

Abstract
NMDA receptor-dependent long-term potentiation (LTP) and depression (LTD) are forms of synaptic plasticity underlying learning and memory that are expressed through increases and decreases, respectively, in dendritic spine size and AMPA receptor (AMPAR) phosphorylation and postsynaptic localization. The A-kinase anchoring protein 79/150 (AKAP79/150) signaling scaffold regulates AMPAR phosphorylation, channel activity, and endosomal trafficking associated with LTP and LTD. AKAP79/150 is targeted to dendritic spine plasma membranes by an N-terminal polybasic domain that binds phosphoinositide lipids, F-actin, and cadherin cell adhesion molecules. However, we do not understand how regulation of AKAP targeting controls AMPAR endosomal trafficking. Here, we report that palmitoylation of the AKAP N-terminal polybasic domain targets it to postsynaptic lipid rafts and dendritic recycling endosomes. AKAP palmitoylation was regulated by seizure activity in vivo and LTP/LTD plasticity-inducing stimuli in cultured rat hippocampal neurons. With chemical LTP induction, we observed AKAP79 dendritic spine recruitment that required palmityolation and Rab11-regulated endosome recycling coincident with spine enlargement and AMPAR surface delivery. Importantly, a palmitoylation-deficient AKAP79 mutant impaired regulation of spine size, endosome recycling, AMPAR trafficking, and synaptic potentiation. These findings emphasize the emerging importance of palmitoylation in controlling synaptic function and reveal novel roles for the AKAP79/150 signaling complex in dendritic endosomes.
Introduction
LTP and LTD at hippocampal synapses underlie spatial learning and formation of declarative memories (Malenka and Bear, 2004). These forms of synaptic plasticity are induced by postsynaptic NMDAR Ca2+ influx in dendritic spines that causes coordinated changes in spine size and AMPAR trafficking to control synaptic strength (Derkach et al., 2007; Newpher and Ehlers, 2009; Kasai et al., 2010). In particular, LTP activates kinases including calcium/calmodulin-dependent protein kinase II, protein kinase C (PKC), and cAMP-dependent protein kinase (PKA) to promote spine enlargement and AMPAR phosphorylation, exocytosis, and lateral movement to synapses to increase synaptic strength (Lee, 2006; Opazo and Choquet, 2011). In contrast, LTD activates phosphatases including protein phosphatases 1, 2A, and 2B [calcineurin (CaN)] to trigger spine shrinkage and AMPAR dephosphorylation, removal from synapses, and endocytosis to decrease synaptic strength. These kinases and phosphatases are localized to glutamate receptors at the postsynaptic density (PSD) by a network of scaffold proteins (Sheng and Hoogenraad, 2007; Sanderson and Dell'Acqua, 2011). Alterations in dendritic spine structure and postsynaptic scaffolding that regulate LTP/LTD are implicated in a number of brain disorders including Alzheimer's, schizophrenia, and autism (Penzes et al., 2011).
AKAP79/150 (human 79/rodent 150) is a scaffold protein encoded by the AKAP5 gene that targets PKA, PKC, and CaN to the PSD to regulate AMPAR phosphorylation, trafficking, and activity associated with LTP and LTD (Tavalin et al., 2002; Smith et al., 2006; Lu et al., 2007, 2008; Tavalin, 2008; Jurado et al., 2010; Sanderson and Dell'Acqua, 2011). Accordingly, AKAP150-null and PKA-anchoring-deficient mice exhibit learning and memory deficits (Tunquist et al., 2008; Weisenhaus et al., 2010). AKAP79/150 is targeted to dendritic spines by an N-terminal basic domain that binds the acidic phospholipid phosphatidylinositol-4,5-bisphosphate (PI4,5P2), F-actin, and cadherin adhesion molecules (Dell'Acqua et al., 1998; Gomez et al., 2002; Gorski et al., 2005; Horne and Dell'Acqua, 2007). The AKAP is linked to both AMPAR and NMDAR by additional binding to the membrane-associated guanylate kinase (MAGUK) scaffolds SAP97 and PSD-95 (Colledge et al., 2000; Xu et al., 2008; Bhattacharyya et al., 2009; Robertson et al., 2009). NMDAR activation of CaN and phospholipase C (PLC) promotes PI4,5P2 hydrolysis and actin depolymerization in spines leading to AMPAR endocytosis and spine shrinkage along with disruption of AKAP interactions with membranes, F-actin, cadherins, and PSD-95 to remove it from spines (Gomez et al., 2002; Gorski et al., 2005; Smith et al., 2006; Horne and Dell'Acqua, 2007). However, we still do not understand how AKAP79/150 targeting is coordinated with dendritic membrane trafficking that controls AMPAR localization and spine size.
S-palmitoylation by palmitoyl acyl transferases (PATs) modifies Cys residues by the addition of the C-16 fatty acid palmitate. Postsynaptic proteins including PSD-95, GRIP1, AMPAR, and NMDAR are palmitoylated and these modifications control trafficking and synaptic function (Fukata and Fukata, 2010). Here, we report that AKAP79/150 palmityolation is controlled by neuronal activity and regulates its targeting to postsynaptic membrane lipid rafts and dendritic endosomes. Importantly, we find that AKAP79/150 palmitoylation regulates spine enlargement, endosome recycling, and AMPAR trafficking pathways associated with LTP.
Materials and Methods
Hippocampal neuron and COS7 cell culture and transfection
COS7 cells were maintained in DMEM (Invitrogen) with 10% FBS at 37°C, 5% CO2. COS7 cells were transfected by CaPO4 or Lipofectamine 2000 (Invitrogen) with GFP/CFP/YFP-tagged AKAP79 WT, C36,129S, AKAP deletion mutants, βSAP97i3(i5), PKA-RIIα, PLCδ-PH, and CaNAα expressed from previously described pEGFP, pECFP, or pEYFP (Clontech) plasmids (Gomez et al., 2002; Oliveria et al., 2003; Horne and Dell'Acqua, 2007; Robertson et al., 2009). TfR-mCherry and superecliptic pHluorin (SEP)-TfR plasmids (Park et al., 2006) were provided by Dr. Michael Ehlers (Howard Hughes Medical Institute/Duke University, Durham, NC; present affiliation, Pfizer Global Research, Groton, CT). pmCherryN1 (for AKAP79 fusion), Rab11DN-YFP, and Rab11WT-CFP plasmids were provided by Dr. Alexander Sorkin (University of Pittsburgh, Pittsburgh, PA). For electrophysiological recordings, imaging G/C/YFP and mCherry, and standard immunostaining, hippocampal neuron cultures were prepared from postnatal day 0–2 Sprague Dawley male and female rats, plated at medium density (300–450 cells/mm2), and maintained in Neurobasal plus B27 (Invitrogen) and 200 μm GlutaMAX (Invitrogen) as previously described (Robertson et al., 2009). For acyl-biotinyl exchange (ABE)/1-biotinamido-4-[4′-(maleimidomethyl)cyclohexanecarboxamido]butane (BMCC) biotinylation assays and GluR1 surface staining, cultures were prepared from embryonic day 18 male and female rats and plated at high density (750–1000 cells/mm2) as previously described (Kang et al., 2004). Neurons were transfected on DIV 10–14 as indicated in the figure legends using Lipofectamine 2000. In some cases, pSilencer-sh150RNAi (provided by Dr. John Scott, Howard Hughes Medical Institute/University of Washington, Seattle, WA) was transfected to suppress AKAP150 expression; pSilencer-sh150RNAi was previously characterized by our laboratory and others for efficient knockdown of rat AKAP150 for 2–12 d in hippocampal neurons and for rescue with human AKAP79 (not recognized by the shRNAi) (Hoshi et al., 2005; Oliveria et al., 2007; Robertson et al., 2009).
