

Abstract
S-glutathionylation is a reversible post-translational modification that continues to gain eminence as a redox regulatory mechanism of protein activity and associated cellular functions. Many diverse cellular proteins such as transcription factors, adhesion molecules, enzymes, and cytokines are reported to undergo glutathionylation, although the functional impact has been less well characterized. De-glutathionylation is catalyzed specifically and efficiently by glutaredoxin (GRx, aka thioltransferase), and facile reversibility is critical in determining the physiological relevance of glutathionylation as a means of protein regulation. Thus, studies with cohesive themes addressing both the glutathionylation of proteins and the corresponding impact of GRx are especially useful in advancing understanding. Reactive oxygen species (ROS) and redox regulation are well accepted as playing a role in inflammatory processes, such as leukostasis and the destruction of foreign particles by macrophages. We discuss in this review the current implications of GRx and/or glutathionylation in the inflammatory response and in diseases associated with chronic inflammation, namely diabetes, atherosclerosis, inflammatory lung disease, cancer, and Alzheimer’s disease, and in viral infections.
Introduction
Glutaredoxin and S-glutathionylation
S-glutathionylation is a post-translational modification whereby the cysteine-sulfhydryl moiety of glutathione (GSH) forms a disulfide bond with a cysteine-sulfhydryl moiety of a protein, and it represents a mechanism of reversible redox regulation of protein activity and cell signaling (Gallogly and Mieyal, 2007; Shelton et al., 2005). S-glutathionylation of some proteins (protein-SSG) seems to persist paradoxically in the reducing environment of the cell, while a signaling stimulus is necessary for glutathionylation of other proteins (Mieyal et al., 2008). Glutaredoxin (GRx) has been well characterized by us and others as an efficient and specific catalyst of de-glutathionylation, responsible for intracellular reversal of protein-SSG (Chrestensen et al., 2000; Gravina and Mieyal, 1993; Thomas et al., 1995; Yang et al., 1998). The catalytic mechanism of protein-SSG formation in cells is still largely unknown, despite the attribution to several enzymes including GRx itself under certain conditions (Gallogly and Mieyal, 2007). Physiological and pathological relevance of GRx and glutathionylation can be exemplified by the recent explosion in PubMed entries, even though the number of works addressing the two with respect to one another has been limited (Figure 1). For example, increased GRx activity was reported to modulate potassium channel gating in diabetic rat hearts (Li et al., 2005), but whether glutathionylation of channel proteins is the underlying mechanism of action was not addressed. Furthermore, the majority of studies reporting protein glutathionylation have not tested its reversibility by GRx, an essential criteria for determining its potential as a regulatory mechanism. Future studies of GRx and glutathionylation will inevitably merge and shed light on sulfhydryl redox regulation of protein activity and cellular functions. We review here the implications of GRx and glutathionylation on inflammatory responses, associated diseases, and viral infections.
Figure 1.
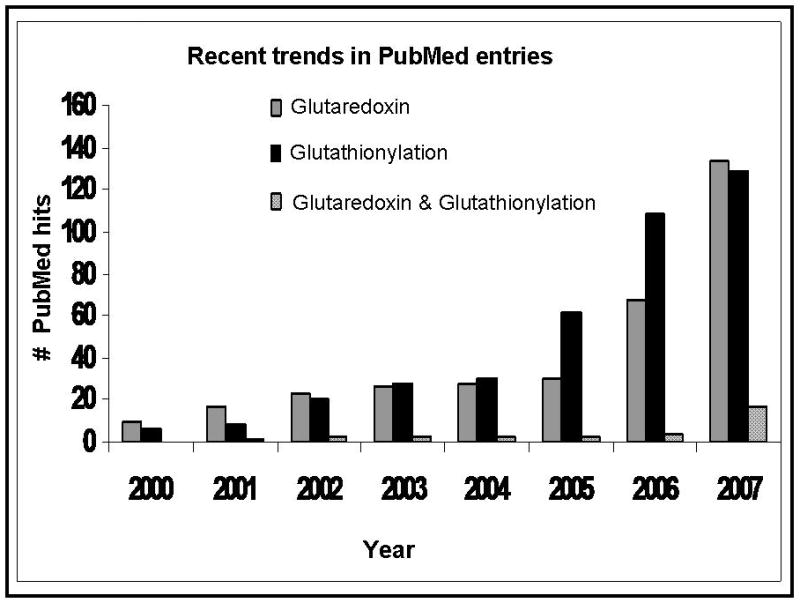
Number of yearly publications involving studies on GRx, on glutathionylation, or on glutaredoxin and glutathionylation as measured in PubMed entries.
Reactive Oxygen Species (ROS), Leukocytes, Cytokines, and Glutaredoxin (GRx)
The balance of inflammatory signaling versus chronic hyper-active inflammatory processes determines whether the outcome is the removal of a danger (e.g. bacterial infection) or progression to disease (e.g. atherosclerosis). Key characterizations of inflammation are production of pro-inflammatory cytokines typically through activation of the NFκB signaling pathway and transmigration of the circulating leukocyte from the blood to tissue (Figure 2). Inflammatory responses are largely intertwined with redox signaling and reactive oxygen species (ROS). ROS are involved in inflammatory signal transduction leading to cytokine production, and ROS contribute to killing the pathogenic agents in the inflammatory cells after phagocytosis. ROS and/or cytokines mediate white blood cell recruitment to the site of injury, promote glutathionylation of many proteins in inflammatory signaling, and possibly activate or induce expression of GRx.
Figure 2.
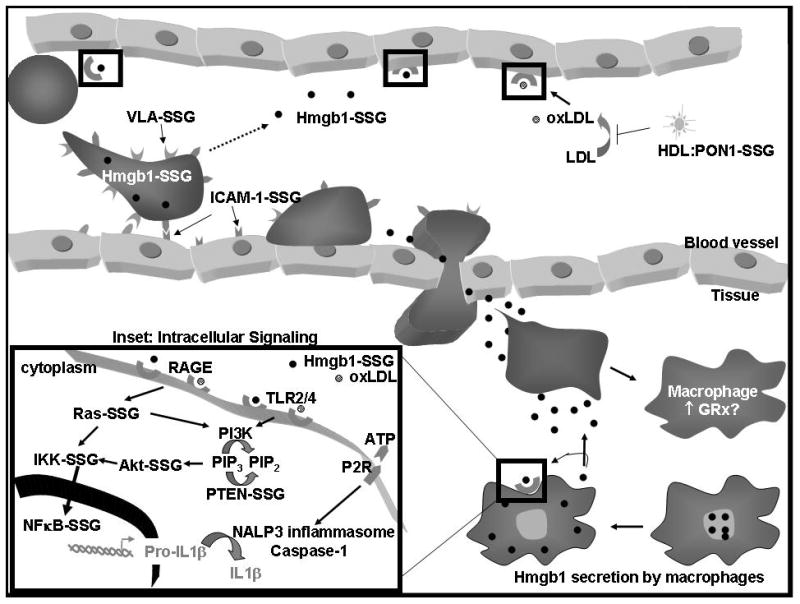
Glutathionylated proteins involved in inflammatory responses. Shown here are leukostasis, diapedesis, and production of pro-inflammatory cytokines in response to Hmgb1 that is secreted from resident macrophages. Nuclear Hmgb1 is transported to the cytosol for active lysosomal secretion by various cells such as macrophages and leukocytes. Ligands (Hmgb1, oxLDL) binding to the RAGE and TLR2/TLR4 receptors (highlighted in black boxes) induce an intracellular signaling cascade shown in the inset. Glutaredoxin (GRx) might also be involved in monocyte differentiation into macrophages.
ROS and cytokine induction of glutaredoxin and de-glutathionylation
ROS were reported to induce GRx expression in human coronary artery smooth muscle cells (CASMCs), (Okuda et al., 2001), and ROS are known to induce cytokine production. TNFα and IL1β are the prototypic pro-inflammatory cytokines, and anti-inflammatory cytokines (e.g. IL-10, IL-13) inhibit pro-inflammatory cytokine production by macrophages. Chemokines are chemotactic cytokines, responsible for the recruitment of circulating leukocytes into sites of injury in tissue and include RANTES, MCP-1, MIP-1α, and MIP-1β, IL-8, lymphotactin, and Fractalkine.
IL-6 has been reported to induce GRx in murine monocytic leukemia-derived M1 cells (Takashima et al., 1999), and IL-4 and IL-13 increased GRx in mouse airway epithelial cells (Reynaert et al., 2007). IFNγ increased GRx with a corresponding decrease in protein glutathionylation in mouse airway epithelial cells (Reynaert et al., 2007). However, the induction of GRx seems to be cytokine-specific because TGFβ1 decreases GRx with a corresponding increase in protein glutathionylation in mouse airway epithelium and human alveolar epithelium (Peltoniemi et al., 2004), (Reynaert et al., 2007) (Table 1). TNFα seems to induce differential changes in GRx and glutathionylation likely dependent on species, cell type, and treatment conditions (Table 1). TNFα leads to increased GRx activity and decreased glutathionylation of pro-caspase 3 in bovine aortic endothelial cells (Pan and Berk, 2007), but TNFα causes no change in GRx in mouse airway epithelium or human alveolar epithelium (Peltoniemi et al., 2004; Reynaert et al., 2007). Increased glutathionylation of ICAM-1 and IKKα in TNFα–treated human pulmonary aortic endothelial cells (Mukherjee et al., 2007) suggests a potential decrease in GRx activity and further adds to the complexity.
Table 1.
The effects of cytokine treatments on glutaredoxin and/or glutathionylation of proteins.
| Cytokine | Treatment | Change | Cell type | Reference |
|---|---|---|---|---|
| IL-6 | 20ng/ml 72hr | ↑ GRx protein | Murine monocytic leukemia-derived M1 cells | Takashima et al. 1999 |
| IL-4 | 20ng/ml 48hr | ↑ GRx activity | Mouse airway epithelium | Reynaert et al. 2007 |
| IL-13 | 10pg/ml 48hr | ↑ GRx activity | Mouse airway epithelium | Reynaert et al. 2007 |
| IFN-γ | 20pg/ml 28hr | ↑GRx activity, ↓PSSG | Mouse airway epithelium | Reynaert et al. 2007 |
| * TGFβ1 | 2ng/ml 72hr, 10ng/ml 48hr | ↓ GRx protein and activity, ↑ PSSG | Human alveolar epithelium, Mouse airway epithelium | Peltoniemi et al. 2004, Reynaert et al. 2007 |
| TNFα | 10ng/ml + 10μg/ml CHX, 3–6hr | ↑ GRx activity, ↓ Pro-caspase 3-SSG | Bovine aortic endothelium | Pan and Berk 2007 |
| TNFα | 50ng/ml 24–72hr, 20ng/ml 48hr | No change in GRx | Mouse airway epithelium, Human alveolar epithelium | Reynaert et al. 2007, Peltoniemi et al. 2004, |
| TNFα | 10ng/ml 6hr | ↑ ICAM-1-SSG | Human pulmonary aortic endothelial cells | Murkherjee et al. 2007 |
| TNFα | 10ng/ml 6hr | ↑ IKKα-SSG | Human pulmonary aortic endothelial cells | Murkherjee et al. 2007 |
TGFβ1 (2ng/ml) had no affect on GRx at 24 and 48 hr (Peltoniemi et al. 2004). Abbreviations: CHX, cycloheximide; protein-SSG, glutathionylated proteins.
