

Abstract
Glutamate is the most abundant excitatory neurotransmitter in the vertebrate central nerve system and plays an important role in synaptic plasticity required for learning and memory. Activation of glutamate ionotropic receptors promptly triggers membrane depolarization and Ca2+ influx, resulting in the activation of several different protein kinases and transcription factors. For example, glutamate-mediated Ca2+ influx activates Ca2+/calmodulin-dependent kinase, protein kinase C, and mitogen activated protein kinases resulting in activation of transcription factors such as cyclic AMP response element binding protein (CREB). Abnormally prolonged exposure to glutamate causes neuronal injury, and such “excitotoxicity” has been implicated in many acute and chronic diseases including ischemic stroke, hypoglycemia, epilepsy, amyotrophic lateral sclerosis, Alzheimer’s, Huntington’s and Parkinson’s diseases. Interestingly, although glutamate-induced Ca2+ influx can cause DNA damage by a mitochondrial reactive oxygen species-mediated mechanism, the Ca2+ simultaneously activates CREB, resulting in up-regulation of the DNA repair and redox protein apurinic/apyrimidinic endonuclease 1. Here, we review connections between physiological or aberrant glutamate receptor activation, Ca2+-mediated signaling, oxidative DNA damage and repair efficiency, and neuronal vulnerability. We conclude that glutamate signaling involves an adaptive cellular stress response pathway that enhances DNA repair capability, thereby protecting neurons against injury and disease.
Keywords: Glutamate, Excitotoxicity, Oxidative DNA damage, APE1, Neurodegenerative diseases
1. Glutamate signaling
Glutamate is one of twenty essential amino acids and is the main excitatory neurotransmitter in the mammalian central nervous system (CNS). Glutamate has been reported to regulate neurogenesis, neurite outgrowth, synaptogenesis and neuron survival (Mattson 2008). The concentration of glutamate is strictly maintained in the CNS, and glutamate released into the synaptic gap is promptly recovered by neurons and/or glial cells (especially astrocytes) via the glutamate transporter 1 (Fig. 1). Glutamate receptors are categorized into two groups, ionotropic and metabotropic. Metabotropic glutamate receptors (mGluRs) mediate slow response by activating different downstream second messenger molecules via heterotrimeric G-protein. The ionotropic glutamate receptors (iGluRs) are ligand-gated ion channels, which permit the flow of Na+ and/or Ca2+ in response to activation. The major function of iGluRs is mediating fast excitatory synaptic transmission.
Figure 1.
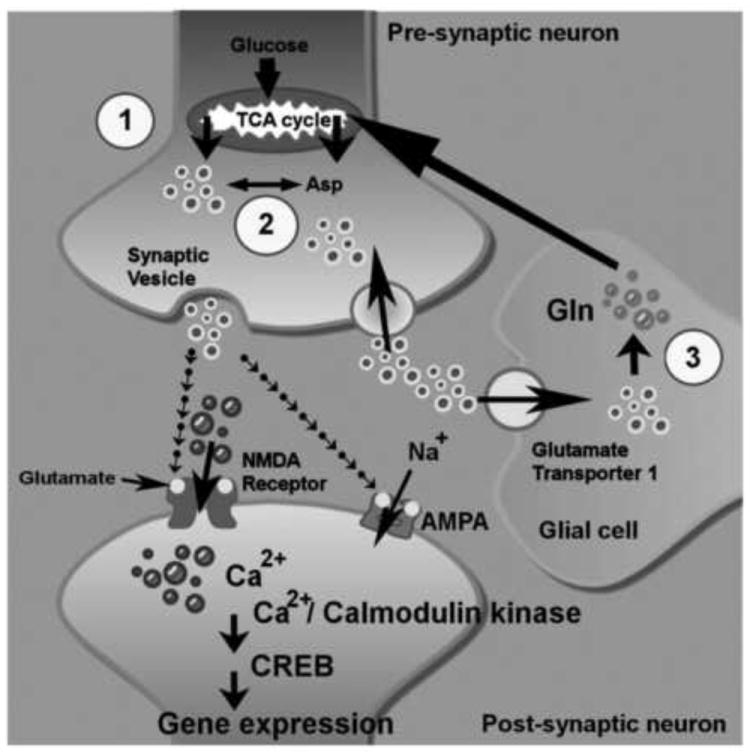
Glutamate synthesis, recycling, and signaling in neuronal cells. The glutamate concentration is well maintained in central nervous system (CNS). Glutamate (light ringed circles) is commonly derived from products of TCA (citric acid cycle) (circle number 1) and converts from Aspartate (Asp) by aspartate transaminase (circle number 2). The released glutamate in the synaptic gap can be promptly sequestered into neurons and/or glial cells via glutamate transporter 1. The recycled glutamate is converted into glutamine by glutaminase in glial cells. Then, glutamine is delivered to neurons and converted into glutamate by glutamate synthetase (circle number 3). The activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA) receptors by glutamate binding trigger Na+ influx and consequently cause membrane depolarization. In the meantime, the depolarized membrane facilitates the Ca2+ influx (dark ringed circles) from activated N-Methyl-d-aspartic acid (NMDA) receptors. The cytosolic Ca2+ acts as a secondary messenger to initiate Ca2+/calmodulin-dependent kinase and CREB-mediated regulation of gene expression.