ABE and BMCC assays of AKAP150 palmitoylation in rat brain and neurons
Palmitoylation of AKAP150 in rat brain and hippocampal neuron extracts was detected using the related ABE and BMCC-biotin labeling methods. The ABE method was performed as published in the study by Kang et al. (2008). Briefly hydroxylamine (HAM) was used to cleave Cys-palmitoyl thioester linkages, and then the resulting free Cys-SH groups were labeled with a thiol-specific biotinylation reagent to allow protein identification by streptavidin precipitation followed by immunoblotting (ABE) or immunoprecipitation followed by streptavidin blotting (BMCC variation). For the BMCC variation, neurons were collected and lysed in Tris buffer (in mm: 150 NaCl, 5 EDTA, 50 Tris, pH 7.4, 20 NEM, 0.02% NaN3, 1% Triton X-100). AKAP150 was immunoprecipitated and rinsed with SDS buffer (in mm: 500 NaCl, 0.1% SDS, 5 EDTA, 50 Tris, pH 7.4, 10 NEM, 0.03% NaN3). The precipitate was rinsed with Tris buffer and treated with HAM (Tris buffer plus 1 m HAM, pH 7.0–7.2) for 1 h at room temperature. Proteins were then rinsed with pH 6.2 Tris buffer and incubated in biotin-BMCC solution (pH 6.2 Tris buffer, 1 μm biotin-BMCC) for 1 h at 4°C. Proteins were washed in pH 6.2 Tris buffer followed by PBS and then analyzed by SDS-PAGE and blotting to nitrocellulose. Palmitoylation was detected using HRP-conjugated streptavidin (Thermo Fisher Scientific) blotting followed by ECL detection. AKAP150 was detected by immunoblotting with ECL detection as previously described (Robertson et al., 2009; Brandao et al., 2012).
3H-palmitate metabolic labeling of human AKAP79 in COS7 cells
COS7 cells were labeled with 1 mCi/ml 3H-palmitic acid (57 Ci/mmol; PerkinElmer Life Sciences) in DMEM at 37°C for 3 h. Cells were lysed in 50 mm HEPES, 1% Triton X-100 buffer containing a protease inhibitor mixture (Sigma-Aldrich). AKAP79-GFP was immunoprecipitated with anti-GFP (Invitrogen) and separated by SDS-PAGE. Samples were split between two gels: one gel for transfer to nitrocellulose and detection of protein levels by anti-GFP immunoblotting with ECL detection and the second gel for detection of 3H-palmitate by fluorography using Amplify (GE Healthcare) and exposure to Hyperfilm (GE Healthcare) at −80°C.
Triton X-100 extraction of cells
Hippocampal neurons were plated on poly-d-lysine-coated glass-bottom, 35 mm microscope dishes (MatTek). Cells transfected with AKAP79-GFP constructs were imaged at 37°C, 5% CO2 (see below), and the positions were marked for reimaging after extraction. The dish was rinsed with 4°C Krebs'–Ringer's–Henseleit (KRH) buffer (in mm: 130 NaCl, 5 KCl, 1.2 NaH2PO4, 10 glucose, 1.8–2 CaCl2, 25 HEPES, pH 7.4, 0.1% BSA) followed by 2 min extraction at 4°C with 0.1% Triton X-100 in KRH. Dishes were then rinsed twice with KRH and cells were reimaged. COS7 cells transfected with AKAP79-GFP were collected in PBS and solubilized in Tris buffer (in mm: 50 Tris, pH 7.4, 150 NaCl, 1 EDTA, 1 EGTA, 0.8% Triton X-100) for 30 min at 4°C. The samples were centrifuged at 13,000 × g for 10 min. Samples of both supernatant and pellet were analyzed by SDS-PAGE and anti-GFP immunoblotting with ECL detection. The percentage of detergent soluble anti-GFP signal in the supernatant (TxS) was determined relative to the total signal detected in the supernatant plus pellet (TxS+TxP).
Synaptic lipid raft fractionation
Whole brains were homogenized (glass/Teflon homogenizer) in fractionation buffer (in mm: 150 NaCl, 1 EDTA, 20 Tris, pH 7.4, 200 μm PMSF, and protease inhibitors). Homogenates were centrifuged at 800 × g for 10 min. The supernatant was collected and centrifuged at 15,000 × g for 15 min to isolate the synaptic membrane pellet fraction. A sample of the supernatant (nonsynaptic fraction) was retained. The pellet was suspended in 2 ml of fractionation buffer containing sucrose (42% final concentration) and Triton X-100 (0.5% final concentration). The samples were rotated for 30 min at 4°C, and then placed on the bottom of a 12 ml centrifuge tube. The samples were overlaid with 8 ml of fractionation buffer containing 35% sucrose and then 2 ml of fractionation buffer containing 16% sucrose. The samples were spun at 35,000 rpm (Beckman; SW-41 rotor) for 18 h at 4°C. Twelve fractions were collected from the top of the gradient. Equal volumes of each fraction were separated by SDS-PAGE, and proteins of interest were detected by immunoblotting. The following primary antibodies were used: rabbit anti-AKAP150 as above (Robertson et al., 2009; Brandao et al., 2012), rabbit anti-PSD-95, mouse anti-flottilin (BD Biosciences), rabbit anti-SNAP25, and chicken anti-transferrin receptor (TfR) (Zymed). Secondary antibodies for quantitative analysis of immunoblots by Odyssey Infrared Imaging System (Li-COR Bioscience) were anti-rabbit IR800 (1:10,000; Rockland), anti-chicken IR800 (1:10,000; LI-COR), and anti-mouse Alexa Fluor 680 secondary antibodies (1:10,000; Invitrogen).
Kainate seizures
Adult female Wistar rats received intraperitoneal injections of kainate (15 mg/kg) or saline vehicle. Rats were observed for seizure activity that was rated according to the Racine scale (Racine, 1972) as follows: stage 0, immobility; stage 1, facial automatism; stage 2, head nodding; stage 3, unilateral/bilateral forelimb clonus; stage 4, bilateral forelimb clonus and rearing; stage 5, rearing, falling, and generalized convulsions. Kainate-treated rats that sustained stage 3 and above seizures were used for the study. Brain tissue was collected 30 min after seizure onset.
Electrophysiology
Whole-cell voltage-clamp recordings were performed at room temperature under infrared-differential contrast microscopy with an Axopatch 200B amplifier (Molecular Devices). Microelectrodes had resistances of 3–6 MΩ. AMPAR mEPSCs were recorded from hippocampal pyramidal neurons at a holding potential of −65 mV using a Cs methylsulfonate internal solution containing the following (in mm): 5 CsCl, 140 CsCH3SO3, 10 HEPES, 10 EGTA, 2 NaATP, 0.3 NaGTP, pH 7.3, in artificial CSF (ACSF) [in mm: 130 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 11 glucose (osmolarity, 320 mOsm) plus 1 μm TTX, 50 μm picrotoxin]. Access resistances were between 15 and 35 MΩ; if the access resistance changed >20%, the recording was discarded. Data were analyzed using Mini Analysis Program (Synaptosoft). For chemical LTP (cLTP), neurons were treated with 100 μm glycine in Mg2+-free ACSF for 10 min without TTX and picrotoxin at 37°C, and then recovered in normal ACSF without TTX and picrotoxin for 25 min at 37°C before recording.
Immunostaining
Standard immunofluorescence.
Neurons and COS7 cells were fixed in 3.7% formaldehyde, permeabilized with 0.2% Triton X-100 (TX-100), and processed as previously described (Robertson et al., 2009) for staining with the following antibodies: 1:500 anti-early endosome antigen 1 (EEA1) (BD Biosciences) and goat anti-mouse Alexa647 (Invitrogen; 1:1000); 1:500 anti-GluR1 (Millipore) and goat anti-rabbit Alexa 647 (Invitrogen; 1:1000); 1:500 anti-GluR2 (Millipore Bioscience Research Reagents) and goat anti-rabbit Alexa 647 (Invitrogen; 1:1000); 1:500 anti-bassoon (Stressgen) and goat anti-mouse Texas Red (Invitrogen; 1:1000); anti-PSD-95 (NeuroMab; 1:500) and goat anti-mouse Alexa 647 (Invitrogen; 1:500).
AMPAR-GluR1 surface labeling.
For experiments with NMDA–chemical LTD (cLTD), neurons on coverslips were treated in Neurobasal containing 10 or 30 μm NMDA plus 20 μm CNQX and 1 μm TTX, for 5 min at 37°C. Cells were then rinsed twice and left in Neurobasal plus 20 μm CNQX, 1 μm TTX, and 100 μm AP-V and harvested after 20 min. For cLTP, cells were treated with glycine (200 μm) in Mg2+-free extracellular solution (in mm: 140 NaCl, 1.3 CaCl2, 5 KCl, 25 HEPES, pH 7.4, 33 glucose, 0.5 μm TTX, 1 μm strychnine, 20 μm bicuculline methiodide) at 37°C for 5 min. Cells were then rinsed twice with room temperature Mg2+-free extracellular solution without glycine, and incubated at 20°C for 20 min. Surface GluR1 subunits were labeled with anti-GluR1 N-terminal domain antibody (Calbiochem) (1:15 dilution) for 20 min at 10°C. Cells were then fixed with 4% paraformaldehyde (5 min, room temperature) and rinsed three times with PBS, and nonspecific sites were blocked with 2.5% BSA in PBS for 30 min at room temperature. Goat anti-rabbit Alexa 568 (Invitrogen; 1:1000) was added for 1 h to detect GluR1. AKAP79-GFP constructs were labeled with chicken anti-GFP (Abcam; 1:1000) followed by goat anti-chicken Alexa 488 (Invitrogen; 1:1000).