Role of extracellular glutaredoxin?
GRx protein has been identified in sputum supernatant and plasma from non-smokers, smokers, and smokers with chronic obstructive pulmonary disease (COPD), and it was elevated in sputum supernatants in patients with COPD exacerbations (Peltoniemi et al., 2006). Samples were immunoprobed for actin and manganese superoxide dismutase to eliminate the possibility that GRx was released from ruptured cells; however, a standard soluble cytosolic marker such as lactate dehydrogenase would have been more conclusive. The identity of cells that secrete GRx into extracellular milieu was not fully investigated, but since macrophages have been reported to contain the majority of pulmonary GRx, they were speculated to be the alveolar site of secretion. In contrast to alveolar macrophages, PMA (phorbol 12-myristate 13-acetate) inhibited GRx secretion from peripheral blood mononuclear cells (PBMC), suggesting that these differentiated monocytes and lymphocytes may not secrete GRx in vivo. GRx has also been reported to be present in human plasma (Lundberg et al., 2004; Nakamura et al., 1998), and to be decreased in plasma of pre-operative cardiac surgery patients (Nakamura et al., 1998). These analyses were conducted with an ELISA assay for GRx, and Nakamura et al. demonstrated that GRx content does not correlate with hemoglobin content, suggesting that lysis of red blood cells does not contribute substantially to plasma GRx. In addition, Lundberg et al. analyzed intra- and extra-cellular GRx in twelve diverse cell lines. GRx was found in all eight cell lines (A549, ECV304, H82, HEC293, HeLa, Jurkat, U1810, and U937), and in the extracellular media of three cell lines (HeLa, Jurkat, and U937) (Lundberg et al., 2004). Together, these reports suggest that if GRx is actively secreted by macrophages it may be tissue specific, and that macrophages may not be the primary source of GRx excreted into the plasma. The function of extracellular GRx, its mechanism of secretion, and translocation stimuli are currently unknown. GRx does not seem to function as a chemokine since it does not attract polymorphonuclear neutrophilic leukocytes (PMNs) and monocytes in a Boyden chamber (Bertini et al., 1999). However, GRx-mediated deglutathionylation of extracellular proteins is conceivable. For example, glutathionylation has been implicated in the regulation of membrane anchored molecules essential for leukostasis (i.e. ICAM-1-SSG and VLA-SSG, see below) as well as secreted and circulating proteins (Hmgb1-SSG and PON-1-SSG respectively, see below). However, it is not known whether the other factors involved in catalytic turnover of GRx are also in the plasma in sufficient quantities, i.e., GSH, GSSG Reductase, and NADPH.
Glutaredoxin in monocyte-macrophage differentiation
IL-6 is known to trigger differentiation of the murine monocytic leukemia-derived M1 cell line into macrophages, and GRx induction via IL-6 corresponded to macrophage phagocytic activity (Takashima et al., 1999). Additionally, 12-myristate 13-acetate (PMA) treatment, known to induce macrophage differentiation of a human monocytic leukemic cell line U937, also elevated GRx mRNA (Takashima et al., 1999). However, GRx protein was reported to be unaltered in peripheral blood mononuclear cells (PBMC) in response to PMA (Lundberg et al., 2004). The apparent discrepancy between these studies could be due to differences in the cells and possibly the stimuli. It would be instructive in future studies to monitor macrophage maturation in stimulated non-cancer cells where GRx was knocked down. Furthermore, determining the activity of GRx in monocytes compared to macrophages will be essential, but the implication that regulation by GRx could be involved in macrophage differentiation is intriguing, particularly in the context of atherosclerosis where monocyte differentiation into macrophages is the hallmark of early plaque development and subsequent macrophage differentiation into foam cells marks disease progression (Bobryshev, 2006) (see below). Circulating blood cells likely differ in the fine tuning of redox modulations compared to differentiated tissue-embedded macrophages, and tissue specificity must be considered. For example, alveolar macrophages are unique in their exposure to high levels of oxygen, and have been distinguished from peritoneal macrophages in release of TNFα, IL-6, NO· and O2·− (Forman and Torres, 2001). Additional studies have highlighted the importance of GRx in differentiated macrophages. Decreased GRx and corresponding increases in protein glutathionylation were associated with increased adriamycin-induced damage of macrophages (Asmis et al., 2005). In accordance with this study, GRx was also shown to be important in macrophage defense against oxLDL (Wang et al., 2006).
Signaling an inflammatory response
Inflammatory signaling can be initiated either by exogenous “non-self” pathogen-associated molecular pattern molecules (PAMPs) such as lipopolysaccharide (LPS) or by endogenous danger signals that are released in response to tissue damage, called damage (or danger)-associated molecular patterns (DAMPs) (also known as alarmins or endokines) (e.g. Hmgb1, ATP, ROS, Sendai virus) (Foell et al., 2007; Harris and Raucci, 2006; Petrilli et al., 2007). Hmgb1 has been reported to be glutathionylated (see below), and is particularly interesting because it can initiate pro-inflammatory cytokine production through both toll-like receptors (TLRs) and the receptor for advanced glycation end products (RAGE) (Figure 2, inset). Signal transduction downstream of TLRs and RAGE is carried out by distinct but intersecting signaling pathways, which contain many mediators that are glutathionylated. In brief, RAGE activates the NFκB upstream activator, IkB kinase (IKK), through Ras whereas the PI3K-Akt pathway mediates TLR signaling (van Beijnum et al., 2008) (Figure 2, inset). Notably, both signaling cascades converge on the activation of the NFκB pathway, which involves a particularly abundant group of proteins that are subject to modulation by glutathionylation. In addition, HS60 and HS70 are examples of TLR2/4 ligands (Seong and Matzinger, 2004) that have been shown to be glutathionylated in T-lymphocytes in response to oxidative stimuli (Fratelli et al., 2002).
The binding of DAMPs or PAMPs to pattern recognition receptors (PRRs) such as the toll like receptors (TLRs) on the membrane of the cell surface leads to activation of the inflammasome and production of pro-IL1β and pro-IL18 via activation of NFκB (see inset in Figure 2) (Church et al., 2008; Mariathasan and Monack, 2007). PAMP activation of TLRs also leads to the production of many other pro-inflammatory cytokines and chemokines such as RANTES, macrophage-derived chemokine (MDC), interferon-gamma-inducible protein (IP-10), MIP1α, and MIP1β, tumor necrosis factor-α (TNF-α), and interleukins IL-1β and IL-12 (Biragyn et al., 2002). A second DAMP or PAMP signal activates an inflammasome via intracellular PRRs called NOD-like receptors (NLRs). Within the known inflammasome family, The NALP3 inflammasome is the best characterized and most promiscuously activated, representing a complex of proteins including the NLR NALP3 and pro-caspase-1 (Church et al., 2008; Petrilli et al., 2007). Activation of the NALP3 inflammasome leads to caspase-1 activation and subsequent cleavage of pro-IL-1β and pro-IL-18 to mature cytokines (Figure 2, inset) (Church et al., 2008; Mariathasan and Monack, 2007).
S-glutathionylation of inflammatory signaling mediators
Hmgb1-SSG
High mobility group protein B1 (Hmgb1) is a nuclear alarmin that is released into the extracellular milieu via cellular necrosis or lysosomal exocytosis after translocation to the cytoplasm in activated monocytes, macrophages, mature dendritic cells, natural killer cells, and endothelial cells (Figure 2) (Harris and Raucci, 2006; van Beijnum et al., 2008). Hmgb1 translocation and secretion has been linked to regulation by post-translational modifications such as methylation, acetylation, and phosphorylation by blockade of the nuclear localization sequence (Harris and Andersson, 2004; van Beijnum et al., 2008). Hmgb1 acts as a chemokine to recruit circulating monocytes, T cells and dendritic cells, induces NFκB activation and subsequent production of pro-inflammatory cytokines, and elicits cross presentation and antigen specific T cell immunity of activated dendritic cells via receptor for advanced glycation end products (RAGE) and toll like receptor 2 and 4 (TLR2, TLR4) (Hagemann et al., 2007; Harris and Andersson, 2004; van Beijnum et al., 2008).
Glutathionylation of Hmgb1 was reported in the nucleus of human and rat retinal pigmented epithelial (RPE) cell lines in response to diamide treatment, and reported to be documented by mass spectral analysis (Hoppe et al., 2006). Evidence of protein glutathionylation in the nucleus is limited but not unprecedented (see NFκB discussion in cancer section). Though the majority of Hmgb1 is typically in the nucleus, its localization is tissue-specific (Harris and Andersson, 2004). Therefore, subsequent analysis of Hmgb1 compartmentalization in the RPE, or an additional control for cytosolic contamination such as lactate dehydrogenase is essential. Because diamide is an artificial oxidant, future studies addressing the glutathionylation status of Hmgb1 (Hmgb1-SSG) in cells stimulated with inflammatory cytokines or lipopolysaccharide (LPS), and the effect of manipulation of GRx activity in situ, will be essential in determining the physiological relevance of Hmgb1-SSG. Also woMutational analysis suggested that Cys106 is necessary for nuclear sequestering of Hmgb1 in Chinese hampster ovary epithelium cells (CHO-K1) (Hoppe et al., 2006), but effects of glutathionylation of the three cysteine residues in Hmgb1 on subcellular localization was not tested. Since active secretion of Hmgb1 seems to be best characterized in activated inflammatory cells such as macrophages, analysis of the glutathionylation status in the subcellular compartments and extracellular milieu of these cells would also be intriguing. Overall, delineating the impact of changes in GRx and/or glutathionylation on Hmgb1 localization, recruitment of T cells, dendritic cells, and monocytes, and binding to the receptor for advanced glycation end product (RAGE) and toll like receptor-4 (TLR4) on dendritic cells and monocytes would uncover potential points of regulation in inflammation. Whether glutathionylation impacts Hmgb1 affinity for RAGE or TLR4 is unknown. Since Hmgb1 leads to anti-tumorigenic T-cell cross presentation and pro-tumorigenic production of inflammatory cytokines, differences in Hmgb1 receptor affinity might be a pathological determinant (Hagemann et al., 2007).