Glutamate activates ionotropic receptors such as alpha-amino-5-methyl-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-Methyl-d-aspartic acid (NMDA) receptors, which regulate neuronal membrane depolarization and Ca2+ influx and are integral in the functional responses of neurons in processes such as learning and memory (Fig 1). The activation of NMDA receptors by glutamate results in Ca2+ influx in postsynaptic neurons, which initiates downstream signaling cascades and regulates gene expression. Promoter analysis indicates that Ca2+ influx through different calcium channels activates distinct signaling pathways that target either the serum response element (SRE) or the calcium response element (CaRE) within the c-fos promoter. The adenosine 3’,5’-monophosphate/cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) is a transcription factor ubiquitously expressed in neurons and is a substrate for depolarization-activated Ca2+-calmodulin-dependent protein kinases (CaMKs) (Fig.2). Calcium influx leads to the rapid induction of a number of immediate-early genes. These observations suggest that Ca2+ can regulate gene expression by multiple signaling pathways including one that involves the Ca2+-dependent phosphorylation of the transcription factor CREB (Fig.2) (Ghosh et al. 1994). CREB residue Ser133 is the major site of phosphorylation by CaMK in vitro and after membrane depolarization in vivo (Sheng et al. 1991). The dimerized phospho-CREB (pCREB) binds to the consensus nucleotide sequence TGACGTCA, cyclic AMP response element (CRE) (Berkowitz et al. 1989). Mutation of Ser133 diminishes the ability of CREB to respond to Ca2+, which suggests that CaMKs may transduce electrical signals to the nucleus and that CREB functions to integrate Ca2+ and cAMP signaling (Gonzalez and Montminy 1989; Sheng, Thompson et al. 1991; Matthews et al. 1994; Wu et al. 2001).
Figure 2.
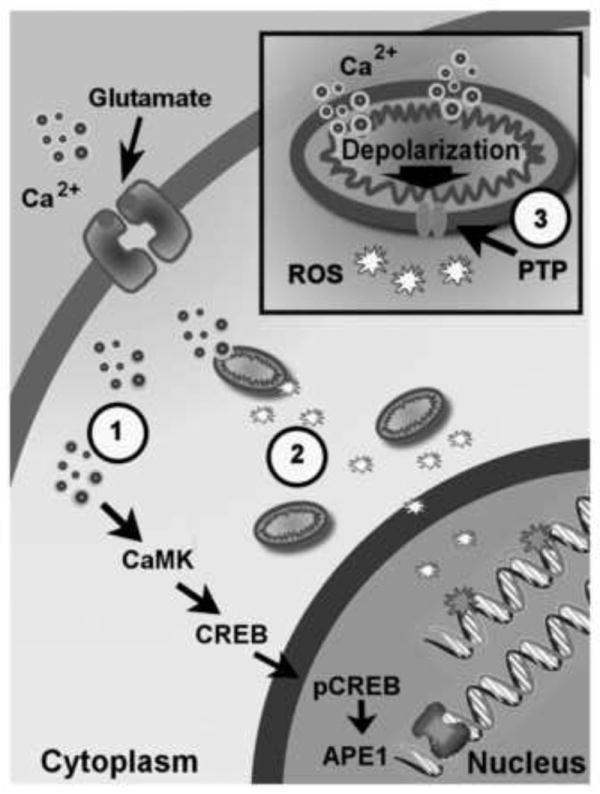
Physiological concentrations of glutamate induces oxidative DNA damage and elevate APE1 expression via the Ca2+/calmodulin-dependent kinase and cyclic AMP response element binding protein (CREB)-mediated signaling. Glutamate-induced oxidative DNA damage is from mitochondrial-generated reactive oxygen species (ROS) which is produced by Ca2+-(small light ringed circles) mediated mitochondrial membrane depolarization. The circle numbered 1 indicates that pre-administration of intracellular Ca2+ chelator, BAPTA-AM, prohibits oxidative DNA damage from glutamate treatment. The circle numbered 2 shows that pre-treatment with the ROS scavenger, MnTMPyP, decreases glutamate-induced oxidative DNA damage. The circle numbered 3 indicates pre-administration of mitochondrial permeable transition pore blocker, cyclosporin A, reducing oxidative DNA damage by confining ROS in mitochondria.
2. Glutamate, oxidative stress and DNA base excision repair
Glutamate signaling induces mitochondrial Ca2+ uptake, and an increase in mitochondrial respiration can result in elevated levels of superoxide and other genotoxic free radicals (Fig.3) (Sengpiel et al. 1998;Chinopoulos et al. 2000). The reactive oxygen species (ROS), together with rapid mitochondrial membrane permeability changes, trigger cell death in a process termed excitotoxicity (Reynolds 1999; Mattson 2003).
Figure 3.
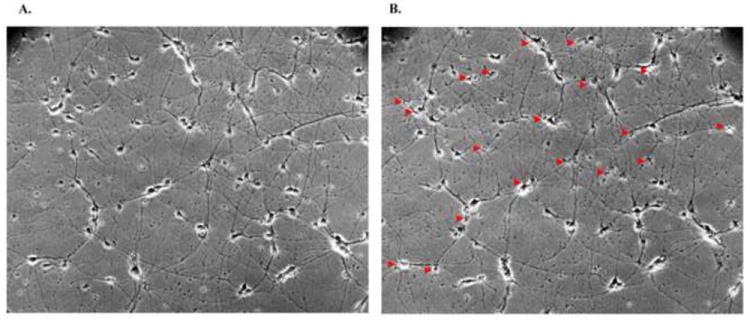
Glutamate-induced neuronal death. Panel A illustrates the rat primary cortical neurons before glutamate treatment. Twenty-four hours after 10 minutes treatment with 100 μM glutamate, panel B shows that death occurred in many of the cortical neurons (arrows).
High concentrations of glutamate can cause neuronal death, which typically involves DNA damage and induction of apoptosis (Kruman et al. 2000; Culmsee et al. 2001). Indeed, treatment with high concentrations of glutamate (≥100 μM) induces excitotoxic cell death in rat primary cerebral cortical cultures (Fig. 3). While high levels of glutamate clearly induce excitotoxic cell death, the relevance of these studies to normal brain function is not clear. Post-mitotic neurons must remain functional for the entire life span of an organism. This longevity is generally incompatible with the notion that neurons are constantly undergoing a significant level of cell death as a result of glutamate-mediated excitotoxicity and DNA damage.