Fluorescence imaging and analysis
Images of transfected COS7 cells and neurons were acquired on Axiovert 200M microscopes (Zeiss) with a 63× objective (1.4 NA; plan-Apo) and either Axiocam HR (Zeiss) or CoolSNAP2 (Photometrics) CCD cameras. Focal plane z-stacks (0.5 μm spacing) were acquired and deconvolved to correct for out-of-focus light, and in some cases 2D maximum intensity projections were generated (Slidebook 4.0–5.0; Intelligent Imaging Innnovations). Either single focal planes or projection images were used in mask analysis of mean intensities and colocalization. For live-cell imaging of AKAP79-G/Y/CFP, neurons on poly-d-lysine- and laminin-coated 35 mm glass-bottom dishes were maintained at 33°C in Tyrode's saline (in mm: 25 HEPES, pH 7.4, 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 0.1% BSA). For NMDA-cLTD, multiple untreated neurons were imaged, treated with 10 μm NMDA for 10 min on the microscope stage, imaged again, and then treated with 30 μm NMDA and imaged again 10 min later. For cLTP treatment, neurons were incubated in Tyrode's without Mg2+ and treated for 10 min with 100 μm glycine at 37°C in an incubator, followed by washing with regular Tyrode's, and live imaging of G/YFP 20–30 min later. For live-cell spinning disc confocal imaging of SEP-TfR exocytosis, neurons were plated on poly-d-lysine- and laminin-coated, no. 1, 25 mm round glass coverslips at a density of 500,000 cells/coverslip. Neurons were transfected from 12 to 14 DIV with SEP-TfR and AKAP79WT-mCherry or C36,129S-mCherry using Lipofectamine 2000 as above. Images were acquired at 35°C on an Axio Observer microscope (Zeiss) with a 63× objective (1.4 NA, plan-Apo) using 488 and 561 nm laser excitation and a CSU-XI spinning disk confocal scan head (Yokogawa) coupled to an Evolve 512 EM-CCD camera (Photometrics) driven by Slidebook 5.0. Before imaging, neurons were incubated in ACSF plus 2 mm MgCl2 for 30 min and maintained at 35°C in a perfusion imaging chamber (P-2; Warner Instruments). Baseline rates of SEP-TfR exocytic events were determined by acquiring z-stacks of 10–14 optical sections (0.5 μm spacing) every 7 s for 4 min. For cLTP stimulation neurons, the bath solution was exchanged by peristaltic pump (Harvard Apparatus) perfusion with ACSF without Mg2+ with 200 μm glycine and 50 μm picrotoxin during 4 min of additional imaging.
Image analysis
Images were analyzed by Northern Eclipse software (EMPIX Imaging) or Slidebook 4.0–5.0 (Intelligent Imaging Innovations) using mask analysis to measure mean fluorescence intensities, spine/shaft mean intensity ratios, spine areas, spine numbers, puncta numbers/10 μm, or integrated intensity colocalization ratios as described previously (Horne and Dell'Acqua, 2007; Robertson et al., 2009). Integrated intensity colocalization ratios for discrete regions of punctate fluorescence 1.5× above the mean were measured from 2D maximum intensity projections of deconvolved z-stacks for neurons or from single xy-focal planes for COS7 cells. However, for analysis of colocalization in single deconvolved xy-focal planes through dendrites in Figure 7, E and F, the threshold for defining puncta was set at 2–2.5× above the mean. Where indicated in the legend of Figure 4, CFP or YFP was cotransfected as cytoplasmic spine filler (images not shown) for use in mask analysis of spine areas and numbers as in the study by Robertson et al. (2009). Analysis of 4D spinning-disc movies was conducted using Slidebook 5.0 by first generating time-lapse series of deconvolved 2D-maximum intensity projection images from the 3D image z-stacks for each time point. SEP-TfR membrane fusion events were then identified by an acute increase in GFP fluorescence more than twofold above the mean intensity of the dendrite. Time-lapse data sets were then divided into pre- (t = 0–4 min) and post-cLTP treatment (t = 4–8 min) image sequences. In the pretreatment images, the number of events per 100 μm length of dendrite per 1 min was calculated to determine the basal rate of TfR-vesicle exocytosis for each neuron as plotted in Figure 10I. The total number of events post-cLTP treatment was divided by the total number of events pretreatment for each neuron to determine the percentage increase in exocytosis in response to cLTP as plotted in Figure 10J. Time-lapse composite channel images and 3D-intensity plots in Figure 10E–H were generated for the SEP-TfR (GFP, 488 nm) channel from the pre- (t = 0–4 min) and post-cLTP treatment (t = 4–8 min) image sequences to show cumulative exocytosis over the indicated 4 min time window. These images are presented in pseudocolor to represent SEP-TfR fluorescence intensity (in arbitrary fluorescence units) on a relative scale from low (blue) to high (red) and in Figure 10, E and F, are overlaid onto monochrome images of AKAP79 WT or C36,129S dendrite fluorescence (mCherry, 561 nm) from the final frame of the time window. SEP-TfR fluorescence intensity images at t = 0 are provided for comparison.
Figure 7.
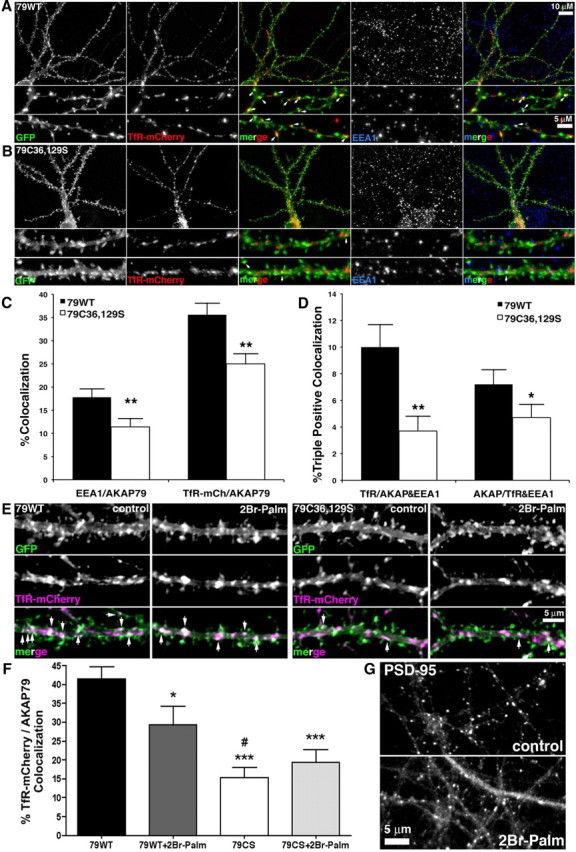
AKAP79 palmitoylation is required for localization to dendritic recycling endosomes in hippocampal neurons. A, B, Maximum intensity 2D projection images from deconvolved 3D image stacks of hippocampal neurons cotransfected (12–14 DIV) with either 79WT (A) or C36,129S-GFP (green) and TfR-mCherry (red) (B) and then immunostained to detect EEA1 (blue). Colocalization of AKAP79-GFP with TfR-mCherry appears yellow in RG merge images (middle panels) and triple colocalization of 79-GFP, TfR-mCherry, and EEA1 appears white in RGB merge images (right panels). The two bottom panels show magnifications of representative dendrites with sites of colocalization indicated by arrows in the RG and RGB merge images. C36,129S-GFP exhibits greatly reduced overlap with TfR-mCherry and EEA1 compared with 79WT-GFP. C, Reduced endosome marker colocalization for C36,129S expressed as percentage of EEA1 or percentage of TfR-mCherry fluorescence colocalized with AKAP79-GFP for 79WT (EEA1/AKAP79, 18 ± 2%; TfR-mCh/AKAP79, 36 ± 3%; n = 16) and C36,129S (EEA1/AKAP79, 11 ± 2%; TfR-mCh/AKAP79, 25 ± 2%; **p < 0.01 by t test to 79WT; n = 16). D, Reduced endosome marker colocalization for C36,129S expressed as percentage of TfR-mCherry fluorescence colocalized with both AKAP79-GFP and EEA1 or percentage of AKAP79-GFP colocalized with both TfR-mCherry and EEA1 for 79WT (TfR/AKAP&EEA1, 10 ± 2%; AKAP/TfR&EEA1, 7 ± 1%; n = 16) and C36,129S (TfR/AKAP&EEA1, 4 ± 1%, **p < 0.01; AKAP/TfR&EEA1, 5 ± 1%, *p < 0.05 by t test to 79WT; n = 16). E, Higher magnification images of single deconvolved focal planes through neuronal dendrites expressing 79WT or C36,129S-GFP (green) and TfR-mCherry (magenta) either before (control) or after 2Br-Palm treatment (20 μm, 16 h). Colocalization of AKAP79-GFP with TfR-mCherry appears white and is indicated by arrows in the RM merge images. F, Quantification of percentage of TfR-mCherry colocalized with AKAP79-GFP demonstrates that 2Br-Palm decreases colocalization with 79WT (control, 42 ± 4%, n = 13; 2Br-Palm, 29 ± 5%, n = 11; *p < 0.05) but not with C36,129S (control, 15 ± 3%, n = 14; 2Br-Palm, 19 ± 4%, n = 14), which is already significantly decreased compared with 79WT (***p < 0.001) and 79WT+2Br-Palm (#p < 0.05). Comparisons by t test. G, PSD-95 immunostaining in control (0.01% DMSO vehicle) and 2Br-Palm-treated neurons showing obvious, but incomplete, declustering of PSD-95 indicating partial inhibition of protein palmitoylation. Error bars indicate SEM.