In addition, the pro-inflammatory properties of Hmgb1 have been indicated in many diseases such as diabetic retinopathy, arthritis, atherosclerosis, systemic lupus erythematosus, cancer, and sepsis (Harris and Raucci, 2006; van Beijnum et al., 2008). Hmgb1 has a 7-fold higher affinity for RAGE than does its classical ligands, advanced glycation end products (AGEs), and independent of Hmgb1, involvement of the RAGE receptor has been found to be predominant in diabetes, amyloidosis, and cancer among other inflammatory diseases.
PON1-SSG
Oxidized LDL (oxLDL) is a well established atherogenic agent, and can activate signaling transduction pathways by binding to TLRs and RAGE (Figure 2 and Figure 2 inset) (Harja et al., 2008; Seong and Matzinger, 2004). PON1 is a paraoxonase found in complex with high density lipoprotein (HDL), prevents oxidation of low density lipoprotein (LDL), and hydrolyzes lipid peroxides in LDL via its arylesterase and lactonase activities (Rozenberg and Aviram, 2006; Singh et al., 2007). Overall, PON1 activity is associated with inflammatory diseases such as coronary artery disease, atherosclerosis, and diabetes (Flekac et al., 2007; Singh et al., 2007); however, the reported activity of PON1 seems to be quite variable, dependent on the nature of the study, and displaying interindividual and racial variation. For example, PON1 activity is decreased in coronary artery disease in plasma of North West Indian Punjabis irrespective of diabetes (Singh et al., 2007); however, activity of PON1 in leukocytes of a non-specific population is decreased in both type 1 and type 2 diabetes (Flekac et al., 2007). Furthermore, both PON1 transgenic mice and PON1 knockout mice suggest that PON1 protects against atherosclerosis (Shih et al., 2000; Tward et al., 2002).
Of the three cysteines in PON1, two can form an intramolecular disulfide, and thiol modification of the third one (Cys 283) seems to be near the active site and mediates PON1 inhibition (Nishio and Watanabe, 1997). Mutational analysis revealed that the presence of Cys 283 of PON1 is essential only for its protection of LDL against oxidation, but modifications to Cys 283 inhibit the LDL oxidation protection, arylesterase activity, and paraoxonase activity (Aviram et al., 1999a). Glutathione (GSH), N-acetyl cysteine (NAC), L-cysteine, and 2-mercaptoethanol preserved PON1 paraoxonase activity in the presence of an inhibitory stimulus (cigarette smoke extract), while DTNB (5,5′-dithiobis(2-nitrobenzoic acid) had an inhibitory effect (Nishio and Watanabe, 1997). Incubation of PON1 with oxidized lipids or oxLDL, all isolated from human serum, led to reductions in PON1 arylesterase activity and protection of LDL from oxidation. The lipid binding to Cys283 of PON1 seems to mediate the observed inhibition (Aviram et al., 1999b).
PON1 that was evolved in E. Coli via several rounds of DNA shuffling and screening and PON1 associated with serum HDL that was extracted from human volunteers were both reported to be inhibited by glutathionylation (Rozenberg and Aviram, 2006). Arylesterase, paraoxonase, and lactonase activities were inhibited upon PON1 incubation with oxidized glutathione (GSSG), and activity could be partially restored after treatment with the non-specific disulfide reducing agent, DTT (dithiolthreitol). Elevated HDL-mediated macrophage cholesterol efflux has been attributed previously to PON1 activity on the ABCA1 transporter (Rosenblat et al., 2005; Rozenberg and Aviram, 2006), and Rozenberg and Aviram demonstrated that GSSG decreased the cholesterol efflux associated with HDL in macrophages, presumably through PON1-SSG formation and concomitant inactivation.. The data thus far suggests potential redox regulation of cholesterol via PON1-SSG and could have vast impact in cardiovascular diseases. However, much more work is needed to determine whether glutathionylation of PON1 occurs in vivo in response to a physiological stimulus, and is reversible by an endogenous catalyst i.e. GRx.
VLA-4-SSG
α4 integrins are essential in homing and recruitment of leukocytes and mobilization of progenitor cells (Pujades et al., 1996). Very late antigen-4 (VLA-4, α4β1) is a leukocyte integrin that binds to vascular cell adhesion molecule-1 (VCAM-1), mucosal addressin cell adhesion molecule-1 (MadCAM-1), and fibronectin (Liu et al., 2008)(Figure 2). All 24 cysteines of the α4 subunit of VLA-4 are found in its extracellular domain (Liu et al., 2008), and mutations of Cys278 and Cys717 inhibited VCAM-1 binding in K562 cells (Pujades et al., 1996). Recently, α4 was shown to be glutathionylated under basal conditions in HL-60 clone 15 cells, a cell line that is known to differentiate into eosinophil-like cells upon treatment with n-butyrate (Liu et al., 2008). Treatment of the cells with H202 with or without exogenous GSH increased glutathionylation of α4 as well as its binding to recombinant VCAM-1, and decreased cell rolling on VCAM-1-coated flow chambers (Liu et al., 2008). Moreover, higher concentrations of H202 decreased VCAM-1 binding (Liu et al., 2008), consistent with the authors previous study demonstrating that H202, sodium nitroprusside (SNP), and GSNO inhibited VCAM-1 binding to HL60 clone 15 cells (Chuang et al., 2003). However, superoxide treatment increased cell binding to VCAM-1, and it was shown to be a primary mediator of cell adhesion at the concentrationstested. Overall, these data emphasize that redox regulation occurs in a stimulus- and concentration-dependent manner.
ICAM-1-SSG
ICAM-1 is a primary inflammatory marker best characterized for its role in the tight adherence of leukocyte integrins to endothelial cells, an essential step in leukocyte extravasation (Figure 2). ICAM-1 expression is induced by cytokines such as IFN-γ, IL-1, TNF-α, and IL-6 (Teppo et al., 2001), and is transcriptionally regulated by NFκB. Also, anti-ICAM-1 antibody cross-linking to ICAM-1 can lead to induction of RANTES, ICAM-1, and VCAM, and IL-8 (Blaber et al., 2003; Clayton et al., 1998; Sano et al., 1998). In addition to its direct role in leukostasis, binding of ICAM-1 can induce internalization of ICAM-1 and its binding partner and recycling of ICAM-1 to the membrane in a unique CAM-mediated endocytosis fashion in endothelial cells (Muro et al., 2005). ICAM-1 was recently suggested to play a role in viral neuroinvasion of West Nile virus and associated encephalitis (Dai et al., 2008). Consistent with this study, ICAM-1 is important in the entry of enteroviruses and rhinoviruses and infectivity of HIV-1 (Dai et al., 2008; Xiao et al., 2001). Soluble forms of ICAM (sICAM) can be translated from differentially spliced mRNA or proteolytic shedding from the extracellular domain of membrane-bound ICAM, and sICAM has been found to be increased in inflammatory diseases (Gho et al., 1999; Kusterer et al., 1998). sICAMs have been proposed to both counteract and exacerbate the pro-inflammatory effects of membrane-bound ICAM-1 (Gho et al., 1999; Kusterer et al., 1998). Glutathionylation of ICAM-1 was detected in human pulmonary aortic endothelial cells (HPAEC), and this was increased by TNFα. The extent of TNFα–induced glutathionylation was attenuated by low doses of N-acetylcysteine (NAC) or mitoquinone-Q (mito-Q) (Mukherjee et al., 2007). Increased glutathionylation of ICAM-1 corresponded to enhanced surface expression of ICAM-1 and monocyte adhesion, suggesting that GRx-mediated de-glutathionylation of ICAM-1 would oppose these effects. Data from other studies have implicated a pro-inflammatory role of intracellular GRx via activation of the IKK/NFκB signaling pathway, leading to enhanced cytokine production and intracellular ICAM-1 production, respectively(Reynaert et al., 2006; Shelton et al., 2007). Regulation of ICAM-1 by glutathionylation is an exciting field of study, having potential effects on viral infectivity, endocytosis, ICAM-1 oligomerization, leukostasis, proteolytic cleavage to sICAM forms, and intracellular signaling. Glutathionylation of sICAM could also affect its binding to leukocyte integrins. ICAM-SSG reversal by GRx and corresponding changes in function in vivo are critical determinants to be examined.
IKK-SSG
The IkB kinase (IKK) is a 600–900kDa multimeric complex, consisting of a core of IKKα, IKKβ, and IKKγ subunits, and the IKK complex can be activated by cytokine receptors and PRRs (Lawrence et al., 2005). The IKK complex activates nuclear translocation and inflammatory signaling of NFκB by phosphorylation of the inhibitory protein, IkB (Figure 2, inset). Individual activities of the catalytic subunits (IKKα and IKKβ) can be quite distinct (Lawrence et al., 2005; Scheidereit, 2006), and while IKKβ is clearly a critical mediator, IKKα has been reported not to have a substantial impact in pro-inflammatory responses of NFκB (Li et al., 1999). However, IKKα has also been reported in separate studies to have both anti- and pro-inflammatory roles (Lawrence et al., 2005; Li et al., 2002; Li et al., 1999). To further complicate the signaling network, IKKα and IKKβ both have been found in the nucleus, but only IKKα is thought to be involved in cytoplasm-nuclear shuttling in response to cytokine stimulation (Anest et al., 2003). Several post-translational modifications such as phosphorylation and ubiquitination are well established to modulate activities of IKK; and glutathionylation of IKKβ was demonstrated to have a regulatory role in expression of pro-inflammatory gene expression driven by NF-κB in lung epithelial cells, reversible by GRx (Reynaert et al., 2006) In addition, glutathionylation of IKKα has been reported in human pulmonary aortic endothelial cells, induced via TNFα treatment (Mukherjee et al., 2007). Thus far, IKKβ has been more thoroughly documented for regulation by GRx, but the overall impact of IKK subunit glutathionylation on inflammatory responses will need to be evaluated with respect to cell type, subcellular localization, and stimulus. It also will interesting to determine the catalytic efficiency (Vmax/Km) for each IKK subunit for turnover by GRx, and how overall IKK activity is affected by changes in GRx. IKK is the major convergent point of many signaling cascades (e.g. Figure 2, inset), and this underscores the important of its regulation. Studies aimed at learning whether the additional proteins in the IKK complex influence the substrate specificity or accessibility of IKK to GRx or whether glutathionylation of the other IKK proteins (α β γ) proteins impact signaling via the IKK complex will be advance understanding of elucidating a complicated regulatory process.