In a recent study we showed that transient activation of glutamate receptors in cultured rat cerebral cortical neurons induces DNA damage that is repaired over a period of several hours (Yang et al. 2010). The neurons were exposed to 20 μM glutamate for 10 minutes, a dose that did not cause significant cytotoxicity. Previous studies of cultured rat cortical and hippocampal neurons have suggested similarly, that glutamate at concentrations from 10 – 50 μM elicits an adaptive response that involves modification of dendritic outgrowth and synaptogenesis, including neuronal morphology changes commonly postulated as increasing memory formation. Conversely, higher concentrations of glutamate of 100 μM or greater can damage or kill neurons (Mattson et al. 1988a; Mattson et al. 1988b). When measured by in vivo microdialysis, the extracellular non-synaptic concentration of glutamate in the brain was within the range of 1-3 μM (Langlais and Zhang 1993; Hazell et al. 1993). However, several different lines of evidence suggest that under physiological conditions, such as during learning and memory encoding in hippocampal neurons or activation of motor system neurons during exercise, glutamate concentrations in the synapse transiently reach concentrations of 100 – 1000 μM. For example, using the rapidly reversible competitive NMDA receptor antagonist AP5, it was estimated that the lowest concentrations of glutamate achieved during basal transmission at neonatal synapses was approximately 170 μM, while in response to stimulation that induced long-term potentiation the glutamate concentration rose much higher (Choi et al. 2000). Another study provided evidence that the glutamate released from one presynaptic vesicle is sufficient to elevate the local glutamate concentration at postsynaptic glutamate receptors to more than 30 μM for at least 1 millisecond (Rao-Mirotznik et al. 1998). Interestingly, the Ca2+ influx triggered by activation of NMDA receptors may engage several adaptive stress response pathways (Marini et al. 1999), with the Ca2+/calmodulin/CREB-induced expression of APE1 being one such pathway (Yang et al. 2010).
When neurons were exposed to a low concentration of glutamate (20 μM) for 10 min, oxidative damage was introduced into the nuclear DNA (Yang et al. 2010). As discussed above, these levels of glutamate are physiological and we found that this level of exposure did not cause significant cell death (Yang et al. 2010). Pre-administration with the intracellular Ca2+ chelator (BAPTA-AM), inhibiting the mitochondrial permeable transition pores (cyclosporin A), or using the ROS scavenger (MnTMPyP), all prevented DNA damage (Fig. 2). These results provide further evidence that oxidative stress is integral for glutamate-mediated DNA damage, and intervention at any of the steps preceding the ROS burst can prevent the DNA damage.
The lack of cytotoxicity observed after physiologically relevant concentrations of glutamate is a result of the efficient repair of glutamate-induced DNA damage, presumably through induction of base excision repair (BER) components. BER is the major pathway responsible for the resolution of non helix-distorting lesions, including oxidized, alkylated, and deaminated nucleotides. Elevated levels of intracellular oxidative damage stimulate BER, culminating in an increase in cellular survival (Chen et al. 1998; Ramana et al. 1998; Cabelof et al. 2002). While apurinic/apyrimidinic endonuclease 1 (APE1), DNA polymerase β (pol β) and an XRCC1/ DNA ligase III complex are generally considered the main central components of BER, there are other protein partners including DNA glycosylases involved in repairing the damaged DNA substrate and enhancing DNA repair processes (Fig. 4).
Figure 4.
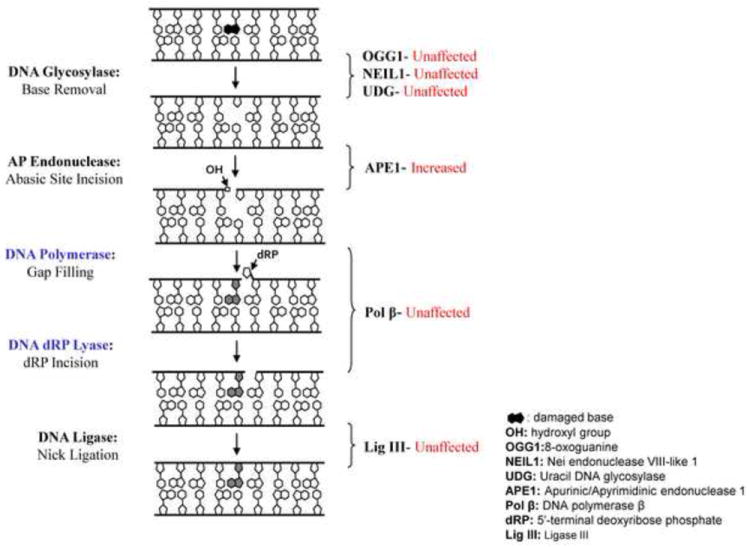
The DNA repair proteins which are or are not affected by glutamate treatment in basic excision repair pathway. The diagram indicates the protein levels and enzyme activities of 8-oxoguanine glycosylase 1 (OGG1), nei endonuclease VIII-like 1 (NEIL1), uracil DNA glycosylase (UDG), DNA polymerase β (Pol β), and ligase III (Lig III) are not affected by glutamate administration.
DNA glycosylases typically catalyze the initial step of BER, removing a damaged base by cleaving the N-glycosidic bond, leaving behind an AP site, which is resolved by APE1. We investigated the activity of several prominent DNA glycosylases, i.e. OGG1, NEIL1 and UDG, as well as APE1, after glutamate exposure (Yang et al. 2010). OGG1, the primary protein for repairing 8-oxoguanine oxidative base lesions paired with cytosine, has been shown to be essential in neuronal maturation (Wang et al. 2010) and protects neurons against oxidative DNA damage (Wong et al. 2008; Liu et al. 2010). Similarly, NEIL1, which repairs a range of oxidative base damages, most notably formamidopyrimidines, and also uracil glycosylase (UNG), the predominant protein for excising uracil in DNA, have both been reported to be active in the human and rodent brain (Endres et al. 2004;Kruman et al. 2004;Rolseth et al. 2008). Glycosylase levels for 8-oxoguanine (OGG1) and uracil were not modulated by glutamate treatment as assessed by immunoblotting (Yang et al. 2010). In contrast, APE1 protein levels, as well as a corresponding ability of cell extracts to incise at AP sites in DNA, were significantly elevated (Fig. 4) after glutamate exposure.