Figure 4.

Palmitoylation and recycling endosome trafficking is required for AKAP79 spine recruitment and spine enlargement in response to cLTP. A, Hippocampal neurons transfected with 79WT-GFP, C36,129S-GFP, YFP, or AKAP150RNAi+YFP (12–14 DIV) imaged for G/YFP before (control; top panels) and 20–30 min after cLTP (100 μm glycine, 0 Mg2+ for 10 min; bottom panels). B, Spine/shaft fluorescence ratios demonstrating that 79WT and C36,129S are both enriched in spines compared with YFP in control neurons (###p < 0.001 by ANOVA compared with YFP) but only 79WT is recruited to spines in response to cLTP: spine/shaft ratios 79WT (control, 1.16 ± 0.03; cLTP, 1.49 ± 0.06; ***p < 0.001 by t test), C36,129S (control, 1.14 ± 0.04; cLTP, 1.14 ± 0.04), YFP (control, 0.74 ± 0.05; cLTP, 0.82 ± 0.06), 150RNAi+YFP (control, 0.79 ± 0.04; cLTP, 0.88 ± 0.03); n = 15–37 neurons. C, 79WT and C36,129S increase mean spine area compared with YFP controls (###p < 0.001 by ANOVA), but cLTP stimulates additional spine enlargement in 79WT (control, 0.73 ± 0.01 μm2; cLTP, 0.82 ± 0.01 μm2) and YFP neurons (control, 0.51 ± 0.01 μm2; cLTP, 0.58 ± 0.01 μm2) (***p < 0.001 to controls by t test) but not C36,129S neurons (control, 0.69 ± 0.01 μm2; cLTP, 0.69 ± 0.02 μm2). n = 209–837 spines from 15–37 neurons. D, Mean spine areas for control and cLTP-treated neurons cotransfected with AKAP150RNAi+YFP, 79WT-YFP+CFP, or 79C36,129S-YFP+CFP. Inhibition of cLTP spine enlargement by AKAP150RNAi (control, 0.49 ± 0.01 μm2; cLTP, 0.52 ± 0.01 μm2; *p < 0.05 by t test) is rescued by 79WT (79WT+150RNAi: control, 0.62 ± 0.02 μm2; cLTP, 0.73 ± 0.02 μm2, ***p < 0.001 by t test) but not C36,129S (79CS+150RNAi: control, 0.59 ± 0.02 μm2; cLTP, 0.62 ± 0.02 μm2). Both 79WT+150RNAi and 79CS+150RNAi enlarged basal spine sizes compared with 150RNAi (###p < 0.001, ##p < 0.01 by ANOVA) similar to Figure 4C. n = 234–458 spines from 13–20 neurons. E, F, Cumulative plots of spine areas (binned by 0.05 μm2 increments) reveal distribution shifts toward larger basal spine sizes for (E) 79WT or C36,129S compared with YFP in control neurons. cLTP caused an additional shift toward larger spine areas compared with their respective controls for YFP and 79WT but not for C36,129S. F, AKAP150RNAi suppressed changes in spine area distribution in response to cLTP and 79WT, but not C36,129S, rescue expression restored a larger cLTP-induced distribution shift. G, BMCC palmitoylation (top panel) of endogenous rat AKAP150 in 14–21 DIV hippocampal neurons 20 min after vehicle (control) or cLTP (200 μm glycine, 0 Mg2+, 5 min). AKAP150 palmitoylation (corrected for AKAP150 levels; bottom panel) increases to 132 ± 12% of control cells following cLTP treatment; n = 5; *p < 0.05 by t test. H, Hippocampal neurons transfected (12–14 DIV) with 79WT-CFP plus YFP or Rab11DN-YFP (S25N mutation) imaged for CFP (YFP images not shown) before (control) and 20–30 min after cLTP (100 μm glycine, 0 Mg2+ for 10 min). I, C36,129S-CFP in hippocampal neurons transfected with YFP (top panel) or Rab11DN-YFP (bottom panel) (YFP images not shown). J, Spine/shaft fluorescence basal spine localization is increased and cLTP-induced recruitment is prevented by blocking recycling endosome trafficking with Rab11DN-YFP: 79WT-CFP+YFP (control, 1.23 ± 0.06; cLTP, 1.5 ± 0.1; **p < 0.01), 79WT-CFP+Rab11DN (control, 1.5 ± 0.1; ##p < 0.01 to 79WT-CFP control; cLTP, 1.6 ± 0.1). Rab11DN-YFP did not change basal spine localization of C36,129S-YFP (79CS-CFP+YFP, 1.3 ± 0.1; 79CS-CFP+Rab11DN-YFP, 1.2 ± 0.1). n = 5–10 neurons. Comparisons by t test. K, Rab11DN increases basal mean spine area and prevents cLTP-stimulated spine enlargement in neurons expressing 79WT: 79WT-CFP+YFP (control, 0.73 ± 0.04 μm2; cLTP, 0.83 ± 0.03 μm2; *p < 0.05); 79WT-CFP+Rab11DN (control, 0.89 ± 0.06 μm2; ##p < 0.01 to 79WT-CFP control; cLTP, 0.83 ± 0.06 μm2). Rab11DN-YFP did not increase basal mean spine area in C36,129S-CFP neurons (79CS-CFP+YFP, 0.65 ± 0.04 μm2; 79CS-CFP+Rab11DN-YFP, 0.58 ± 0.03 μm2). n = 70–138 spines from 5–10 neurons. Comparisons by t test. L, Quantification of spine/shaft fluorescence ratios showing that 79WT-YFP basal spine localization and cLTP recruitment was normal in Rab11WT-CFP-expressing neurons: 79WT-YFP+CFP (control, 1.17 ± 0.05; cLTP, 1.38 ± 0.07; **p < 0.01 by t test), 79WT-YFP+Rab11WT-CFP (control, 1.23 ± 0.06; cLTP, 1.45 ± 0.1; *p < 0.05 by t test). n = 8–12 neurons. M, Rab11WT-CFP expression increased basal mean spine area slightly in 79WT-YFP neurons but additional cLTP-induced spine enlargement was still observed: 79WT-YFP+CFP (control, 0.58 ± 0.02 μm2; cLTP, 0.69 ± 0.02 μm2; **p < 0.01), 79WT-YFP+Rab11WT-CFP (control, 0.64 ± 0.02 μm2; #p < 0.05 to 79WT-YFP control; cLTP, 0.72 ± 0.03 μm2; *p < 0.05); n = 168–267 spines from 8–12 neurons. Error bars indicate SEM.
Figure 10.