NFκB-SSG
NFκB can be activated directly in an IKK-independent (atypical) fashion or activated indirectly by signaling through IKK-dependent (canonical, non-canonical, and atypical) pathways (Perkins, 2007). Production of pro-inflammatory cytokines is largely attributed to NFκB activation, and often considered independently of NFκB-regulated apoptotic events. In vitro glutathionylation of p50 (p50-SSG) was shown to inhibit its DNA binding (Pineda-Molina et al., 2001). Our research group found that GRx restores DNA binding activity of nuclear NFκB isolated from hypoxic pancreatic cancer cells treated with N-acetylcysteine (NAC), suggesting inhibition of NFκB via glutathionylation of p50, p65, or a transcriptional co-activator/repressor (Qanungo et al., 2007). Consistent with these studies, GRx was reported to enhance NIK-mediated activation of NFκB in HEK293 cells (Hirota et al., 2000). S-glutathionylation also has been reported to inhibit at least 11 other proteins involved in signaling pathways leading to activation of NFκB, including the ubiquitin-activating (E1) and ubiquitin-carrier (E2) enzymes (Jahngen-Hodge et al., 1997; Obin et al., 1998), 20S proteasome (Demasi et al., 2003), Ras (Adachi et al., 2004; Clavreul et al., 2006a; Clavreul et al., 2006b), Akt (Murata et al., 2003), PTEN (Cruz et al., 2007), and IKKα/IKKβ (Mukherjee et al., 2007; Reynaert et al., 2006). The complexity and cross-talk of signaling pathways provide multiple check points that support proper cell function, but also challenge the elucidation of regulatory mechanisms, especially because the control points differ among cell types and cellular conditions. Glutathionylation of NFκB itself or its upstream signaling intermediates is an important consideration in cell regulation and production of pro-inflammatory cytokines, making this a potential focal point of therapeutic intervention in inflammatory diseases, as discussed further (below).
Ras-SSG
Ras is an important signaling mediator of the NFκB pathway as well as in Akt signaling (Figure 2 inset). Angiotensin II and oxLDL lead to glutathionylation and concomitant activation of Ras with subsequent Akt activation (Adachi et al., 2004; Clavreul et al., 2006a; Clavreul et al., 2006b). Furthermore, overexpression of GRx inhibits the Ras activation pathway, presumably via de-glutathionylation of Ras-SSG.
Akt-SSG and PTEN-SSG
ROS is known to activate the PI3K-Akt pathway, and extracellular ATP leads to generation of ROS in macrophages via the purinergic receptors (P2X and P2Y) (Cruz et al., 2007; Foell et al., 2007). Akt activity is promoted by PI3K-catalyzed PIP3 formation, and inhibited by conversion of PIP3 back to PIP2, mediated byPTEN (phosphatase and tensin homologue deleted from chromosome 10) (Figure 2 inset). ATP-driven formation of ROS activated the PI3K/Akt pathway and IL-1β production in alveolar macrophages, and this response corresponded to an increase in glutathionylation of PTEN (Cruz et al., 2007). Glutathionylation was interpreted to inhibit PTEN activity, thereby enhancing activation of the PI3K/Akt pathway. Akt itself was previously suggested to be regulated by GRx (Murata et al., 2003; Wang et al., 2007), and additional Akt-interacting proteins PP2A and ASK-1 have been implicated in regulation by glutathionylation and binding to GRx, respectively. Learning the extent of regulation by glutathionylation of each signaling mediator is necessary to reveal individual contributions to overall control of the activity of the PI3K/Akt pathway. Nevertheless, PI3K/Akt signaling is a prevalent inflammatory pathway, and regulation by glutathionylation has significant implications.
Implications of S-glutathionylation in Inflammatory Diseases
We will discuss in brief the role of glutaredoxin and S-glutathionylation in a number of diseases involving inflammatory mediators, namely diabetes, atherosclerotic cardiovascular disease, inflammatory lung disease, cancer and, Alzheimer’s neurodegenerative disease. For an additional review, see (Mieyal et al., 2008).
Diabetes
Chronic elevated blood glucose levels lead to the metabolic inflammatory disease called diabetes, involving tissue damage associated with accumulation of advanced glycation end products (AGEs), activation of protein kinase C (PKC), activation of the polyol pathway, and activation of the hexosamine pathway (Brownlee, 2001). Superoxide is the predominant ROS implicated in propagation of these pathways (Brownlee, 2001). AGEs are particularly relevant in the context of this review since much attention is focused on the RAGE receptor. In addition, high glucose, ROS, and oxLDL are implicated in stimulation of adhesion molecules and chemokines, with subsequent macrophage accumulation (Tesch, 2007). OxLDL also has been reported to mediate Ras glutathionylation in insulin resistance (Clavreul et al., 2006b). IκB kinase (IKK) activation is greatly involved in insulin resistance, and in particular, IKKβ mediates anti-inflammatory and anti-diabetic effects of aspirin and aspirin-derivatives (Arkan et al., 2005; Shoelson et al., 2003). NFκB signaling is activated in tissues of diabetic animals including the retina (Romeo et al., 2002; Shelton et al., 2007; Zheng et al., 2004), kidney (Schmid et al., 2006), and liver (Cai et al., 2005).
The PKC pathway is a target of glutathionylation, and GRx was reported to reactivate PKC-α, which was glutathionylated in NIH3T3 cells (Ward et al., 2000). In vitro studies demonstrated the affects of glutathionylation on specific isoforms of PKC (Chu et al., 2001).. Furthermore, GRx is increased in the heart and eye of diabetic rat models ((Li et al., 2005; Shelton et al., 2007) (Figure 3), suggesting that GRx likely plays an important regulatory role in redox signaling in diabetes. For a detailed review of protein glutathionylation (aldose reductase, SERCA, RyR, PTP1B, Ras, MEKK, c-Jun, Akt, IKK, NFκB, etc.) and GRx (regulation of potassium channels and insulin exocytosis) in insulin secretion and signaling, see (Mieyal et al., 2008).
Figure 3.
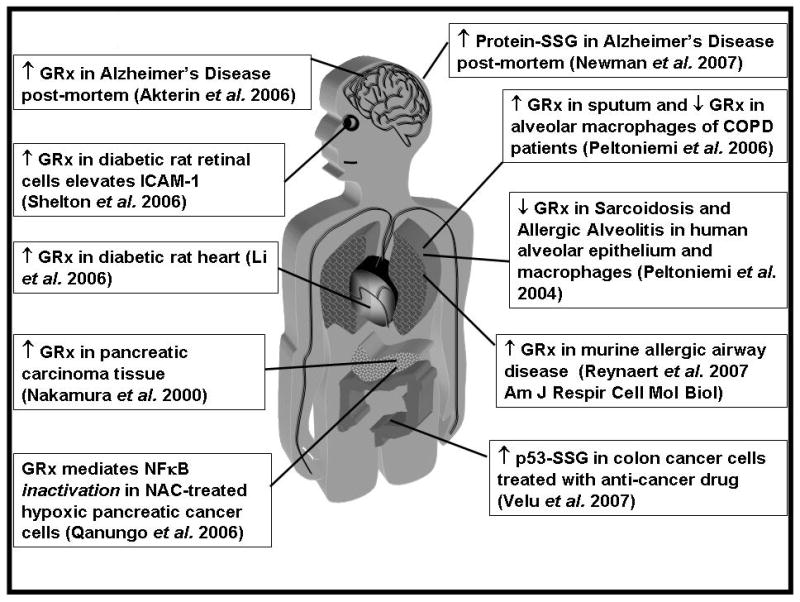
Changes in glutaredoxin and protein glutathionylation in disease. COPD is chronic obstructive pulmonary disease
Atherosclerosis
Atherosclerosis is the prototypic cardiovascular disease involving inflammation, and atherosclerotic plaques induce signaling pathways in endothelial cells via TLR2 and TLR4 (Seong and Matzinger, 2004). As mentioned above, monocyte migration through the endothelium and concomitant differentiation into macrophages are hallmarks of the early stages of atherosclerosis. Further differentiation of macrophages into foam cells is accompanied by lipid uptake and accumulation, and foam cell aggregation constitutes the core of atherosclerotic plaques. oxLDL enhances monocyte recruitment and is taken up by macrophages, promoting foam cell development, and oxLDL can be found in established macrophage foam cells within atherosclerotic plaques (Bobryshev, 2006). GRx has been identified in endothelial cells, adventitia fibroblasts, and medial smooth muscle cells in both atherosclerotic and non-atherosclerotic coronary arteries of post-mortem humans, and localized predominantly in to infiltrated macrophages of atheroscle-rotic lesions (Okuda et al., 2001). OxLDL seems to mediate glutathionylation of Ras and subsequent Akt signaling in endothelial cells (Clavreul et al., 2006b). Ras is activated by glutathionylation, and this is reversible by GRx (Adachi et al., 2004). An extensive review of protein glutathionylation (actin, GAPDH, complex I, complex II, SERCA, RyR, α-KGDH, and Ras) in a broader scope of cardiovascular diseases can be found in (Mieyal et al., 2008).
Inflammatory lung diseases
Lung tissue is exposed to more oxygen than most other parts of the body, suggesting that redox regulation would play a critical role in physiological and pathological processes of the pulmonary system. Briefly discussed here are the potential roles of GRx and reversible protein-SSG formation in COPD, Asthma, CF, UIP, and Sarcoidosis and Allergic Alveolitis.
COPD
The most common irritant of chronic obstructive pulmonary disease (COPD), cigarette smoke, leads to accumulation and activation of macrophage and neutrophils, and the subsequent tissue damage in the lung is irreversible (Peltoniemi et al., 2006). GRx was decreased in alveolar macrophages in progression of COPD severity, and GRx content was correlated with lung function (Figure 3) (Peltoniemi et al., 2006). The decreases in GRx observed in whole lung homogenates were similar to those in the inflammatory cells, indicating that GRx is mostly localized in macrophages (Peltoniemi et al., 2006). The authors speculated that the decrease in GRx could be a result of downregulation by TGF-β, or to secretion of GRx. In contrast to decreases of intracellular GRx, it was reported to be elevated in sputum supernatants in patients with COPD exacerbations (Figure 3); however identifying the source of this extracellular GRx is anticipated to be a subject of future studies.