Increased levels of APE1 are suggested to be neuroprotective (Vasko et al. 2005) and of particular interest when investigating the toxicity of glutamate on neurons. Recently, two factors – glutamate and the neuropeptide adenylate cyclase-activating polypeptide – have both been identified as regulators of APE1 levels in neurons (Yang et al. 2010; Stetler et al. 2010). Both glutamate and adenylate cyclase-activating polypeptide activated CREB via phosphorylation by different kinases. CREB plays a key role in regulating APE1 gene expression (Fig.3). The APE1 protein is versatile and besides its function as the predominant abasic site endonuclease, APE1 is also a redox effector factor (Ref-1). In its redox activator capacity, APE1 controls different cellular processes, such as apoptosis and proliferation, through a stimulatory interaction with different transcription factors including NF-κB and p53 (Tell et al. 2005). Hence, APE1 may have different functions after glutamate activation. For instance, the redox function of APE1 may be required to restore a balance between the antioxidant systems after elevated levels of ROS induced by glutamate, while the endonuclease function of APE1 might facilitate the repair of the glutamate-induced DNA damage. This notion was recently addressed by using a human APE1 construct with a single point mutation in the APE1 domain: the resultant protein lacked the endonuclease function but retained full redox capacity. Importantly, the mutant APE1 did not have the neuroprotective function as previously reported with wild type APE1 (Stetler et al. 2010). The results suggested that the DNA processing function of APE1 is indispensable for the protection of neurons from glutamate-induced oxidative damage. Finally, APE1 has been shown to stimulate DNA polymerase β (Liu et al. 2007), hence the induction of APE1 by glutamate may lead to downstream increases in associated DNA repair proteins.
Interestingly, activation of NF-κB in neurons has been shown to protect them against insults that induce oxidative stress and excitotoxicity (Mattson et al. 1997; Yu et al. 1999). NF-κB is activated in response to glutamate (Jiang et al. 2003), and one important gene target of NF-κB is manganese superoxide dismutase (MnSOD or SOD2), a mitochondrial antioxidant enzyme that suppresses production of ROS that can cause DNA damage (Mattson et al. 1997). Studies of NF-κB-deficient cells have provided evidence that NF-κB up-regulates DNA repair pathways (Wang et al. 2009), although the specific gene targets involved have not been established. In addition, the redox chaperone activity of APE1 enhances NF-κB-mediated gene expression, which may be facilitated by a physical association with target transcription factors (Ando et al. 2008). Collectively, the available data suggest that NF-κB up-regulates targets such as MnSOD that reduce DNA damage, while simultaneously enhancing DNA repair, thereby protecting neurons against excitotoxicity.
DNA polymerase β is the predominant polymerase in neuronal tissue in contrast to actively dividing tissue, which also engages DNA polymerases in repair that have a dual role in replication (Raji et al. 2002). Further, DNA polymerase β null mice die immediately after birth, apparently due to defective neurogenesis characterized by apoptotic cell death in the developing central and peripheral nervous systems (Sugo et al. 2000). DNA polymerase β is also moderately elevated after glutamate exposure (Yang et al. 2010). This trend correlates with previous reports that DNA polymerase β is induced in a protective response to genotoxic stress (Cabelof et al. 2002; Cabelof et al. 2003; Sykora and Snow 2008; Unnikrishnan et al. 2010). It is apparent that neurons are able to efficiently regulate BER; however, it remains to be seen what affect the decline in BER capacity attributed to aging may have on glutamate genotoxicity.
3. Glutamate and neurodegeneration
Many studies have reported that glutamate excitotoxicity increases oxidative stress in both in vitro and in vivo models, and accumulating evidence indicates that glutamate-induced oxidative stress contributes to neuronal death in neurodegenerative diseases (Mattson 2003). Aging and disease-specific processes, such as amyloid accumulation in Alzheimer’s disease and cerebral ischemia in stroke, perturb glutamate homeostasis and increase sensitivity of neurons to glutamate which can produce chronic excitotoxicity. There is considerable evidence that BER capacity declines with age, raising the notion that glutamate-induced oxidative stress could pose a much greater burden in aging neurons (Rao et al. 2001; Raji et al. 2002; Intano et al. 2003; Krishna et al. 2005). Further, if the glutamate system is dysregulated during aging, or if additional oxidative stress is placed on neurons, then the neurodegenerative process would be accelerated. This notion is supported by etiologies from prominent diseases associated with aging and suggests that increased oxidative stress caused by glutamate dysregulation may play a pivotal role in pushing an aging neuron beyond the point of being able to up-regulate its repair systems to compensate. We next review the role of glutamate in four neurodegenerative diseases, all sharing two common factors, an increase in oxidative stress and DNA damage, although each stem from different underlying cause(s).
Alzheimer’s disease (AD) is the most common neurodegenerative disorder in the elderly population. Glutamate excitotoxicity undoubtedly has a role in AD, and likely accelerates disease progression (Choi 1988; Facheris et al. 2004; Gonsette 2008; Dong et al. 2009; Lau and Tymianski 2010). Amyloid β-peptide (Aβ) tends to aggregate with both itself and other proteins, forming inter-cellular senile plaques potentially increasing the vulnerability of neurons. Studies of mutations in the β-amyloid precursor protein (APP) and presenilin 1 (PS1) that cause inherited early-onset forms of AD have provided considerable evidence for a pivotal role for Aβ in the disease process (for review see (Mattson 2004)). Because APP is axonally transported and present in high amounts in pre-synaptic terminals, Aβ is generated and aggregates at synaptic terminals. During the process of self-aggregation, Aβ induces membrane-associated oxidative stress which can render neurons vulnerable to excitotoxicity (Mattson et al. 1992). By increasing ROS production, Aβ can cause damage to both nuclear (Kruman et al. 2002) and mitochondrial (Bozner et al. 1997) DNA in neurons. Analysis of postmortem brain tissue samples have demonstrated increased oxidative DNA damage and deficient BER in patients with mild cognitive impairment and AD, in comparison with age-matched neurologically normal subjects (Weissman et al. 2007). One contributing factor to impaired BER in neurons in AD is suggested by the reduced activity of neuronal circuits involved in learning and memory (Gleichmann et al. 2010; Kapogiannis and Mattson 2010). In this aspect, glutamatergic signaling up-regulates BER protecting neurons against degeneration.