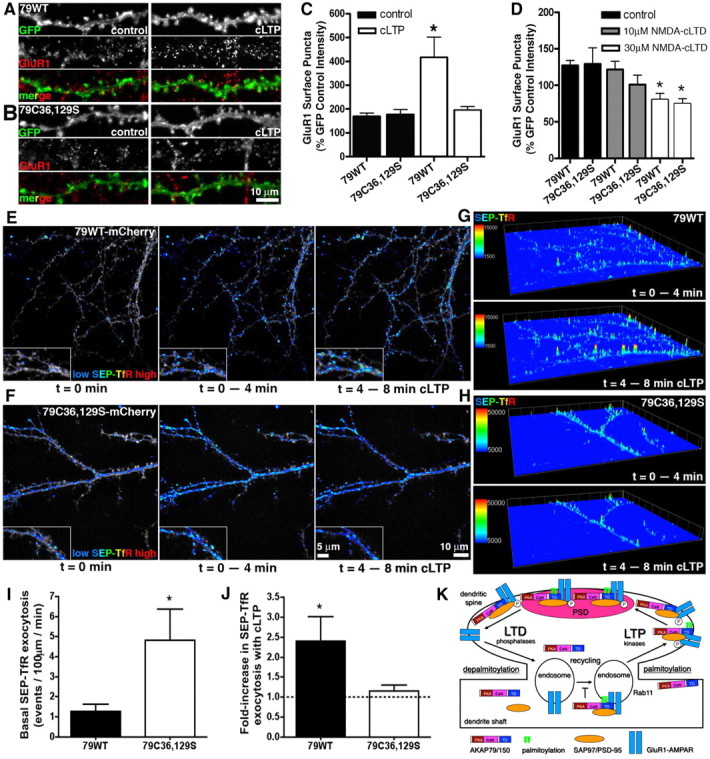
AKAP79 palmitoylation is required for increases in GluR1 surface expression and exocytosis from recycling endosomes in response to cLTP. A, B, Hippocampal neurons cotransfected (14–21 DIV) with AKAP150RNAi and 79WT (A) or C36,129S-GFP (B) and left untreated (control) or treated with cLTP (200 μm glycine, 0 Mg2+, 20 μm bicuculline, 5 min). Surface GluR1 receptors were labeled 20 min later. C, GluR1 dendritic surface puncta mean intensity expressed as a percentage of untreated GFP control cells (images not shown) increases in response to cLTP in 79WT (control, 169 ± 14%; cLTP, 416 ± 84%; *p < 0.05 by t test) but not in C36,129S neurons (control, 176 ± 20%; cLTP, 195 ± 16%). n = 9–12 neurons. D, NMDA-cLTD treatment (10 or 30 μm, 5 min) leads to normal GluR1 internalization in both 79WT and C36,129S neurons (images not shown): Quantification of GluR1 puncta intensity for 79WT (control, 127 ± 7%; 10 μm NMDA, 121 ± 11%; 30 μm NMDA, 80 ± 9%; *p < 0.05 by ANOVA) and C36,129S (control, 129 ± 22%; 10 μm NMDA, 101 ± 13%; 30 μm NMDA, 75 ± 7%; *p < 0.05 by ANOVA); n = 9–12 neurons. E, F, Images of initial dendritic SEP-TfR fluorescence intensity (pseudocolor, low-blue to high-red) at t = 0 and then accumulated intensity due to exocytosis (time-composite image) over t = 0–4 min of basal conditions and then over t = 4–8 min of subsequent cLTP stimulation (200 μm glycine, 0 Mg2+, 50 μm picrotoxin) in neurons transfected (12–14 DIV) with 79WT (E) or C36,129S-mCherry (monochrome underlay) (F). G, H, Three-dimensional time-composite plots of dendritic SEP-TfR fluorescence intensity (z-dimension; pseudocolor, low-blue to high-red) showing accumulated recycling exocytosis events for basal conditions (t = 0–4 min) and cLTP stimulation (t = 4–8 min) in neurons expressing 79WT (G) and C36,129S (H). Pseudocolor scale bars in arbitrary fluorescence units. I, Elevated basal SEP-TfR exocytosis rate measured as events/100 μm of dendrite/min for C36,129S (4.8 ± 1.5; n = 8; *p < 0.05 by t test) compared with 79WT (1.3 ± 0.4; n = 11). J, cLTP stimulation of SEP-TfR exocytosis measured as fold increase over the basal rate for 79WT (2.4 ± 0.6; n = 11; *p < 0.05 by one-sample t test) but not C36,129S (1.2 ± 0.2; n = 8). K, Proposed model for regulation of AKAP79/150 palmitoylation and endosome trafficking in dendrites during LTD and LTP. Palmitoylation (green) of the AKAP targeting domain (TD) (blue) and the PKA (red) and CaN (magenta) anchoring domains are indicated. PKC binds to the AKAP TD (data not shown). Interactions with the MAGUK scaffold proteins SAP97 and PSD-95 (orange) and AMPAR-GluR1 (blue) are also indicated. Palmitoylated AKAP is present in the PSD, extrasynaptic plasma membrane, and endosomes where it may limit basal recycling and GluR1 synaptic localization. Depalmitoylated AKAP is present only in the extrasynaptic plasma membrane where it still may promote basal GluR1 phosphorylation. Palmitoylated AKAP and phosphorylated GluR1 are delivered to spines at extrasynaptic locations by recycling endosome exocytosis during cLTP and then recruited into the PSD. AKAP is removed from the PSD and depalmitoylated to uncouple it from both the plasma membrane and endosomes following cLTD. GluR1 is dephosphorylated and removed from the extrasynaptic membrane by endocytosis. Additional roles for PLC-catalyzed PI4,4P2 hydrolysis and CaN-dependent actin depolymerization in AKAP79/150 spine removal are not depicted. Error bars indicate SEM.
Animal care and use
All experiments using both male and female rats were performed under protocols approved by the Institutional Animal Care and Use Committees of the University of Colorado, Denver, or University of British Columbia.
Statistical analysis
Group comparisons to control were performed in Prism (GraphPad) using one-way ANOVA followed by Dunnet's or Bonferroni's post hoc analysis. Pairwise and one-sample comparisons were performed in Prism or Excel (Microsoft) using Student's t test as indicated in Results and the figure legends. Data are expressed as mean ± SEM (error bars). Binned cumulative distributions were generated in Prism. Significance is reported as p < 0.05.
Results
AKAP79/150 is palmitoylated on two conserved Cys residues within its N-terminal targeting domain
AKAP150 was identified as a palmitoylated protein in a proteomic screen of rat brain synaptic membranes (Kang et al., 2008). Here, we map the sites of palmitoylation to two conserved Cys residues in the N-terminal basic targeting domain (Fig. 1A,B). Rat AKAP150 differs from its human ortholog AKAP79 (both products of the AKAP5 gene) primarily due to the insert of a repetitive sequence with no known function just N-terminal to the conserved CaN anchoring domain (Fig. 1B). Thus, we confirmed palmitoylation of AKAP79-GFP expressed in COS7 cells by metabolic labeling with 3H-palmitate (Fig. 1C). To map palmitoylation sites, we screened a series of AKAP79 fragments (Fig. 1B) and found that only the N-terminal targeting domain (1–153) was palmitoylated (Fig. 1C) with both the A and C basic subdomains containing likely palmitoylation sites (Fig. 1A,B). Accordingly, mutation of either of two conserved Cys residues to Ser, C36S in subdomain A or C129S in subdomain C, reduced AKAP79 palmitoylation (Fig. 1D), but only the double C36,129S mutant eliminated palmitoylation (Fig. 1E). As a control, mutation of two nonconserved Cys resides (C62S or C150S) did not impact palmitoylation (Fig. 1D). C36 is very close to two Arg residues (R39 and R40) that mediate PKC anchoring to the A subdomain (Faux et al., 1999), but an R39,40A double mutant that inhibits PKC anchoring, did not alter palmitoylation (Fig. 1E).
Figure 1.

AKAP79 is palmitoylated on two conserved Cys residues in the N-terminal polybasic membrane targeting domain. A, Sequence alignments of the AKAP79/150 A and C basic targeting subdomains from multiple species showing the locations of C36 and C129 in green. Basic residues are in blue and other highly conserved residues are in black, bold. B, Diagram of selected AKAP79 mutants used for mapping palmitoylation sites. The locations of the A–C basic targeting subdomains (blue), C36 and C129 (green), CaN anchoring domain (purple), and PKA anchoring (red) are indicated. C, COS7 cells transfected with GFP-tagged AKAP79 constructs and labeled with 3H-palmitate detected by fluorography. Palmitoylation (top panel) of the N-terminal 1–153 targeting domain fragment is similar to full-length AKAP79 with no detectable palmitoylation observed for 154–427. Levels of AKAP79 proteins detected by anti-GFP immunoblotting (bottom panel). D, Palmitoylation (top panel) is reduced by C36S and C129S (but not C62S or C150S) mutations within the AKAP79 N-terminal targeting domain. Levels of AKAP79 proteins detected by anti-GFP immunoblotting (bottom panel). E, Palmitoylation (top panel) is prevented by the C36,129S double point mutation but is not impacted by the R39,40A mutation that inhibits nearby PKC binding to AKAP79. Levels of AKAP79 proteins detected with anti-GFP immunoblotting are shown (bottom panel).
