMed] [Google Scholar]
- Chen Y., Schier A. F. (2001). The zebrafish Nodal signal Squint functions as a morphogen. Nature 411, 607–610 [DOI] [PubMed] [Google Scholar]
- Collignon J., Varlet I., Robertson E. J. (1996). Relationship between asymmetric nodal expression and the direction of embryonic turning. Nature 381, 155–158 [DOI] [PubMed] [Google Scholar]
- Constam D. B. (2009). Running the gauntlet: an overview of the modalities of travel employed by the putative morphogen Nodal. Curr. Opin. Genet. Dev. 19, 302–307 [DOI] [PubMed] [Google Scholar]
- Feistel K., Blum M. (2008). Gap junctions relay FGF8-mediated right-sided repression of Nodal in rabbit. Dev. Dyn. 237, 3516–3527 [DOI] [PubMed] [Google Scholar]
- Hadjantonakis A. K., Pisano E., Papaioannou V. E. (2008). Tbx6 regulates left/right patterning in mouse embryos through effects on nodal cilia and perinodal signaling. PLoS ONE 3, e2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada H., Meno C., Watanabe D., Saijoh Y. (2002). Establishment of vertebrate left-right asymmetry. Nat. Rev. Genet. 3, 103–113 [DOI] [PubMed] [Google Scholar]
- Hashimoto H., Rebagliati M., Ahmad N., Muraoka O., Kurokawa T., Hibi M., Suzuki T. (2004). The Cerberus/Dan-family protein Charon is a negative regulator of Nodal signaling during left-right patterning in zebrafish. Development 131, 1741–1753 [DOI] [PubMed] [Google Scholar]
- Hou J., Yashiro K., Okazaki Y., Saijoh Y., Hayashizaki Y., Hamada H. (2004). Identification of a novel left-right asymmetrically expressed gene in the mouse belonging to the BPI/PLUNC superfamily. Dev. Dyn. 229, 373–379 [DOI] [PubMed] [Google Scholar]
- Kanai Y., Kanai-Azuma M., Noce T., Saido T. C., Shiroishi T., Hayashi Y., Yazaki K. (1996). Identification of two Sox17 messenger RNA isoforms, with and without the high mobility group box region, and their differential expression in mouse spermatogenesis. J. Cell Biol. 133, 667–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai-Azuma M., Kanai Y., Gad J. M., Tajima Y., Taya C., Kurohmaru M., Sanai Y., Yonekawa H., Yazaki K., Tam P. P., et al. (2002). Depletion of definitive gut endoderm in Sox17-null mutant mice. Development 129, 2367–2379 [DOI] [PubMed] [Google Scholar]
- Kawasumi A., Nakamura T., Iwai N., Yashiro K., Saijoh Y., Belo J. A., Shiratori H., Hamada H. (2011). Left-right asymmetry in the level of active Nodal protein produced in the node is translated into left-right asymmetry in the lateral plate of mouse embryos. Dev. Biol. 353, 321–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I., Saunders T. L., Morrison S. J. (2007). Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell 130, 470–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Good J. A., Joubin K., Giraldez A. J., Ben-Haim N., Beck S., Chen Y., Schier A. F., Constam D. B. (2005). Nodal stability determines signaling range. Curr. Biol. 15, 31–36 [DOI] [PubMed] [Google Scholar]
- Levin M., Mercola M. (1998). Gap junctions are involved in the early generation of left-right asymmetry. Dev. Biol. 203 90–105 [DOI] [PubMed] [Google Scholar]
- Levin M., Mercola M. (1999). Gap junction-mediated transfer of left-right patterning signals in the early chick blastoderm is upstream of Shh asymmetry in the node. Development 126, 4703–4714 [DOI] [PubMed] [Google Scholar]
- Mahlapuu M., Ormestad M., Enerback S., Carlsson P. (2001). The forkhead transcription factor Foxf1 is required for differentiation of extra-embryonic and lateral plate mesoderm. Development 128, 155–166 [DOI] [PubMed] [Google Scholar]
- Marjoram L., Wright C. (2011). Rapid differential transport of Nodal and Lefty on sulfated proteoglycan-rich extracellular matrix regulates left-right asymmetry in Xenopus. Development 138, 475–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J., Somlo S., Makova S., Tian X., Brueckner M. (2003). Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell 114, 61–73 [DOI] [PubMed] [Google Scholar]
- Meno C., Saijoh Y., Fujii H., Ikeda M., Yokoyama T., Yokoyama M., Toyoda Y., Hamada H. (1996). Left-right asymmetric expression of the TGF beta-family member lefty in mouse embryos. Nature 381, 151–155 [DOI] [PubMed] [Google Scholar]
- Mercola M. (2003). Left-right asymmetry: nodal points. J. Cell Sci. 116, 3251–3257 [DOI] [PubMed] [Google Scholar]
- Nakamura T., Mine N., Nakaguchi E., Mochizuki A., Yamamoto M., Yashiro K., Meno C., Hamada H. (2006). Generation of robust left-right asymmetry in the mouse embryo requires a self-enhancement and lateral-inhibition system. Dev. Cell 11, 495–504 [DOI] [PubMed] [Google Scholar]
- Niederreither K., McCaffery P., Drager U. C., Chambon P., Dolle P. (1997). Restricted expression and retinoic acid-induced downregulation of the retinaldehyde dehydrogenase type 2 (RALDH-2) gene during mouse development. Mech. Dev. 62, 67–78 [DOI] [PubMed] [Google Scholar]