Asthma
Similar to COPD, allergen-induced inflammation in the lung is a common component of asthma, and eosinophil and neutrophil generation of ROS is robust (Reynaert et al., 2007). A number of different changes in GRx were observed in different contexts. First, GRx was found to be decreased in the murine lung with allergic airway disease and in human alveolar epithelium in response to TGFβ (Peltoniemi et al., 2004; Reynaert et al., 2007). Secondly, treatment with IL-4, IL-13, and IFNγ led to increased GRx activity in murine airway epithelium, and finally, TNFα was found not to have an effect on GRx (see above, Table 1, (Reynaert et al., 2007). Accordingly, INFγ and TGFβ led to an increase and decrease in protein glutathionylation, respectively. In contrast to the results for human cells by Peltoniemi et al., Reynaert et al. (Reynaert et al., 2007) found significant expression of GRx in murine bronchial epithelium, and these authors speculated that the differences could be attributed to species specificity (Reynaert et al., 2007). Therefore, it will be interesting to see how cytokines such as IL-4, IL-13, and INFγ affect GRx and protein glutathionylation in analogous studies in human tissue. Furthermore, GRx and glutathionylation of IKK were shown to be involved in NFκB-mediated cytokine production (Reynaert et al., 2006).
UIP, Sarcoidosis and Allergic alveolitis
GRx was documented in macrophages in human lung with three inflammatory diseases, namely Sarcoidosis, Allergic Alveolitis, and Usual Interstitial Pneumonia (UIP) (Peltoniemi et al., 2004). GRx in human lung tissue was reported to be nearly exclusively localized to alveolar macrophages, with much less content in bronchial epithelium (Peltoniemi et al., 2004). GRx was decreased in alveolar macrophages and bronchial epithelial cells in lungs of smokers with either Sarcoidosis or Allergic Alveolitis (Peltoniemi et al., 2004). UIP also decreased GRx immunostaining in epithelium atop fibroblast foci and in other fibrotic areas (Peltoniemi et al., 2004). GRx was reported to be present in alveolar macrophages, but no changes in response to UIP were indicated (Peltoniemi et al., 2004).
Cystic Fibrosis
Cystic Fibrosis (CF) lung disease increases the risk of airway infection and subsequent inflammation, characterized by infiltrating inflammatory cells, increased cytokines, and increased ROS production (De Rose, 2002). Underlying the pathophysiology of CF is a dysfunction in the cystic fibrosis transmembrane conductance regulator (CFTR), leading to altered ion transport in the lung. CFTR activity was reported to be inhibited by glutathionylation at Cys 1344 and restored by E. Coli GRx-mediated deglutathionylation (Wang et al., 2005), and human GRx was reported to give similar results in part of the study. Though interesting, several unknowns need to be discovered before evaluating the potential therapeutic utility of modulating reversible glutathionylation of the CFTR. For example, studies using human GRx under physiological conditions (i.e., non- CFTR-overexpressing cells) would be recommended. Alternatively, identifying increases in CFTR expression levels and changes in CFTR-SSG status in CF patients could validate the current model.
Cancer
Cancer treatments damage and kill cells, leading to the release of chemokines, cytokines, and factors such as the alarmin Hmgb1 (Apetoh et al., 2007; Hagemann et al., 2007) (see descriptions above of Hmgb1-SSG in other contexts). Inflammatory cells are essential in clearing damaged cancer cells and cell debris, and macrophages can constitute up to 50% of tumor masses (Hagemann et al., 2007). Hmgb1 may trigger tumor-promoting pro-inflammatory cytokine production from macrophage and dendritic cells (Hagemann et al., 2007). However, Hmgb1 release from dying cancer cells was shown to be an essential component of the T cell anti-tumor antigen cross presentation immune response via TLR4 (Apetoh et al., 2007). The effect of glutathionylation and GRx on Hmgb1 localization, chemotaxis, and receptor binding (discussed above) are critical in evaluating the utility of redox manipulation in cancer therapeutics. In addition, the study linking regulation of NFκB activity by GRx-mediated glutathionylation was conducted in the context of hypoxic pancreatic cancer cells (see above, (Qanungo et al., 2007)), implicating NFκB and modulation of its glutathionylation status as a regulatory mechanism in cancer. Consistent with this concept, a previous study reported elevated levels of GRx in pancreatic carcinoma tissue (Figure 3, (Nakamura et al., 2000)). Together, these studies support the role of NFκB activation (which is inhibited by GRx-reversible glutathionylation) in producing pro-survival factors in pancreatic cancer. These limited examples already demonstrate profound implications for cancer regulation via GRx and glutathionylation. See (Mieyal et al., 2008) for a review of other examples implicated in cancer, including protein kinase C (PKC), PI3K-Akt and associated proteins, protein tyrosine phosphatase (PTP), c-Jun N-terminal kinase (JNK), H-Ras, NFκB pathway proteins, c-Jun, p53, and AP-1, pro-caspase 3, and Ku.
Alzheimer’s Disease
An established feature of Alzheimer’s disease (AD) is β-amyloid toxicity, and accumulation of A-beta plaques, neurofibrillary tan gles, and reactive microglia are thought to underlie neuroinflammation (Akiyama et al., 2000; Shepherd et al., 2007). In addition, β-amyloid is a ligand for RAGE (van Beijnum et al., 2008). Elevated GRx has been reported in Alzheimer’s disease of post-mortem brains (Figure 3), and A-beta accumulation in a cell culture model leads to oxidation of GRx which is unable to bind ASK, promoting activation of ASK, (Akterin et al., 2006). In a separate proteomics study, increased glutathionylation was reported for α-enolase, GAPDH, deoxyhemoglobin, and α-Crystallin B, and concomitant inhibition of GAPDH and enolase activities were reported in AD brain (Newman et al., 2007). The increase in both GRx and glutathionylation reported in these two studies is an apparent inconsistency if conditions favor GRx-mediated deglutathionylation; however, in the event that glutathionyl-radical generating conditions prevail, then GRx may promte glutathionylation of proteins (Qanungo et al., 2007; Starke et al., 2003).
S-glutathionylation and glutaredoxin in viral infections
Viruses are commonly known for compromising the immune system by depleting CD4+ helper T cells, but also elicit classical inflammatory responses. For example, viruses such as the polio virus, rubella virus, cytomegalovirus, enterovirus, and Coxsackie virus lead to myocarditis or inflammation of the myocardium. The inflammatory events associated with HIV as well as hepatitis C virus (HCV) have been correlated with the increased risk of the pro-inflammatory disease of atherosclerosis (Eugenin et al., 2008; Madden et al., 2008; Melendez et al., 2008; Reingold et al., 2008). HIV-infected individuals have increased concentration of serum lipids, soluble ICAM-1, soluble VCAM-1, insulin resistance, soluble tumor necrosis factor-alpha (sTNFR2), fibrinogen, and pro-inflammatory cytokines (TNFα, IL-6, IL-1β) and chemokines (MIP1α, MIP1β, and RANTES) (Appay and Sauce, 2008; Madden et al., 2008; Melendez et al., 2008). Furthermore, HIV can invade lymphocytes by viral fusion with their chemokine receptors CCR5 and CXCR4 acting as co-receptors with CD4 or by endocytosis upon binding to CCR5, CXCR4, or CD4 (Eugenin et al., 2008). CCR5 ligands, macrophage inflammatory protein (MIP)-1α and β and RANTES, have been shown to inhibit viral entry (Kasama et al., 2006).
Once inside the host cell, the viral RNA is converted to DNA and is then integrated into the host genetic material. Viral replication utilizes the host machinery, and new viral particles are enveloped for transport to the next host cell. HIV-1 proteases are aspartyl proteases critical for viral maturation, and are potentially regulated by S-glutathionylation (Davis et al., 1996; Davis et al., 1997). S-glutathionylation of Cys67-SSG leads to activation, and S-glutathionylation of Cys95-SSG (or glutathionylation of both Cys95 and Cys67) inhibits protease dimerization and proteolysis (Davis et al., 1997). GRx preferentially deglutathionylates Cys95-SSG, restoring proteolytic activity. Thus, selective deglutathionylation of Cys95-SSG on HIV-1 that is glutathionylated on both Cys95 and Cy67 would lead to a super-active protease (Davis et al., 1997). Hence, GRx taken up from host cells may be important in regulating maturation and/or propagation of the virus under oxidative stress conditions of the viral infection. Analogous to HIV, Human T-cell leukemia virus type 1 (HTLV-1) is a retrovirus that utilizes a HTLV-1 protease that is also inhibited by glutathionylation (Davis et al., 2003). In addition, GRx-catalyzed de-glutathionylation rescues the activity of HTLV-1 protease (Davis et al., 2003). Thus, GRx promotes activity of HIV-1 and HTLV-1 proteases, suggesting GRx may represent a therapeutic target in anti-viral treatments.
Sendai virus was recently shown to induce GRx-catalyzed deglutathionylation of IRF3 and corresponding transcriptional activation (Prinarakis et al., 2008). Interferon regulatory factor 3 (IRF3) is a transcription factor that regulates production of interferons. Activated IRF3 dimerizes and translocates to the nucleus where it partners with CBP/p300 co-activators to regulate expression of genes such as IFNs that contain interferon-stimulated response elements (ISREs) in their promoter regions (Prinarakis et al., 2008). Ablation of viral induced-IFNs in GRx knockdown cells led to modest decreases in NFκB activation and minimal inhibition of IL-8 (Prinarakis et al., 2008). The mechanism of regulation by glutathionylation of IRF3 does not seem to involve IRF3 dimerization, nuclear translocation, or phosphorylation but rather binding to CBP and possibly DNA binding (Prinarakis et al. 2008). Reduction of IRF3 by DTT fails to mimic reduction by GRx in restoring IRF3 binding to CBP (Prinarakis et al., 2008). This result accentuates the fact that DTT reduces other disulfide bonds besides glutathione-containing mixed disulfides, and it highlights that DTT cannot substitute for GRx as a diagnostic tool for qualitative and quantitative analysis of protein-SSG. Mutational analysis was reported to reveal potential glutathionylation of all six cysteines in IRF3, and glutathionylation of four of the cysteines (C-222, C-347, C-289, and C-371) each decreased IRF3 transcription (Prinarakis et al., 2008). Additional studies would be beneficial to elucidate the glutathionylation status of IRF3 in the nucleus, and to discover what impact glutathionylation of IRF3 has on its nuclear translocation or nuclear sequestration.