AKAP79 palmitoylation confers resistance to detergent extraction from cell membranes consistent with lipid raft association
Since C36 and C129 are embedded in basic regions that control membrane targeting through PI4,5P2 and protein–protein interactions (Dell'Acqua et al., 1998; Gomez et al., 2002; Gorski et al., 2005), we examined C36,129S for impacts on targeting in hippocampal neurons. Interestingly, 79WT and C36,129S-GFP showed similar localization to the somato-dendritic plasma membrane including spines (Fig. 2A,E,F; control panels). Palmitoylation frequently promotes association with detergent-resistant lipid rafts. Accordingly, while ∼65% of the 79WT-GFP fluorescence in neurons resisted extraction with TX-100, C36,129S-GFP was almost completely extracted by TX-100 (Fig. 2A). This increased TX-100 solubility of C36,129S compared with 79WT was confirmed by extraction of COS7 cells (Fig. 2B,C). Thus, while AKAP79 palmitoylation on C36 and 129S is not absolutely required for membrane and spine targeting, it likely increases the strength of AKAP79 membrane attachment by promoting association with lipid rafts. [During the course of this work, an independent study (Delint-Ramirez et al., 2011) reported that AKAP79 palmitoylation on C36 and C129 is required for lipid raft association and contributes to plasma membrane targeting along with phosphoinositide binding in HEK-293 cells.]
Figure 2.

AKAP79 palmitoylation confers resistance to Triton X-100 extraction and opposes removal from dendritic spines with NMDA-cLTD stimulation. A, Hippocampal neurons transfected with 79WT or C36,129S-GFP (12–14 DIV) live-imaged before (top panels) and after (bottom panels) 2 min extraction with 0.1% TX-100 on ice. TX-100 extracted 36 ± 5% of 79WT-GFP fluorescence (normalized to before values; *p < 0.05 to before by ANOVA) and 95 ± 1% of 79C36,129S-GFP fluorescence (***p < 0.001 to before; ###p < 0.001 to 79WT after by ANOVA). B, COS7 transfected with GFP-tagged 79WT and C36,129S solubilized in 0.8% cold Triton X-100 followed by centrifugation to separate detergent-soluble material in the supernatant (TxS) from the insoluble in the pellet (TxP). C, Quantification of AKAP79-GFP immunoreactivity in TxS-supernatant samples as a percentage of the total immunoreactivity (TxS+TxP): AKAP79WT, 57 ± 5%; 79C36,129S, 81 ± 3%; GFP, 86 ± 3%. n = 5; **p < 0.01 by ANOVA compared with 79WT. D, Endogenous rat AKAP150 in 14–21 DIV hippocampal neurons shows decreased palmitoylation (BMCC method, top panel, normalized to amount of AKAP150 precipitated, bottom panel) 20 min following NMDA-cLTD (30 μm NMDA, 3 min; 25 ± 7% of control; n = 7; *p < 0.05 by t test) or 20 h after treatment with 2Br-Palm (100 μm; palmitoylation undetectable). E–G, Compared with 79WT, C36,129S is more easily removed from dendritic spines by NMDA-cLTD. Live-cell imaging of 14 DIV hippocampal neuron dendrites expressing 79WT (E) or C36,129S-GFP (F) before treatment (control), after 10 min in 10 μm NMDA, and then after 10 min in 30 μm NMDA. G, Normalized spine/shaft mean GFP fluorescence ratios showing increased sensitivity to low-dose NMDA-induced removal from spines for C36,129S (control, 1.12 ± 0.04, n = 14; 10 μm NMDA, 0.55 ± 0.02, n = 19) compared with 79WT (control, 1.23 ± 0.03, n = 15; 10 μm NMDA, 0.95 ± 0.08, n = 6; 30 μm NMDA, 0.65 ± 0.03, n = 16). ***p < 0.001 compared with respective controls and ###p < 0.001 with 79WT, 10 μm NMDA, by ANOVA for 79WT and by t test for C36,129S. Error bars indicate SEM.
NMDA-LTD induces AKAP79/150 depalmitoylation to promote removal from dendritic spines
We previously showed that NMDAR agonist activation that chemically induces LTD (NMDA-cLTD) causes translocation of AKAP79/150 away from dendritic membranes and spines through pathways that involve PLC-catalyzed PI4,5P2 hydrolysis and CaN-dependent F-actin reorganization (Gomez et al., 2002; Horne and Dell'Acqua, 2007); however, we wanted to determine whether palmitoylation also regulates AKAP translocation in response to NMDA. Using the biotin-BMCC variation of the ABE method to detect palmityolation (see Materials and Methods), we found that NMDA-cLTD leads to partial, but rapid (20 min) depalmitoylation of endogenous AKAP150 in hippocampal neurons (Fig. 2D). For comparison, much longer treatment (20 h) with the PAT inhibitor 2-bromo-palmitate (2Br-Palm) was required to induce more complete AKAP150 depalmitoylation (Fig. 2D). Consistent with depalmitoylation promoting NMDA-induced AKAP translocation from spines, in live-cell imaging experiments we measured 79-GFP mean spine/shaft fluorescence ratios (Horne and Dell'Acqua, 2007) to show that C36,129S moved prominently from spines with 10 μm NMDA (Fig. 2F,G), whereas comparable redistribution of 79WT required 30 μm NMDA (Fig. 2E,G). Importantly, 79WT and C36,129S showed similar levels of basal spine enrichment measured by spine/shaft fluorescence ratios of ∼1.20 (Fig. 2G) compared with a cytoplasmic YFP (∼0.7; see Fig. 4A,B). Thus, NMDA-induced AKAP translocation from spines is promoted by depalmitoylation in addition to PLC-mediated depletion of PI4,5P2 and actin depolymerization as previously shown (Gomez et al., 2002; Horne and Dell'Acqua, 2007).
AKAP79/150 palmitoylation and synaptic lipid raft association increase following seizure activity in vivo
The results above indicate that AKAP palmitoylation stabilizes it in membranes through association with lipid rafts and is regulated by NMDAR activity. To explore activity-regulated palmitoylation in vivo, we induced seizures in rats by injection of kainate, isolated synaptic lipid rafts using sucrose density gradient centrifugation, and then analyzed AKAP150 distribution by immunoblotting. AKAP150 palmitoylation detected by the ABE method was increased (∼75%) 30 min after kainate (Fig. 3A). In control rats, AKAP150 sedimented in synaptic lipid raft fractions 1–3 peaking with the palmitoylated raft marker Flotillin in fraction 2 (Fig. 3B,C) and was also detected in nonraft synaptic membrane fractions 11–12 and the nonsynaptic supernatant (not run on the gradient) that contains cytoplasm and internal membranes (Fig. 3B,D). PSD-95, which is also palmitoylated and is a postsynaptic binding partner of AKAP150, showed a similar fractionation pattern as AKAP150, but PSD-95 was not detected in the nonsynaptic fraction (Fig. 3B,E). In contrast, the TfR, a nonraft enriched transmembrane protein, exhibited an equal distribution across fractions (Fig. 3G). Following kainate seizures, AKAP150 levels in raft fractions increased while levels in nonraft and nonsynaptic fractions decreased (Fig. 3B,D). PSD-95 also shifted its distribution from nonraft to raft fractions following seizures (Fig. 3B,E). Importantly, distribution of the raft marker Flotillin did not change with kainate (Fig. 3B,C). As an additional control, SNAP25, a palmitoylated protein present in both axons and dendrites that is not known to bind AKAP or PSD-95, enriched in lipid raft fraction 1 after kainate but did not closely codistribute with AKAP150 or PSD-95 on the gradient in control or kainate (Fig. 3B,F). Membrane distribution of the nonraft TfR did not change with kainate (Fig. 3G). Overall, these results demonstrate that seizures promote AKAP150 palmitoylation and movement to postsynaptic lipid rafts.
Figure 3.