Parkinson’s disease (PD) is another neurodegenerative disease commonly afflicting the elderly. It is distinguished by symptoms including resting tremor, bradykinesia and rigidity. There are a number of genetic mutations known to cause PD including mutations in genes encoding α-synuclein, parkin, DJ1 and LRRK2 (Hardy et al. 2006). The parkin and DJ1 proteins play roles in the regulation and stability of the excitatory glutamate synapses, enhancing the plasticity of glutamate synapses and may protect neurons against glutamate excitotoxicity (Wang et al. 2008; Dong et al. 2009). Oxidative DNA damage has been found in the substantia nigra region of the PD brain, which is heavily affected in PD disease progression (Alam et al. 1997; Zhang et al. 1999), suggesting that oxidative DNA damage may play a pathogenic role in the neurodegeneration of dopaminergic neurons in PD. In an alternate hypothesis, loss of dopaminergic neurons in the PD substantia nigra pars compacta (SNpc) may be a result of a disruption of glutamate homeostasis (Facheris et al. 2004; Gonsette 2008; Dong et al. 2009; Meredith et al. 2009). Using a PD mouse model, Meredith et al. (Meredith et al. 2009) showed elevated extracellular levels of glutamate in the SNpc when compared to control animals. The increase in glutamate levels in the PD mice was suggested to be a result of attenuated clearance from the synapses. The authors also linked increased levels of apoptosis and autophagy to glutamate associated excitotoxicity in the PD mouse model (Meredith et al. 2009). However, links between glutamate signaling, DNA damage and degeneration of dopaminergic neurons in PD remain to be established.
Huntington’s disease (HD) is a genetically inherited autosomal-dominant neurodegenerative disorder caused by trinucleotide (CAG) repeat expansions in the huntingtin gene. The expression of mutant huntingtin enhances NMDA activity and sensitizes type 1 inositol 1,4,5-trisphosphate receptors, causing a disturbance in calcium homeostasis (Bezprozvanny and Hayden 2004; Zhang et al. 2008). The disrupted calcium homeostasis induces mitochondrial dysfunction resulting in reduction of ATP concentration and impairment of mitochondrial energy metabolism associated with generation of ROS (Brouillet et al. 1999). Post mortem brain samples of HD patients have higher levels of oxidative DNA damage (8-Oxo-dG) in both nuclei and mitochondria (Browne et al. 1997; Polidori et al. 1999). The increased levels of 8-oxo-7,8-dihydroguanine (8-Oxo-dG) were also found in HD mouse models (Bogdanov et al. 2001; De et al. 2008). Huntingtin may promote accumulation of DNA damage by impairing repair of double strand breaks (Enokido et al. 2010). Paradoxically, data suggest that the DNA repair enzyme OGG1 plays a role in the expansion of CAG repeats in the huntingtin gene, thereby exacerbating the transgenerational disease process (Kovtun et al. 2007).
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder linked to progressive motor neuron loss. The hallmark of ALS is motor neuron degeneration, which may result, in part, from over activation of glutamate receptors in motor neurons (Corona et al. 2007). Motor neurons from Cu/Zn-SOD mutant mice (ALS model) are sensitive to glutamate toxicity associated with ROS production and display elevated intracellular calcium levels and mitochondrial dysfunction (Kruman et al. 1999). Substantial increases in oxidative DNA damage also have been observed in the nuclear DNA from the spinal cord, frontal cortex, striatum and cerebellum from an ALS mouse model (Aguirre et al. 2005).
Collectively, glutamate-induced oxidative stress and DNA damage has important pathogenic roles in many neurodegenerative disorders (Rolig and McKinnon 2000; Facheris et al. 2004; Dong et al. 2009). The neurodegeneration associated with the previously discussed diseases and potentially with normal aging is a slow insidious process. Central to the progression of age- and disease-related neurodegeneration is the accumulation of oxidative DNA damage attributed to a decline in DNA repair capacity. The role of glutamate related excitotoxicity in these chronic disorders is uncertain. However, from the acute cellular induction of DNA repair capacity in response to glutamate mediated DNA damage, we can predict that activities such as exercise, reading and leaning may be beneficial in maintaining neural DNA repair capacity at a high level though out life potentially abrogating the DNA damage from oxidative stress associated with chronic diseases such as Alzheimer’s.
4. Conclusions
Emerging evidence suggest that physiological levels of activation of synaptic glutamate receptors can up-regulate DNA repair systems, thereby increasing the resiliency of the neurons and resulting in reduced vulnerability to age-related degeneration and acute injury. We have found that glutamate signaling can enhance BER by a mechanism involving Ca2+-mediated activation of CREB, which induces the expression of APE1 (Yang, et. al,2010). The calcium – CREB pathway has previously been shown to play a major role in synaptic plasticity and learning and memory (Sheng et al. 1991; Ghosh et al. 1994; Bito et al. 1996; Hongpaisan et al. 2003; Carlezon, Jr. et al. 2005). The changes in gene expression may be involved in many aspects of the brain, including development, neurogenesis, neurite outgrowth, synaptogenesis, synaptic plasticity, and learning and long-term memory (Mattson 2003; Mattson 2008). Less well studied are the protective effects of glutamate-mediated DNA repair. It is apparent that physiologically relevant levels of glutamate are effectively increasing BER efficiency in neurons. However, with a compromised DNA repair system or increased oxidative stress, the neuron can be rapidly overwhelmed leading to cell death. A better understanding of molecular mechanisms by which neurotransmitters affect DNA damage and repair may lead to the development of new therapeutic approaches for neurodegenerative disorders.
Research Highlights.