- Nonaka S., Tanaka Y., Okada Y., Takeda S., Harada A., Kanai Y., Kido M., Hirokawa N. (1998). Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 95, 829–837 [DOI] [PubMed] [Google Scholar]
- Oki S., Hashimoto R., Okui Y., Shen M. M., Mekada E., Otani H., Saijoh Y., Hamada H. (2007). Sulfated glycosaminoglycans are necessary for Nodal signal transmission from the node to the left lateral plate in the mouse embryo. Development 134, 3893–3904 [DOI] [PubMed] [Google Scholar]
- Osada S. I., Saijoh Y., Frisch A., Yeo C. Y., Adachi H., Watanabe M., Whitman M., Hamada H., Wright C. V. (2000). Activin/nodal responsiveness and asymmetric expression of a Xenopus nodal-related gene converge on a FAST-regulated module in intron 1. Development 127, 2503–2514 [DOI] [PubMed] [Google Scholar]
- Pearce J. J., Penny G., Rossant J. (1999). A mouse cerberus/Dan-related gene family. Dev. Biol. 209, 98–110 [DOI] [PubMed] [Google Scholar]
- Pfister S., Jones V. J., Power M., Truisi G. L., Khoo P. L., Steiner K. A., Kanai-Azuma M., Kanai Y., Tam P. P., Loebel D. A. (2011). Sox17-dependent gene expression and early heart and gut development in Sox17-deficient mouse embryos. Int. J. Dev. Biol. 55, 45–58 [DOI] [PubMed] [Google Scholar]
- Rankin C. T., Bunton T., Lawler A. M., Lee S. J. (2000). Regulation of left-right patterning in mice by growth/differentiation factor-1. Nat. Genet. 24, 262–265 [DOI] [PubMed] [Google Scholar]
- Saijoh Y., Adachi H., Sakuma R., Yeo C. Y., Yashiro K., Watanabe M., Hashiguchi H., Mochida K., Ohishi S., Kawabata M., et al. (2000). Left-right asymmetric expression of lefty2 and nodal is induced by a signaling pathway that includes the transcription factor FAST2. Mol. Cell 5, 35–47 [DOI] [PubMed] [Google Scholar]
- Saijoh Y., Oki S., Ohishi S., Hamada H. (2003). Left-right patterning of the mouse lateral plate requires nodal produced in the node. Dev. Biol. 256, 160–172 [DOI] [PubMed] [Google Scholar]
- Sakamoto Y., Hara K., Kanai-Azuma M., Matsui T., Miura Y., Tsunekawa N., Kurohmaru M., Saijoh Y., Koopman P., Kanai Y. (2007). Redundant roles of Sox17 and Sox18 in early cardiovascular development of mouse embryos. Biochem. Biophys. Res. Commun. 360, 539–544 [DOI] [PubMed] [Google Scholar]
- Shaw R. M., Fay A. J., Puthenveedu M. A., von Zastrow M., Jan Y. N., Jan L. Y. (2007). Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 128, 547–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M. M., Wang H., Leder P. (1997). A differential display strategy identifies Cryptic, a novel EGF-related gene expressed in the axial and lateral mesoderm during mouse gastrulation. Development 124, 429–442 [DOI] [PubMed] [Google Scholar]
- Sulik K., Dehart D. B., Iangaki T., Carson J. L., Vrablic T., Gesteland K., Schoenwolf G. C. (1994). Morphogenesis of the murine node and notochordal plate. Dev. Dyn. 201, 260–278 [DOI] [PubMed] [Google Scholar]
- Tanaka C., Sakuma R., Nakamura T., Hamada H., Saijoh Y. (2007). Long-range action of Nodal requires interaction with GDF1. Genes Dev. 21, 3272–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y., Okada Y., Hirokawa N. (2005). FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 435, 172–177 [DOI] [PubMed] [Google Scholar]
- Thomas T., Jordan K., Simek J., Shao Q., Jedeszko C., Walton P., Laird D. W. (2005). Mechanisms of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. J. Cell Sci. 118, 4451–4462 [DOI] [PubMed] [Google Scholar]
- Tremblay K. D., Hoodless P. A., Bikoff E. K., Robertson E. J. (2000). Formation of the definitive endoderm in mouse is a Smad2-dependent process. Development 127, 3079–3090 [DOI] [PubMed] [Google Scholar]
- Tsiairis C. D., McMahon A. P. (2009). An Hh-dependent pathway in lateral plate mesoderm enables the generation of left/right asymmetry. Curr. Biol. 19, 1912–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermot J., Pourquie O. (2005). Retinoic acid coordinates somitogenesis and left-right patterning in vertebrate embryos. Nature 435, 215–220 [DOI] [PubMed] [Google Scholar]
- Viotti M., Niu L., Shi S. H., Hadjantonakis A. K. (2012). Role of the gut endoderm in relaying left-right patterning in mice. PLoS Biol. 10, e1001276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson D. G. (1998). In Situ Hybridization – A Practical Approach. Oxford: Oxford University Press; [Google Scholar]
- Yan Y. T., Gritsman K., Ding J., Burdine R. D., Corrales J. D., Price S. M., Talbot W. S., Schier A. F., Shen M. M. (1999). Conserved requirement for EGF-CFC genes in vertebrate left-right axis formation. Genes Dev. 13, 2527–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka H., Meno C., Koshiba K., Sugihara M., Itoh H., Ishimaru Y., Inoue T., Ohuchi H., Semina E. V., Murray J. C., et al. (1998). Pitx2, a bicoid-type homeobox gene, is involved in a lefty-signaling pathway in determination of left-right asymmetry. Cell 94, 299–305 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}