Increased AKAP150 palmitoylation and rat brain postsynaptic lipid raft localization in vivo following kainate seizures. A, Rats were treated with vehicle (control; left two lanes) or with kainate (right two lanes) to induce seizure activity, and after 30 min whole brains were processed for ABE palmitoylation (top panel). Samples without HAM were used as assay controls. Quantification corrected for AKAP150 levels (bottom panel) confirmed increased AKAP150 palmitoylation after kainate seizures (176 ± 9% of control; n = 3; *p < 0.05 by t test). B, Whole-brain synaptic membrane fractions from control and kainate-treated rats were separated by sucrose density gradient centrifugation to isolate lipid rafts and analyzed by immunoblotting as indicated. Lipid raft fractions 1–3 were detected with anti-Flotillin antibody. Following kainate seizures, AKAP150 and PSD-95 redistribute into lipid raft fractions. C–G, Quantifications of percentages of proteins in lipid raft fractions 1–3 (as a percentage of total immunoreactivity detected). All data points are mean ± SEM and analyzed by t test, n = 3–4. C, Flotillin (control, 81 ± 7%; kainate, 72 ± 3%). D, AKAP150 (control, 43 ± 7%; kainate, 65 ± 4%; *p < 0.05). E, PSD-95 (control, 45 ± 3%; kainate, 76 ± 4%; *p < 0.05). F, SNAP25 (control, 38 ± 2%; kainate, 57 ± 2%; *p < 0.05). G, TfR (control, 14 ± 2%; kainate, 19 ± 5%).
AKAP79 palmitoylation is required for recruitment to dendritic spines and spine enlargement linked to LTP
The above results for seizures and cLTD prompted us to investigate whether AKAP palmitoylation and postsynaptic localization are regulated by LTP. Using chemical stimulation that induces NMDAR-dependent LTP (Lu et al., 2001) (cLTP) (10 min in 100 μm glycine, 0 Mg2+) and triggers spine enlargement, AMPAR dendritic exocytosis, and AMPAR synaptic insertion in cultured neurons (Park et al., 2004, 2006; Yudowski et al., 2007; Lin et al., 2009; Fortin et al., 2010; Kennedy et al., 2010), we found that 79WT-GFP moved to spines as shown by an increase in the spine/shaft ratio 25 min after cLTP (Fig. 4A,B). Similar to our previous work on spine development (Robertson et al., 2009), 79WT-GFP overexpression increased basal spine sizes above those in YFP controls as measured by increased mean spine area (Fig. 4C) and a right-shifted distribution of spine areas on a cumulative plot (Fig. 4E); however, cLTP induced additional spine enlargement in both 79WT and YFP neurons (Fig. 4C,E). AKAP79 spine enrichment following cLTP was not simply due to spine enlargement increasing cytosolic spine fluorescence because YFP spine/shaft ratios did not significantly increase with cLTP (Fig. 4B). While C36,129S-GFP increased basal spine size above YFP controls, no additional spine enlargement or recruitment of C36,129S to spines was seen in response to cLTP (Fig. 4A,C,E). Similarly, acute (<48 h) RNAi knockdown of AKAP150 largely prevented cLTP-induced spine enlargement (Fig. 4A,D,F), and these inhibitory effects of 150RNAi were rescued by 79WT but not C36,129S (Fig. 4D,F). These results indicate that palmitoylated AKAP79 is required for cLTP-induced AKAP spine recruitment and spine enlargement. Accordingly, endogenous AKAP150 palmitoylation detected by the BMCC method increased ∼30% after cLTP (Fig. 4G).
Rab11-dependent recycling endosome trafficking regulates AKAP79 spine localization
Previous work by Ehlers and colleagues (Park et al., 2004, 2006) demonstrated that both spine enlargement and synaptic potentiation linked to LTP depend on exocytosis from dendritic recycling endosomes that are regulated by the small GTPase Rab11. Using a dominant-negative Rab11 mutant (Rab11DN-YFP) known to interfere with these events, we found that 79WT-CFP recruitment to spines during cLTP depends on Rab11 trafficking (Fig. 4H,J). Consistent with previous studies, neurons expressing Rab11DN and 79WT also failed to increase spine sizes following cLTP (Fig. 4H,K). Interestingly, control 79WT+Rab11DN-expressing neurons showed larger spine sizes and significantly higher basal AKAP spine enrichment compared with 79WT alone (Fig. 4H,K), but these changes were not observed for C36,129S+Rab11DN (Fig. 4I–K). In contrast to Rab11DN, Rab11WT-CFP+79WT-YFP expression did not alter basal AKAP79 spine/shaft ratio (Fig. 4L) and increased spine size only slightly (∼9%; Fig. 4M). In addition, we still observed significant cLTP-induced AKAP79 spine recruitment and spine enlargement in 79WT+Rab11WT neurons (Fig. 4L,M). Thus, endosome recycling that is disrupted by Rab11DN specifically regulates spine targeting of palmitoylated AKAP79 in coordination with spine size.
Palmitoylation is required for localization of the AKAP79 signaling complex in endosomes but not the plasma membrane
AKAP79/150 is generally considered to be a peripheral plasma membrane protein, but the above findings raise the possibility that AKAP trafficking is also associated with endosomes. In COS7 cells transfected with 79WT-GFP, we used live-cell labeling with the recycling cargo transferrin conjugated to Texas Red (Tfr-TxRd) followed by fixation and immunostaining with EEA1 to assess AKAP79 localization. In addition to prominent 79WT-GFP localization in plasma membrane cell–cell contacts and ruffles (∼88% of total; Fig. 5A,C), we observed a smaller, but appreciable amount of 79WT-GFP in punctate structures (∼12% of total; Fig. 5A,C) that showed colocalization with EEA1 (∼85% of total EEA1) or Tfr-TxRd (∼70% of total Tfr-TxRd) (Fig. 5A,C,D). In some endosomes, 79WT-GFP colabeled only with Tfr-TxRd (Fig. 5A, yellow arrows), but in most endosomes colabeling was seen with Tfr-TxRd and EEA1 (Fig. 5A, white arrows). Strikingly, C36,129S-GFP targeted to the plasma membrane but showed essentially no colocalization with Tfr-TxRd or EEA1 in endosomes (Fig. 5B–D).
Figure 5.

AKAP79 palmitoylation is required for endosomal but not plasma membrane localization. A, B, Images of COS7 cells expressing 79WT (A) or C36,129S-GFP (green) (B) live-labeled with Tfr-TxRd (red) and then fixed and immunostained for EEA1-Alexa 647 (blue). The insets show magnifications of endosomes. Colocalization of 79-GFP with Tfr-TxRd is yellow in RG merge panels (middle). Triple colocalization of 79-GFP, Tfr-TxRd, and EEA1 is white in the RGB merge panels (right). 79WT-GFP shows localization to both plasma membrane and endosomes, while C36,129S-GFP is only found at the plasma membrane. C, Loss of C36,129S compared with 79WT-GFP fluorescence (expressed as percentage of total GFP fluorescence) colocalized with EEA1 (AKAP79/EEA1: 79WT, 12.3 ± 4%; C36,129S, 2 ± 1%; *p < 0.05) or Tfr-TxRd (AKAP79/Tfr: 79WT, 11.8 ± 5; C36,129S, 2 ± 1%; *p < 0.05); n = 10 cells. Comparisons by t test to 79WT. D, Loss of EEA1 (EEA1/AKAP79: 79WT, 85 ± 5%; C36,129S, 11 ± 3%; ***p < 0.001) or Tfr-TxRd fluorescence (Tfr/AKAP79: 79WT, 69 ± 8; 79C36,129S, 9 ± 4%; ***p < 0.001) colocalized (expressed as percentage of total fluorescence) with C36,129S compared with 79WT-GFP; n = 10 cells. Comparisons by t test. E, COS7 cells expressing 79WT-GFP (green) treated with 100 μm 2Br-Palm for 6 h and then labeled with Tfr-TxRd (red) and EEA1 (blue). Site of residual AKAP colocalization with Tfr-TxRd (yellow arrowheads) plus EEA1 (white arrowhead) are indicated on the RGB merge panel magnification inset. F, Quantification demonstrating decreased EAA1 or Tfr-TxRd fluorescence colocalized with C36S (EEA1, 23 ± 5%; Tfr, 23 ± 5%), C129S (EEA1, 26 ± 8%; Tfr, 25 ± 9%), C36,129S (EEA1, 10 ± 2%; Tfr, 10 ± 3%), and 79WT+2Br-Palm (EEA1, 22 ± 6%; Tfr, 22 ± 5%) compared with 79WT (73 ± 5%; Tfr, 64 ± 5%) by ANOVA, ***p < 0.001. C36S, C129S, and 2Br-Palm did not remove AKAP79 from endosomes as completely as C36,129S compared by t test, #p < 0.05; n = 12–27 cells. G, AKAP79WT-CFP (magenta) colocalizes with PI4,5P2 (PLCδ-PH-YFP, green) in plasma membrane ruffles of COS7 cells (seen as white in merge panel) but not in most endosomes (indicated by magenta arrows in the merge panel magnification inset). Error bars indicate SEM.