Summary
There is an expanding awareness that inflammatory events play an integral role in nearly every known disease, including diabetes, cancer, cardiovascular diseases, inflammatory lung disease, rheumatoid arthritis, Muckle-Wells syndrome, inflammatory bowel disease, sepsis, Crohn’s disease, lupus, psoriasis, asthma and chronic bronchitis, amyloidosis and spongiform encephalopathy, cirrhosis, hepatitis, cystic fibrosis, periodontitis, ischemia/reperfusion injury, gout, familial and cold autoinflammatory syndrome, etc. Many inflammatory responses are related according to mediators (oxLDL, Hmgb-1, AGE, S100 proteins, and β-amyloid) that activate receptors in common, e.g., TLR or RAGE; and the signaling cascades emanating from these receptors are largely subject to regulation by glutathionylation. A listing of the key proteins discussed throughout this review is provided in Table 2. As shown, glutathionylation regulates proteins with vastly different cellular functions such as adhesion molecules, enzymes, cytokines, and transcription factors. Further studies are encouraged that will evaluate whether reversible glutathionylation of these identified proteins actually plays a role in regulation of cellular functions. In particular, does glutathionylation or de-glutathionylation of these proteins occur within the cell under natural conditions in response to physiological stimuli and lead to functional changes that are reversible by GRx?
Table 2A.
Proteins implicated in regulation by glutathionylation that are involved in inflammatory processes.
Proteins of inflammation implicated in regulation by glutathionylation
| Protein | Function | Affect of S-glutathionylation | Stimuli | Milieu | GRx tested | Reference |
|---|---|---|---|---|---|---|
| α-4VLA | Leukocyte integrin | ↑ Binding to VCAM, ↓ Cell rolling | H2O2 +/− GSH | Eosinophils | No | Liu et al. 2008 |
| Hmgb1 | Alarmin/DAMP | N/A | Diamide | RPE cells | Yes | Hoppe et al. 2006 |
| HIV-1 protease | Aspartic proteinase for viral maturation | ↓ proteolysis | GSH + guanidine | in vitro | Yes | Davis et al. 1997 |
| HTLV-1 protease | Aspartic proteinase for viral maturation | ↓ proteolysis | GSH + guanidine | in vitro | Yes | Davis et al. 2003 |
| ICAM-1 | Adhesion molecule | N/A | TNFα +/− NAC and Mito-Q | Pulmonary aortic endothelial cells | No | Mukerjee et al. 2007 |
| IKKα | Kinase | N/A; Linked to ↑ IkBα phosphorylation | TNFα +/− NAC and Mito-Q | Jurkat cells; Pulmonary endothelial cells | No | Mukerjee et al. 2007 |
| IKKβ | Kinase | ↓ IkB phosphorylation and NFκB activation | TNFα + H2O2 | Airway epithelial cells | Yes | Reynaert et al. 2006 |
Abbreviations: AII, Angiotensin II; GSSG, glutathione disulfide; HEK, human embryonic kidney; Mito-Q, mitoquinone-Q; NAC, N-acetyl cysteine; RPE, retinal pigmented epithelium
GRx has been reported to be increased in many inflammatory diseases (Figure 3), but whether this induction is reflective of a homeostatic attempt to protect the body from physiological dysfunction, or an attribute of the disease progression is a topic of speculation requiring further experimental examination. Knowledge of multiple aspects of disease that correspond to changes in glutaredoxin will also be important. Collectively, the findings on glutaredoxin are not comprehensive enough to determine whether targeting it would be therapeutic effective. Most likely therapeutic manipulation of glutaredoxin would have to be disease specific and localized, because of its tissue- and cell-specific roles under different conditions. For example, inhibition of glutaredoxin would appear to be a meaningful therapeutic approach for AIDS, because GRx appears to mediate activation of the HIV-1 protease. Also localized inhibition of GRx in the retina may block its pro-inflammatory role in activation of the NFκB pathway. In contrast, glutaredoxin appears to mediate glutathionylation and inactivation of NFκB in hypoxic pancreatic cancer cells treated with N-acetyl cysteine, suggesting that enhancing GRx activity in this case would promote apoptosis of these cancer cells. Likewise, the paradoxical increase in glutaredoxin along with an increase in protein glutathionylation (reported in separate studies) in Alzheimer’s disease requires further studies to determine whether GRx is responsible for the increased protein-SSG and whether these events contribute to disease progression, thus identifying GRx as a target for inhibition. Glutaredoxin has been reported to change in different directions in different models of inflammatory lung disease, but whether the different results are due to artificial aspects of the models needs to be resolved. The rapidly evolving nature of humans and disease may keep us forever chasing a satisfactory mechanism for proper inflammatory balance to ward off infection but avoid self-mutilation. Nevertheless, glutaredoxin-regulated glutathionylation likely represents one important mechanism by which inflammatory pathways and signaling mediators are modulated, and thus further attention to this system is warranted to discover novel means of therapeutic intervention.
Table 2B.
Proteins of inflammation implicated in regulation by glutathionylation
| Protein | Function | Affect of S-glutathionylation | Stimuli | Milieu | GRx tested | Reference |
|---|---|---|---|---|---|---|
| IRF3 | Transcription factor | ↓ CBP binding, ↓ IFN production | Indigenous | HEK293 cells | Yes | Prinarakis et al. 2008 |
| NFκB (p50) | Transcription factor | ↓ DNA Binding | GSSG | In vitro | No | Pineda-Molina et al. 2001 |
| PKC | Kinase | ↓ Peptide phosphorylation | Diamide | NIH3T3 cells | No | Ward et al. 2000 |
| PON-1 | Esterase | ↓ paraoxonase, arylesterase, and lactonase activities | GSSG | Evolved PON1, Human HDL-PON1 | No | Rozenberg & Aviram 2006 |
| PTEN | Phosphatase | ↓ Phosphatase activity (speculative) | ATP | Alveolar Macrophages | No | Cruz et al. 2007 |
| Ras | G protein | ↑ Protein synthesis, p38 and Akt activation, | All, OxLDL, peroxynitrite | Smooth muscle and Endothelial cells | Yes | Chevruel et al. 2006; Chevruel et al. 2006; Adachi et al. 2004 |
Acknowledgments
This work was supported in part by NIH grants 1 R01 AG024413 and 1 PO1 AG 15885 (JJM), a Merit Review grant from the Department of Veteran’s Affairs (JJM), and NIH training grant 5 T32 EY07157 (MDS).
Abbreviations
- AGE
advanced glycation end products
- COPD
chronic obstructive pulmonary disease
- DAMP
damage-associated molecular patterns (also known as danger-associated molecular patterns)
- DTT
dithiolthreitol
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- GRx
glutaredoxin (also known as thioltransferase)
- GSH
glutathione
- GSSG
glutathione disulfide
- GSNO
S-nitrosylated glutathione
- H2O2
hydrogen peroxide
- HDL
high density lipoprotein
- HIV-1
Human immunodeficiency virus type 1
- Hmgb1
high mobility group protein B1
- HTLV-1
Human T-cell leukemia virus type 1
- ICAM-1
intercellular adhesion molecule-1
- IFN
inter-feron
- IL
interleukin
- IRF3
interferon regulatory factor 3
- IKK
IκB kinase
- LDL
low density lipoprotein
- LPS
lipopolysaccharide
- Mito-Q
mitoquinone-Q
- NAC
N-acetyl cysteine
- NEM
N-ethyl maleimide
- NLR
NOD-like receptor
- NFκB
nuclear factor κB
- PAMP
pathogen-associated molecular pattern molecule
- PI3K
phosphatidylinositol 3-kinase
- PIP2
phosphatidyl-inositol biphosphate
- PIP3
phosphatidylinositol 3,4,5-triphosphate
- PBMC
peripheral blood mononuclear cells
- PON1
paraoxonase
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- PRR
pattern-recognition receptor
- protein-SSG
protein-glutathione mixed disulfide (S-gluta-thionylated protein)
- PTP
protein tyrosine phosphatase
- PTEN
phosphatase and tensin homologue deleted from chromosome 10
- RAGE
receptor for advanced glycation end product
- RPE
retinal pigmented epithelium
- RyR
ryanodine receptor
- ROS
reactive oxygen species
- SNP
sodium nitroprusside
- SOD
superoxide dismutase
- TDOR
thiol disulfide oxidoreductase
- TLR
toll like receptor
- TNFα
tumor necrosis factor alpha
- UIP
usual interstitial pneumonia
- VLA-4
very late antigen-4
- VCAM
vascular cell adhesion molecule
- VSMC
vascular smooth muscle cell
References
- Adachi T, Pimentel DR, Heibeck T, Hou X, Lee YJ, Jiang B, et al. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akterin S, Cowburn RF, Miranda-Vizuete A, Jimenez A, Bogdanovic N, Winblad B, et al. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer’s disease. Cell Death Differ. 2006;13:1454–1465. doi: 10.1038/sj.cdd.4401818. [DOI] [PubMed] [Google Scholar]
- Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- Appay V, Sauce D. Immune activation and inflammation in HIV-1 infection: causes and consequences. J Pathol. 2008;214:231–241. doi: 10.1002/path.2276. [DOI] [PubMed] [Google Scholar]
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- Asmis R, Wang Y, Xu L, Kisgati M, Begley JG, Mieyal JJ. A novel thiol oxidation-based mechanism for adriamycin-induced cell injury in human macrophages. FASEB J. 2005;19:1866–1868. doi: 10.1096/fj.04-2991fje. [DOI] [PubMed] [Google Scholar]
- Aviram M, Rosenblat M, Billecke S, Erogul J, Sorenson R, Bisgaier CL, et al. Human serum paraoxonase (PON 1) is inactivated by oxidized low density lipoprotein and preserved by antioxidants. Free Radic Biol Med. 1999a;26:892–904. doi: 10.1016/s0891-5849(98)00272-x. [DOI] [PubMed] [Google Scholar]
- Aviram M, Rosenblat M, Billecke S, Erogul J, Sorenson R, Bisgaier CL, et al. Human serum paraoxonase (PON 1) is inactivated by oxidized low density lipoprotein and preserved by antioxidants. Free Radic Biol Med. 1999b;26:892–904. doi: 10.1016/s0891-5849(98)00272-x. [DOI] [PubMed] [Google Scholar]
- Bertini R, Howard OM, Dong HF, Oppenheim JJ, Bizzarri C, Sergi R, et al. Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J Exp Med. 1999;189:1783–1789. doi: 10.1084/jem.189.11.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298:1025–1029. doi: 10.1126/science.1075565. [DOI] [PubMed] [Google Scholar]