Glutamate at physiological doses induces DNA damage
This DNA damage is rapidly repaired
APendonuclease is induced in the process
Glutamate dysregulation may lead to defects in DNA repair
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Jenq-Lin Yang, Email: yangj2@mail.nih.gov.
Peter Sykora, Email: sykorap@mail.nih.gov.
Mark P. Mattson, Email: mattsonm@grc.nia.nih.gov.
Reference List
- Aguirre N, Beal MF, Matson WR, Bogdanov MB. Increased oxidative damage to DNA in an animal model of amyotrophic lateral sclerosis. Free Radic Res. 2005;39:383–388. doi: 10.1080/10715760400027979. [DOI] [PubMed] [Google Scholar]
- Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- Ando K, Hirao S, Kabe Y, Ogura Y, Sato I, Yamaguchi Y, Wada T, Handa H. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008;36:4327–4336. doi: 10.1093/nar/gkn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz LA, Riabowol KT, Gilman MZ. Multiple sequence elements of a single functional class are required for cyclic AMP responsiveness of the mouse c-fos promoter. Mol Cell Biol. 1989;9:4272–4281. doi: 10.1128/mcb.9.10.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Hayden MR. Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun. 2004;322:1310–1317. doi: 10.1016/j.bbrc.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante RJ, Beal MF. Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J Neurochem. 2001;79:1246–1249. doi: 10.1046/j.1471-4159.2001.00689.x. [DOI] [PubMed] [Google Scholar]
- Bozner P, Grishko V, LeDoux SP, Wilson GL, Chyan YC, Pappolla MA. The amyloid beta protein induces oxidative damage of mitochondrial DNA. J Neuropathol Exp Neurol. 1997;56:1356–1362. doi: 10.1097/00005072-199712000-00010. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Conde F, Beal MF, Hantraye P. Replicating Huntington’s disease phenotype in experimental animals. Prog Neurobiol. 1999;59:427–468. doi: 10.1016/s0301-0082(99)00005-2. [DOI] [PubMed] [Google Scholar]
- Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Zhou Z, Neher E, Sakmann B. Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA and kainate receptor subtypes. J Physiol. 1995;485(Pt 2):403–418. doi: 10.1113/jphysiol.1995.sp020738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabelof DC, Raffoul JJ, Yanamadala S, Guo Z, Heydari AR. Induction of DNA polymerase beta-dependent base excision repair in response to oxidative stress in vivo. Carcinogenesis. 2002;23:1419–1425. doi: 10.1093/carcin/23.9.1419. [DOI] [PubMed] [Google Scholar]
- Cabelof DC, Yanamadala S, Raffoul JJ, Guo Z, Soofi A, Heydari AR. Caloric restriction promotes genomic stability by induction of base excision repair and reversal of its age-related decline. DNA Repair (Amst) 2003;2:295–307. doi: 10.1016/s1568-7864(02)00219-7. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem. 2000;75:889–907. doi: 10.1046/j.1471-4159.2000.0750889.x. [DOI] [PubMed] [Google Scholar]
- Chen KH, Yakes FM, Srivastava DK, Singhal RK, Sobol RW, Horton JK, Van HB, Wilson SH. Up-regulation of base excision repair correlates with enhanced protection against a DNA damaging agent in mouse cell lines. Nucleic Acids Res. 1998;26:2001–2007. doi: 10.1093/nar/26.8.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Tretter L, Rozsa A, dam-Vizi V. Exacerbated responses to oxidative stress by an Na(+) load in isolated nerve terminals: the role of ATP depletion and rise of [Ca(2+)](i) J Neurosci. 2000;20:2094–2103. doi: 10.1523/JNEUROSCI.20-06-02094.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Choi S, Klingauf J, Tsien RW. Postfusional regulation of cleft glutamate concentration during LTP at ‘silent synapses’. Nat Neurosci. 2000;3:330–336. doi: 10.1038/73895. [DOI] [PubMed] [Google Scholar]
- Corona JC, Romo LB, Tapia R. Glutamate excitotoxicity and therapeutic targets for amyotrophic lateral sclerosis. Expert Opin Ther Targets. 2007;11:1415–1428. doi: 10.1517/14728222.11.11.1415. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J Neurochem. 2001;77:220–228. doi: 10.1046/j.1471-4159.2001.t01-1-00220.x. [DOI] [PubMed] [Google Scholar]
- De LG, Russo MT, Degan P, Tiveron C, Zijno A, Meccia E, Ventura I, Mattei E, Nakabeppu Y, Crescenzi M, Pepponi R, Pezzola A, Popoli P, Bignami M. A role for oxidized DNA precursors in Huntington’s disease-like striatal neurodegeneration. PLoS Genet. 2008;4:e1000266. doi: 10.1371/journal.pgen.1000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30:379–387. doi: 10.1038/aps.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Biniszkiewicz D, Sobol RW, Harms C, Ahmadi M, Lipski A, Katchanov J, Mergenthaler P, Dirnagl U, Wilson SH, Meisel A, Jaenisch R. Increased postischemic brain injury in mice deficient in uracil-DNA glycosylase. J Clin Invest. 2004;113:1711–1721. doi: 10.1172/JCI20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enokido Y, Tamura T, Ito H, Arumughan A, Komuro A, Shiwaku H, Sone M, Foulle R, Sawada H, Ishiguro H, Ono T, Murata M, Kanazawa I, Tomilin N, Tagawa K, Wanker EE, Okazawa H. Mutant huntingtin impairs Ku70-mediated DNA repair. J Cell Biol. 2010;189:425–443. doi: 10.1083/jcb.200905138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facheris M, Beretta S, Ferrarese C. Peripheral markers of oxidative stress and excitotoxicity in neurodegenerative disorders: tools for diagnosis and therapy? J Alzheimers Dis. 2004;6:177–184. doi: 10.3233/jad-2004-6210. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Ginty DD, Bading H, Greenberg ME. Calcium regulation of gene expression in neuronal cells. J Neurobiol. 1994;25:294–303. doi: 10.1002/neu.480250309. [DOI] [PubMed] [Google Scholar]