- Blaber R, Stylianou E, Clayton A, Steadman R. Selective regulation of ICAM-1 and RANTES gene expression after ICAM-1 ligation on human renal fibroblasts. J Am Soc Nephrol. 2003;14:116–127. doi: 10.1097/01.asn.0000040595.35207.62. [DOI] [PubMed] [Google Scholar]
- Bobryshev YV. Monocyte recruitment and foam cell formation in atherosclerosis. Micron. 2006;37:208–222. doi: 10.1016/j.micron.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrestensen CA, Starke DW, Mieyal JJ. Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J Biol Chem. 2000;275:26556–26565. doi: 10.1074/jbc.M004097200. [DOI] [PubMed] [Google Scholar]
- Chu F, Ward NE, O’Brian CA. Potent inactivation of representative members of each PKC isozyme subfamily and PKD via S-thiolation by the tumor-promotion/progression antagonist glutathione but not by its precursor cysteine. Carcinogenesis. 2001;22:1221–1229. doi: 10.1093/carcin/22.8.1221. [DOI] [PubMed] [Google Scholar]
- Chuang KP, Tsai WS, Wang YJ, Shieh CC. Superoxide activates very late antigen-4 on an eosinophil cell line and increases cellular binding to vascular cell adhesion molecule-1. Eur J Immunol. 2003;33:645–655. doi: 10.1002/eji.200323446. [DOI] [PubMed] [Google Scholar]
- Church LD, Cook GP, McDermott MF. Primer: inflammasomes and interleukin 1beta in inflammatory disorders. Nat Clin Pract Rheumatol. 2008;4:34–42. doi: 10.1038/ncprheum0681. [DOI] [PubMed] [Google Scholar]
- Clavreul N, Adachi T, Pimental DR, Ido Y, Schoneich C, Cohen RA. S-glutathiolation by peroxynitrite of p21ras at cysteine-118 mediates its direct activation and downstream signaling in endothelial cells. FASEB J. 2006a;20:518–520. doi: 10.1096/fj.05-4875fje. [DOI] [PubMed] [Google Scholar]
- Clavreul N, Bachschmid MM, Hou X, Shi C, Idrizovic A, Ido Y, et al. S-glutathiolation of p21ras by peroxynitrite mediates endothelial insulin resistance caused by oxidized low-density lipoprotein. Arterioscler Thromb Vasc Biol. 2006b;26:2454–2461. doi: 10.1161/01.ATV.0000242791.28953.4c. [DOI] [PubMed] [Google Scholar]
- Clayton A, Evans RA, Pettit E, Hallett M, Williams JD, Steadman R. Cellular activation through the ligation of intercellular adhesion molecule-1. J Cell Sci. 1998;111 (Pt 4):443–453. doi: 10.1242/jcs.111.4.443. [DOI] [PubMed] [Google Scholar]
- Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Wang P, Bai F, Town T, Fikrig E. Icam-1 participates in the entry of west nile virus into the central nervous system. J Virol. 2008;82:4164–4168. doi: 10.1128/JVI.02621-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DA, Brown CA, Newcomb FM, Boja ES, Fales HM, Kaufman J, et al. Reversible oxidative modification as a mechanism for regulating retroviral protease dimerization and activation. J Virol. 2003;77:3319–3325. doi: 10.1128/JVI.77.5.3319-3325.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DA, Dorsey K, Wingfield PT, Stahl SJ, Kaufman J, Fales HM, et al. Regulation of HIV-1 protease activity through cysteine modification. Biochemistry. 1996;35:2482–2488. doi: 10.1021/bi951525k. [DOI] [PubMed] [Google Scholar]
- Davis DA, Newcomb FM, Starke DW, Ott DE, Mieyal JJ, Yarchoan R. Thioltransferase (glutaredoxin) is detected within HIV-1 and can regulate the activity of glutathionylated HIV-1 protease in vitro. J Biol Chem. 1997;272:25935–25940. doi: 10.1074/jbc.272.41.25935. [DOI] [PubMed] [Google Scholar]
- De Rose V. Mechanisms and markers of airway inflammation in cystic fibrosis. Eur Respir J. 2002;19:333–340. doi: 10.1183/09031936.02.00229202. [DOI] [PubMed] [Google Scholar]
- Demasi M, Silva GM, Netto LE. 20 S proteasome from Saccharomyces cerevisiae is responsive to redox modifications and is S-glutathionylated. J Biol Chem. 2003;278:679–685. doi: 10.1074/jbc.M209282200. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Morgello S, Klotman ME, Mosoian A, Lento PA, Berman JW, et al. Human Immunodeficiency Virus (HIV) Infects Human Arterial Smooth Muscle Cells in Vivo and in Vitro: Implications for the Pathogenesis of HIV-Mediated Vascular Disease. Am J Pathol. 2008;172:1100–1111. doi: 10.2353/ajpath.2008.070457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flekac M, Skrha J, Zidkova K, Lacinova Z, Hilgertova J. Paraoxonase 1 gene polymorphisms and enzyme activities in diabetes mellitus. Physiol Res. 2007 doi: 10.33549/physiolres.931285. [DOI] [PubMed] [Google Scholar]
- Foell D, Wittkowski H, Roth J. Mechanisms of disease: a ‘DAMP’ view of inflammatory arthritis. Nat Clin Pract Rheumatol. 2007;3:382–390. doi: 10.1038/ncprheum0531. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Torres M. Redox signaling in macrophages. Mol Aspects Med. 2001;22:189–216. doi: 10.1016/s0098-2997(01)00010-3. [DOI] [PubMed] [Google Scholar]
- Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci U S A. 2002;99:3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007 doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Gho YS, Kleinman HK, Sosne G. Angiogenic activity of human soluble inter-cellular adhesion molecule-1. Cancer Res. 1999;59:5128–5132. [PubMed] [Google Scholar]
- Gravina SA, Mieyal JJ. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry. 1993;32:3368–3376. doi: 10.1021/bi00064a021. [DOI] [PubMed] [Google Scholar]
- Hagemann T, Balkwill F, Lawrence T. Inflammation and cancer: a double-edged sword. Cancer Cell. 2007;12:300–301. doi: 10.1016/j.ccr.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J Clin Invest. 2008;118:183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris HE, Andersson U. Mini-review: The nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol. 2004;34:1503–1512. doi: 10.1002/eji.200424916. [DOI] [PubMed] [Google Scholar]
- Harris HE, Raucci A. Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep. 2006;7:774–778. doi: 10.1038/sj.embor.7400759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Matsui M, Murata M, Takashima Y, Cheng FS, Itoh T, et al. Nucleoredoxin, glutaredoxin, and thioredoxin differentially regulate NF-kappaB, AP-1, and CREB activation in HEK293 cells. Biochem Biophys Res Commun. 2000;274:177–182. doi: 10.1006/bbrc.2000.3106. [DOI] [PubMed] [Google Scholar]
- Hoppe G, Talcott KE, Bhattacharya SK, Crabb JW, Sears JE. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res. 2006;312:3526–3538. doi: 10.1016/j.yexcr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Jahngen-Hodge J, Obin MS, Gong X, Shang F, Nowell TR, Jr, Gong J, et al. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J Biol Chem. 1997;272:28218–28226. doi: 10.1074/jbc.272.45.28218. [DOI] [PubMed] [Google Scholar]
- Kasama T, Yajima N, Matsukura S, Adachi M. Macrophage inflammatory protein 1 and CCR5 as attractive therapeutic targets for HIV infection. Recent Patents Anti -Infect Drug Disc. 2006;1:275–280. doi: 10.2174/157489106778777655. [DOI] [PubMed] [Google Scholar]
- Kusterer K, Bojunga J, Enghofer M, Heidenthal E, Usadel KH, Kolb H, et al. Soluble ICAM-1 reduces leukocyte adhesion to vascular endothelium in ischemia-reperfusion injury in mice. Am J Physiol. 1998;275:G377–G380. doi: 10.1152/ajpgi.1998.275.2.G377. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, et al. IKKalpha, IKKbeta, and NEMO/IKKgamma are each required for the NF-kappa B-mediated inflammatory response program. J Biol Chem. 2002;277:45129–45140. doi: 10.1074/jbc.M205165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Xu Z, Li S, Rozanski GJ. Redox regulation of Ito remodeling in diabetic rat heart. Am J Physiol Heart Circ Physiol. 2005;288:H1417–H1424. doi: 10.1152/ajpheart.00559.2004. [DOI] [PubMed] [Google Scholar]
- Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, et al. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SY, Tsai MY, Chuang KP, Huang YF, Shieh CC. Ligand binding of leukocyte integrin very late antigen-4 involves exposure of sulfhydryl groups and is subject to redox modulation. Eur J Immunol. 2008;38:410–423. doi: 10.1002/eji.200737556. [DOI] [PubMed] [Google Scholar]
- Lundberg M, Fernandes AP, Kumar S, Holmgren A. Cellular and plasma levels of human glutaredoxin 1 and 2 detected by sensitive ELISA systems. Biochem Biophys Res Commun. 2004;319:801–809. doi: 10.1016/j.bbrc.2004.04.199. [DOI] [PubMed] [Google Scholar]
- Madden E, Lee G, Kotler DP, Wanke C, Lewis CE, Tracy R, et al. Association of antiretroviral therapy with fibrinogen levels in HIV-infection. AIDS. 2008;22:707–715. doi: 10.1097/QAD.0b013e3282f560d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- Melendez MM, McNurlan MA, Mynarcik DC, Khan S, Gelato MC. Endothelial adhesion molecules are associated with inflammation in subjects with HIV disease. Clin Infect Dis. 2008;46:775–780. doi: 10.1086/527563. [DOI] [PubMed] [Google Scholar]
- Mieyal JJ, Gallogly MM, Qanungo S, Sabens SA, Shelton MD. Molecular Mechanisms and Clinical Implications of Reversible Protein S-Glutathionylation. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2089. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee TK, Mishra AK, Mukhopadhyay S, Hoidal JR. High concentration of antioxidants N-acetylcysteine and mitoquinone-Q induces intercellular adhesion molecule 1 and oxidative stress by increasing intracellular glutathione. J Immunol. 2007;178:1835–1844. doi: 10.4049/jimmunol.178.3.1835. [DOI] [PubMed] [Google Scholar]
- Murata H, Ihara Y, Nakamura H, Yodoi J, Sumikawa K, Kondo T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J Biol Chem. 2003;278:50226–50233. doi: 10.1074/jbc.M310171200. [DOI] [PubMed] [Google Scholar]
- Muro S, Gajewski C, Koval M, Muzykantov VR. ICAM-1 recycling in endothelial cells: a novel pathway for sustained intracellular delivery and prolonged effects of drugs. Blood. 2005;105:650–658. doi: 10.1182/blood-2004-05-1714. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Bai J, Nishinaka Y, Ueda S, Sasada T, Ohshio G, et al. Expression of thioredoxin and glutaredoxin, redox-regulating proteins, in pancreatic cancer. Cancer Detect Prev. 2000;24:53–60. [PubMed] [Google Scholar]
- Nakamura H, Vaage J, Valen G, Padilla CA, Bjornstedt M, Holmgren A. Measurements of plasma glutaredoxin and thioredoxin in healthy volunteers and during open-heart surgery. Free Radic Biol Med. 1998;24:1176–1186. doi: 10.1016/s0891-5849(97)00429-2. [DOI] [PubMed] [Google Scholar]
- Newman SF, Sultana R, Perluigi M, Coccia R, Cai J, Pierce WM, et al. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a pro-teomics approach. J Neurosci Res. 2007;85:1506–1514. doi: 10.1002/jnr.21275. [DOI] [PubMed] [Google Scholar]
- Nishio E, Watanabe Y. Cigarette smoke extract inhibits plasma paraoxonase activity by modification of the enzyme’s free thiols. Biochem Biophys Res Commun. 1997;236:289–293. doi: 10.1006/bbrc.1997.6961. [DOI] [PubMed] [Google Scholar]
- Obin M, Shang F, Gong X, Handelman G, Blumberg J, Taylor A. Redox regulation of ubiquitin-conjugating enzymes: mechanistic insights using the thiol-specific oxidant diamide. FASEB J. 1998;12:561–569. doi: 10.1096/fasebj.12.7.561. [DOI] [PubMed] [Google Scholar]
- Okuda M, Inoue N, Azumi H, Seno T, Sumi Y, Hirata K, et al. Expression of glutaredoxin in human coronary arteries: its potential role in antioxidant protection against atherosclerosis. Arterioscler Thromb Vasc Biol. 2001;21:1483–1487. doi: 10.1161/hq0901.095550. [DOI] [PubMed] [Google Scholar]
- Pan S, Berk BC. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circ Res. 2007;100:213–219. doi: 10.1161/01.RES.0000256089.30318.20. [DOI] [PubMed] [Google Scholar]
- Peltoniemi M, Kaarteenaho-Wiik R, Saily M, Sormunen R, Paakko P, Holmgren A, et al. Expression of glutaredoxin is highly cell specific in human lung and is decreased by transforming growth factor-beta in vitro and in interstitial lung diseases in vivo. Hum Pathol. 2004;35:1000–1007. doi: 10.1016/j.humpath.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Peltoniemi MJ, Rytila PH, Harju TH, Soini YM, Salmenkivi KM, Ruddock LW, et al. Modulation of glutaredoxin in the lung and sputum of cigarette smokers and chronic obstructive pulmonary disease. Respir Res. 2006;7:133. doi: 10.1186/1465-9921-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007;19:615–622. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Pineda-Molina E, Klatt P, Vazquez J, Marina A, Garcia dL, Perez-Sala D, et al. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- Prinarakis E, Chantzoura E, Thanos D, Spyrou G. S-glutathionylation of IRF3 regulates IRF3-CBP interaction and activation of the IFNbeta pathway. EMBO J. 2008 doi: 10.1038/emboj.2008.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujades C, Teixido J, Bazzoni G, Hemler ME. Integrin alpha 4 cysteines 278 and 717 modulate VLA-4 ligand binding and also contribute to alpha 4/180 formation. Biochem J. 1996;313 (Pt 3):899–908. doi: 10.1042/bj3130899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qanungo S, Starke DW, Pai HV, Mieyal JJ, Nieminen AL. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J Biol Chem. 2007;282:18427–18436. doi: 10.1074/jbc.M610934200. [DOI] [PubMed] [Google Scholar]
- Reingold JS, Wanke C, Kotler DP, Lewis CE, Tracy R, Heyms Filig ES, et al. Association of HIV Infection and HIV/HCV Coinfection With C-Reactive Protein Levels: The Fat Redistribution and Metabolic Change in HIV Infection (FRAM) Study. J Acquir Immune Defic Syndr. 2008 doi: 10.1097/QAI.0b013e3181685727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulation of glutaredoxin-1 expression in a mouse model of allergic airway disease. Am J Respir Cell Mol Biol. 2007;36:147–151. doi: 10.1165/rcmb.2006-0259RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo G, Liu WH, Asnaghi V, Kern TS, Lorenzi M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51:2241–2248. doi: 10.2337/diabetes.51.7.2241. [DOI] [PubMed] [Google Scholar]
- Rosenblat M, Vaya J, Shih D, Aviram M. Paraoxonase 1 (PON1) enhances HDL-mediated macrophage cholesterol efflux via the ABCA1 transporter in association with increased HDL binding to the cells: a possible role for lysophosphatidylcholine. Atherosclerosis. 2005;179:69–77. doi: 10.1016/j.atherosclerosis.2004.10.028. [DOI] [PubMed] [Google Scholar]
- Rozenberg O, Aviram M. S-Glutathionylation regulates HDL-associated paraoxonase 1 (PON1) activity. Biochem Biophys Res Commun. 2006;351:492–498. doi: 10.1016/j.bbrc.2006.10.059. [DOI] [PubMed] [Google Scholar]
- Sano H, Nakagawa N, Chiba R, Kurasawa K, Saito Y, Iwamoto I. Cross-linking of intercellular adhesion molecule-1 induces interleukin-8 and RANTES production through the activation of MAP kinases in human vascular endothelial cells. Biochem Biophys Res Commun. 1998;250:694–698. doi: 10.1006/bbrc.1998.9385. [DOI] [PubMed] [Google Scholar]
- Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25:6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- Schmid H, Boucherot A, Yasuda Y, Henger A, Brunner B, Eichinger F, et al. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes. 2006;55:2993–3003. doi: 10.2337/db06-0477. [DOI] [PubMed] [Google Scholar]
- Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- Shelton MD, Kern TS, Mieyal JJ. Glutaredoxin regulates nuclear factor kappa-B and intercellular adhesion molecule in Muller cells: Model of diabetic retinopathy. J Biol Chem. 2007;282:12467–12474. doi: 10.1074/jbc.M610863200. [DOI] [PubMed] [Google Scholar]
- Shepherd CE, Grace EM, Mann DM, Halliday GM. Relationship between neuronal loss and ‘inflammatory plaques’ in early onset Alzheimer’s disease. Neuropathol Appl Neurobiol. 2007;33:328–333. doi: 10.1111/j.1365-2990.2006.00816.x. [DOI] [PubMed] [Google Scholar]
- Shih DM, Xia YR, Wang XP, Miller E, Castellani LW, Subbanagounder G, et al. Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit in-creased lipoprotein oxidation and atherosclerosis. J Biol Chem. 2000;275:17527–17535. doi: 10.1074/jbc.M910376199. [DOI] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Yuan M. Inflammation and the IKK[beta]//I[kappa]B//NF-[kappa]B axis in obesity- and diet-induced insulin resistance. Int J Obes Relat Metab Disord. 2003;27:S49–S52. doi: 10.1038/sj.ijo.0802501. [DOI] [PubMed] [Google Scholar]
- Singh S, Venketesh S, Verma JS, Verma M, Lellamma CO, Goel RC. Paraoxonase (PON1) activity in north west Indian Punjabis with coronary artery disease & type 2 diabetes mellitus. Indian J Med Res. 2007;125:783–787. [PubMed] [Google Scholar]
- Starke DW, Chock PB, Mieyal JJ. Glutathionethiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J Biol Chem. 2003;278:14607–14613. doi: 10.1074/jbc.M210434200. [DOI] [PubMed] [Google Scholar]
- Takashima Y, Hirota K, Nakamura H, Nakamura T, Akiyama K, Cheng FS, et al. Differential expression of glutaredoxin and thioredoxin during monocytic differentiation. Immunol Lett. 1999;68:397–401. doi: 10.1016/s0165-2478(99)00087-5. [DOI] [PubMed] [Google Scholar]
- Teppo AM, von WE, Honkanen E, Ahonen J, Gronhagen-Riska C. Soluble intercellular adhesion molecule-1 (sICAM-1) after kidney transplantation: the origin and role of urinary sICAM-1? Transplantation. 2001;71:1113–1119. doi: 10.1097/00007890-200104270-00018. [DOI] [PubMed] [Google Scholar]
- Tesch GH. Role of macrophages in complications of type 2 diabetes. Clin Exp Pharmacol Physiol. 2007;34:1016–1019. doi: 10.1111/j.1440-1681.2007.04729.x. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Poland B, Honzatko R. Protein sulfhydryls and their role in the anti-oxidant function of protein S-thiolation. Arch Biochem Biophys. 1995;319:1–9. doi: 10.1006/abbi.1995.1261. [DOI] [PubMed] [Google Scholar]
- Tward A, Xia YR, Wang XP, Shi YS, Park C, Castellani LW, et al. Decreased atherosclerotic lesion formation in human serum paraoxonase transgenic mice. Circulation. 2002;106:484–490. doi: 10.1161/01.cir.0000023623.87083.4f. [DOI] [PubMed] [Google Scholar]
- van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1) Angiogenesis. 2008;11:91–99. doi: 10.1007/s10456-008-9093-5. [DOI] [PubMed] [Google Scholar]
- Wang J, Pan S, Berk BC. Glutaredoxin mediates Akt and eNOS activation by flow in a glutathione reductase-dependent manner. Arterioscler Thromb Vasc Biol. 2007;27:1283–1288. doi: 10.1161/ATVBAHA.107.144659. [DOI] [PubMed] [Google Scholar]
- Wang W, Oliva C, Li G, Holmgren A, Lillig CH, Kirk KL. Reversible silencing of CFTR chloride channels by glutathionylation. J Gen Physiol. 2005;125:127–141. doi: 10.1085/jgp.200409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Qiao M, Mieyal JJ, Asmis LM, Asmis R. Molecular mechanism of glutathione-mediated protection from oxidized low-density lipoprotein-induced cell injury in human macrophages: role of glutathione reductase and glutaredoxin. Free Radic Biol Med. 2006;41:775–785. doi: 10.1016/j.freeradbiomed.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Ward NE, Stewart JR, Ioannides CG, O’Brian CA. Oxidant-induced S-glutathiolation inactivates protein kinase C-alpha (PKC-alpha): a potential mechanism of PKC isozyme regulation. Biochemistry. 2000;39:10319–10329. doi: 10.1021/bi000781g. [DOI] [PubMed] [Google Scholar]
- Xiao C, Bator CM, Bowman VD, Rieder E, He Y, Hebert B, et al. Interaction of coxsackievirus A21 with its cellular receptor, ICAM-1. J Virol. 2001;75:2444–2451. doi: 10.1128/JVI.75.5.2444-2451.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Jao S, Nanduri S, Starke DW, Mieyal JJ, Qin J. Reactivity of the human thioltransferase (glutaredoxin) C7S, C25S, C78S, C82S mutant and NMR solution structure of its glutathionyl mixed disulfide intermediate reflect catalytic specificity. Biochemistry. 1998;37:17145–17156. doi: 10.1021/bi9806504. [DOI] [PubMed] [Google Scholar]
- Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53:2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]