- Gleichmann M, Zhang Y, Wood WH, III, Becker KG, Mughal MR, Pazin MJ, van PH, Kobilo T, Zonderman AB, Troncoso JC, Markesbery WR, Mattson MP. Molecular changes in brain aging and Alzheimer’s disease are mirrored in experimentally silenced cortical neuron networks. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsette RE. Neurodegeneration in multiple sclerosis: the role of oxidative stress and excitotoxicity. J Neurol Sci. 2008;274:48–53. doi: 10.1016/j.jns.2008.06.029. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson’s disease and parkinsonism. Ann Neurol. 2006;60:389–398. doi: 10.1002/ana.21022. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Butterworth RF, Hakim AM. Cerebral vulnerability is associated with selective increase in extracellular glutamate concentration in experimental thiamine deficiency. J Neurochem. 1993;61:1155–1158. doi: 10.1111/j.1471-4159.1993.tb03635.x. [DOI] [PubMed] [Google Scholar]
- Hongpaisan J, Winters CA, Andrews SB. Calcium-dependent mitochondrial superoxide modulates nuclear CREB phosphorylation in hippocampal neurons. Mol Cell Neurosci. 2003;24:1103–1115. doi: 10.1016/j.mcn.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Intano GW, Cho EJ, McMahan CA, Walter CA. Age-related base excision repair activity in mouse brain and liver nuclear extracts. J Gerontol A Biol Sci Med Sci. 2003;58:205–211. doi: 10.1093/gerona/58.3.b205. [DOI] [PubMed] [Google Scholar]
- Jiang X, Zhu D, Okagaki P, Lipsky R, Wu X, Banaudha K, Mearow K, Strauss KI, Marini AM. N-methyl-D-aspartate and TrkB receptor activation in cerebellar granule cells: an in vitro model of preconditioning to stimulate intrinsic survival pathways in neurons. Ann N Y Acad Sci. 2003;993:134–145. doi: 10.1111/j.1749-6632.2003.tb07522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2010 doi: 10.1016/S1474-4422(10)70277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna TH, Mahipal S, Sudhakar A, Sugimoto H, Kalluri R, Rao KS. Reduced DNA gap repair in aging rat neuronal extracts and its restoration by DNA polymerase beta and DNA-ligase. J Neurochem. 2005;92:818–823. doi: 10.1111/j.1471-4159.2004.02923.x. [DOI] [PubMed] [Google Scholar]
- Kruman II, Culmsee C, Chan SL, Kruman Y, Guo Z, Penix L, Mattson MP. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci. 2000;20:6920–6926. doi: 10.1523/JNEUROSCI.20-18-06920.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Kumaravel TS, Lohani A, Pedersen WA, Cutler RG, Kruman Y, Haughey N, Lee J, Evans M, Mattson MP. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J Neurosci. 2002;22:1752–1762. doi: 10.1523/JNEUROSCI.22-05-01752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Pedersen WA, Springer JE, Mattson MP. ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp Neurol. 1999;160:28–39. doi: 10.1006/exnr.1999.7190. [DOI] [PubMed] [Google Scholar]
- Kruman II, Schwartz E, Kruman Y, Cutler RG, Zhu X, Greig NH, Mattson MP. Suppression of uracil-DNA glycosylase induces neuronal apoptosis. J Biol Chem. 2004;279:43952–43960. doi: 10.1074/jbc.M408025200. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX. Extracellular glutamate is increased in thalamus during thiamine deficiency-induced lesions and is blocked by MK-801. J Neurochem. 1993;61:2175–2182. doi: 10.1111/j.1471-4159.1993.tb07457.x. [DOI] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Liu D, Croteau DL, Souza-Pinto N, Pitta M, Tian J, Wu C, Jiang H, Mustafa K, Keijzers G, Bohr VA, Mattson MP. Evidence that OGG1 glycosylase protects neurons against oxidative DNA damage and cell death under ischemic conditions. J Cereb Blood Flow Metab. 2010 doi: 10.1038/jcbfm.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Prasad R, Beard WA, Kedar PS, Hou EW, Shock DD, Wilson SH. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J Biol Chem. 2007;282:13532–13541. doi: 10.1074/jbc.M611295200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini AM, Ueda Y, June CH. Intracellular survival pathways against glutamate receptor agonist excitotoxicity in cultured neurons. Intracellular calcium responses. Ann N Y Acad Sci. 1999;890:421–437. doi: 10.1111/j.1749-6632.1999.tb08021.x. [DOI] [PubMed] [Google Scholar]
- Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight GS. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol. 1994;14:6107–6116. doi: 10.1128/mcb.14.9.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Med. 2003;3:65–94. doi: 10.1385/NMM:3:2:65. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Dou P, Kater SB. Outgrowth-regulating actions of glutamate in isolated hippocampal pyramidal neurons. J Neurosci. 1988a;8:2087–2100. doi: 10.1523/JNEUROSCI.08-06-02087.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-kappaB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Lee RE, Adams ME, Guthrie PB, Kater SB. Interactions between entorhinal axons and target hippocampal neurons: a role for glutamate in the development of hippocampal circuitry. Neuron. 1988b;1:865–876. doi: 10.1016/0896-6273(88)90134-1. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Totterdell S, Beales M, Meshul CK. Impaired glutamate homeostasis and programmed cell death in a chronic MPTP mouse model of Parkinson’s disease. Exp Neurol. 2009;219:334–340. doi: 10.1016/j.expneurol.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K, Kalivas PW. Group II metabotropic glutamate receptors (mGlu2/3) in drug addiction. Eur J Pharmacol. 2010;639:115–122. doi: 10.1016/j.ejphar.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti F, Bruno V, Copani A, Casabona G, Knopfel T. Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders? Trends Neurosci. 1996;19:267–271. doi: 10.1016/S0166-2236(96)20019-0. [DOI] [PubMed] [Google Scholar]
- Polidori MC, Mecocci P, Browne SE, Senin U, Beal MF. Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci Lett. 1999;272:53–56. doi: 10.1016/s0304-3940(99)00578-9. [DOI] [PubMed] [Google Scholar]
- Raji NS, Krishna TH, Rao KS. DNA-polymerase alpha, beta, delta and epsilon activities in isolated neuronal and astroglial cell fractions from developing and aging rat cerebral cortex. Int J Dev Neurosci. 2002;20:491–496. doi: 10.1016/s0736-5748(02)00079-5. [DOI] [PubMed] [Google Scholar]
- Ramana CV, Boldogh I, Izumi T, Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc Natl Acad Sci U S A. 1998;95:5061–5066. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao KS, Annapurna VV, Raji NS. DNA polymerase-beta may be the main player for defective DNA repair in aging rat neurons. Ann N Y Acad Sci. 2001;928:113–120. doi: 10.1111/j.1749-6632.2001.tb05641.x. [DOI] [PubMed] [Google Scholar]
- Rao-Mirotznik R, Buchsbaum G, Sterling P. Transmitter concentration at a three-dimensional synapse. J Neurophysiol. 1998;80:3163–3172. doi: 10.1152/jn.1998.80.6.3163. [DOI] [PubMed] [Google Scholar]
- Reynolds IJ. Mitochondrial membrane potential and the permeability transition in excitotoxicity. Ann N Y Acad Sci. 1999;893:33–41. doi: 10.1111/j.1749-6632.1999.tb07816.x. [DOI] [PubMed] [Google Scholar]
- Ribeiro FM, Paquet M, Cregan SP, Ferguson SS. Group I metabotropic glutamate receptor signalling and its implication in neurological disease. CNS Neurol Disord Drug Targets. 2010;9:574–595. doi: 10.2174/187152710793361612. [DOI] [PubMed] [Google Scholar]
- Rolig RL, McKinnon PJ. Linking DNA damage and neurodegeneration. Trends Neurosci. 2000;23:417–424. doi: 10.1016/s0166-2236(00)01625-8. [DOI] [PubMed] [Google Scholar]
- Rolseth V, Runden-Pran E, Luna L, McMurray C, Bjoras M, Ottersen OP. Widespread distribution of DNA glycosylases removing oxidative DNA lesions in human and rodent brains. DNA Repair (Amst) 2008;7:1578–1588. doi: 10.1016/j.dnarep.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengpiel B, Preis E, Krieglstein J, Prehn JH. NMDA-induced superoxide production and neurotoxicity in cultured rat hippocampal neurons: role of mitochondria. Eur J Neurosci. 1998;10:1903–1910. doi: 10.1046/j.1460-9568.1998.00202.x. [DOI] [PubMed] [Google Scholar]
- Sheng M, Thompson MA, Greenberg ME. CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- Stetler RA, Gao Y, Zukin RS, Vosler PS, Zhang L, Zhang F, Cao G, Bennett MV, Chen J. Apurinic/apyrimidinic endonuclease APE1 is required for PACAP-induced neuroprotection against global cerebral ischemia. Proc Natl Acad Sci U S A. 2010;107:3204–3209. doi: 10.1073/pnas.1000030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J. 2000;19:1397–1404. doi: 10.1093/emboj/19.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykora P, Snow ET. Modulation of DNA polymerase beta-dependent base excision repair in cultured human cells after low dose exposure to arsenite. Toxicol Appl Pharmacol. 2008;228:385–394. doi: 10.1016/j.taap.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM. Glutamatergic mechanisms in different disease states: overview and therapeutical implications -- an introduction. Amino Acids. 2002;23:147–152. doi: 10.1007/s00726-001-0120-8. [DOI] [PubMed] [Google Scholar]
- Unnikrishnan A, Prychitko TM, Patel HV, Chowdhury ME, Pilling AB, Ventrella-Lucente LF, Papakonstantinou EV, Cabelof DC, Heydari AR. Folate deficiency regulates expression of DNA polymerase beta in response to oxidative stress. Free Radic Biol Med. 2010 doi: 10.1016/j.freeradbiomed.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasko MR, Guo C, Kelley MR. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair (Amst) 2005;4:367–379. doi: 10.1016/j.dnarep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Wang J, Jacob NK, Ladner KJ, Beg A, Perko JD, Tanner SM, Liyanarachchi S, Fishel R, Guttridge DC. RelA/p65 functions to maintain cellular senescence by regulating genomic stability and DNA repair. EMBO Rep. 2009;10:1272–1278. doi: 10.1038/embor.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Osenbroch P, Skinnes R, Esbensen Y, Bjoras M, Eide L. The OGG1 DNA Glycosylase is Essential for Mitochondrial Maturation During Differentiation of Neural Stem Cells. Stem Cells. 2010 doi: 10.1002/stem.542. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chandran JS, Cai H, Mattson MP. DJ-1 is essential for long-term depression at hippocampal CA1 synapses. Neuromolecular Med. 2008;10:40–45. doi: 10.1007/s12017-008-8023-4. [DOI] [PubMed] [Google Scholar]
- Weissman L, Jo DG, Sorensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, Bohr VA. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35:5545–5555. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AW, McCallum GP, Jeng W, Wells PG. Oxoguanine glycosylase 1 protects against methamphetamine-enhanced fetal brain oxidative DNA damage and neurodevelopmental deficits. J Neurosci. 2008;28:9047–9054. doi: 10.1523/JNEUROSCI.2557-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci U S A. 2001;98:2808–2813. doi: 10.1073/pnas.051634198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JL, Takahashi T, Keijzers G, Mattson MP, Bohr VA. Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated Up-regulation of APE1. J Biol Chem. 2010 doi: 10.1074/jbc.M109.082883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP. Lack of the p50 subunit of nuclear factor-kappaB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci. 1999;19:8856–8865. doi: 10.1523/JNEUROSCI.19-20-08856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li Q, Graham RK, Slow E, Hayden MR, Bezprozvanny I. Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington’s disease. Neurobiol Dis. 2008;31:80–88. doi: 10.1016/j.nbd.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol. 1999;154:1423–1429. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]