Treatment with 2Br-Palm reduced colocalization of 79WT with EEA1 and Tfr-TxRd (Fig. 5E,F) similar to single C36S and C129S mutations (Fig. 5F), but none of these manipulations prevented endosome localization as effectively as C36,129S (Fig. 5F). Early endosomes are rich in PI3P (bound by EEA1) but are depleted of PI4,5P2 compared with the plasma membrane; thus, palmitoylation could be required for AKAP targeting to endosomes but not the plasma membrane where greater electrostatic interactions with PI4,5P2 are possible. To test this idea, we coexpressed 79WT-CFP with the pleckstrin homology (PH) domain of PLCδ fused to YFP as a specific probe for PI4,5P2. Similar to our previous studies (Gomez et al., 2002), 79WT-CFP colocalized in plasma membrane ruffles with PLCδ-PH-YFP; however, AKAP79 was also present in numerous endosomes without PLCδ-PH consistent with a lack of PI4,5P2 (Fig. 5G).
We next explored whether palmitoylated AKAP79 could assemble signaling complexes containing PKA, CaN, and MAGUKs in endosomes. Consistent with our previous work (Gomez et al., 2002; Oliveria et al., 2003), 79WT-CFP colocalized with PKA-RII-YFP, CaNA-YFP, or SAP97i3-YFP at the plasma membrane (Fig. 6). However, we also observed a sizeable percentage of EEA1 endosomes (40–60%) that contained 79WT-CFP and either PKA-RII-YFP (Fig. 6A,B), CaNA-YFP (Fig. 6C,D), or SAP97i3-YFP (Fig. 6E,F). The C36,129S-CFP mutant eliminated endosome colocalization of EEA1 with PKA (Fig. 6A,B) and CaN (Fig. 6C,D) but retained its ability to target PKA and CaN to the plasma membrane. Likewise, C36,129S promoted colocalization with SAP97i3 at the plasma membrane but led to a significant, although not complete, loss of SAP97i3 localization with EEA1 (Fig. 6E,F). These results indicate that palmitoylated AKAP79 promotes targeting of PKA, CaN, and SAP97 to endosomes, although SAP97 has some AKAP-independent association.
Figure 6.

Palmitoylation controls AKAP79 assembly of signaling complexes containing PKA, CaN, and SAP97 at endosomes. A, COS7 cells expressing 79WT or C36,129S-CFP (blue) and PKA-RIIα-YFP (green) fixed and immunostained for EEA1 (red) reveals plasma membrane colocalization of AKAP79 and PKA (turquoise in merge panels) and endosome colocalization of all three proteins (insets) for 79WT (white in merge panels) but only plasma membrane AKAP79/PKA colocalization for C36,129S. B, Quantification of percentage of EEA1 fluorescence colocalized with AKAP79-CFP or PKA-RII-YFP for 79WT (79-CFP, 38 ± 5%; PKA-RII-YFP, 54 ± 10%) and C36,129S (79-CFP, 4 ± 2%; PKA-RII-YFP, 4 ± 1%; ***p < 0.001 by t test) reveals loss of PKA endosome targeting for C36,129S. n = 6–7 cells. C, COS7 cells expressing 79WT or C36,129S-CFP (blue) and CaNAα-YFP (green) fixed and immunostained for EEA1 (red) reveals plasma membrane colocalization of AKAP79 and CaN (turquoise in merge panels) and endosome colocalization of all three proteins (insets) for 79WT (white in merge panels) but only plasma membrane AKAP79/CaN colocalization for C36,129S. D, Quantification of percentage of EEA1 fluorescence colocalized with AKAP79-CFP or CaNA-YFP for 79WT (79-CFP, 46 ± 12%; CaNA-YFP, 48 ± 15%) and C36,129S (79-CFP, 7 ± 2%; CaNA-YFP, 6 ± 2%; ***p < 0.001 by t test) reveals loss of CaN endosome targeting for C36,129S; n = 5–9 cells. E, COS7 cells expressing 79WT or C36,129S-CFP (blue) and βSAP97i3-YFP (green) fixed and immunostained for EEA1 (red) reveals plasma membrane colocalization of AKAP79 and SAP97 (turquoise in merge panels) and endosome colocalization of all three proteins (insets) for 79WT (white in merge panels) but mainly plasma membrane AKAP79/SAP97 colocalization for C36,129S. F, Quantification of percentage of EEA1 fluorescence colocalized with AKAP79-CFP or SAP97i3-YFP for 79WT (79-CFP, 43 ± 14%; SAP97i3-YFP, 43 ± 14%) and C36,129S (79-CFP, 6 ± 2%; SAP97i3-YFP, 17 ± 2%; *p < 0.05, **p < 0.01 by t test) reveals reduced SAP97 endosome targeting for C36,129S; n = 7–10 cells. Error bars indicate SEM.
To establish whether palmitoylation controls AKAP79 targeting to dendritic endosomes, we transfected hippocampal neurons with AKAP79-GFP and with the TfR tagged with mCherry and immunostained for EEA1 (Fig. 7A,B). 79WT-GFP showed many sites of punctate colocalization with TfR-mCherry in 2D maximum-intensity projections of 3D-deconvolved image stacks in dendrite shafts and in some cases in spines (Fig. 7A; arrows in RG merge magnification panels). Indeed, ∼35% of TfR-mCherry endosomes contained 79WT-GFP (Fig. 7C). A smaller percentage of endosomes (∼17%) were positive for EEA1 and 79WT-GFP (Fig. 7C) and roughly one-half of these (8–10%) also contained TfR-mCherry (Fig. 7D; A, arrows in RGB merge magnification panels). Overall, it appears that AKAP79 in dendrites, like in COS7 cells, is present in early and recycling endosomes but perhaps more distinctly segregated with TfR in recycling endosomes. C36,129-GFP exhibited normal targeting to dendrite plasma membranes and spines (as seen above in Figs. 2, 4) but very few sites of overlap with EEA1 and TfR-mCherry (Fig. 7B) with significant reductions in the measures of endosome localization (Fig. 7C,D).
Dendritic endosomal colocalization values for C36,129S did not drop as completely as in COS7 cells, which is not surprising given that these endosomes are contained within thin dendritic process and spines that are micrometers to submicrometers in dimension. Considering the inherent resolution limits of conventional fluorescence microscopy, especially in the axial dimension (∼0.5 μm), some incidental overlap of plasma membrane and endosomes is unavoidable in dendrites and spines especially in projection images. To examine this issue further, we analyzed small in-focus regions of dendrites from only single deconvolved focal planes to better separate specific from incidental overlap of AKAP and TfR (Fig. 7E). Accordingly, we found a greater percentage reduction in TfR-mCherry colocalization for C36,129S compared with 79WT (∼64%; Fig. 7F) than measured above for projection images (∼30%). In addition, following treatment with 2Br-Palm, TfR colocalization decreased (∼31%) for 79WT but not for C36,129S. This 2Br-Palm treatment caused an obvious, but incomplete, loss of PSD-95 clustering (Fig. 7G) that depends on palmityolation (El-Husseini et al., 2002), indicating likely only partial inhibition of PAT activity and AKAP palmitoylation. However, neurons treated for longer times with higher doses of 2Br-Palm to induce more complete depalmitoylation of AKAP150 (as in Fig. 2D) showed adverse changes in overall dendritic morphology making colocalization analyses problematic (data not shown). Based on all our analyses, we conclude that the C36,129S palmityolation-deficient mutant, like in COS7 cells, is unable to localize to recycling endosomes in neuronal dendrites.
AKAP79 palmitoylation is required to limit basal synaptic incorporation of GluR2-lacking AMPARs and to maintain synaptic potentiation following cLTP
The failure of C36,129S to localize to recycling endosomes explains its lack of delivery to spines with cLTP and suggests that palmitoylated AKAP79 is co-delivered to spines with AMPARs and other Rab11-regulated cargoes that are necessary for spine enlargement and potentiation. To explore a role for AKAP79 palmitoylation in AMPAR potentiation, we recorded miniature EPSCs (mEPSCs) from neurons expressing GFP, 79WT, or C36,129S-GFP (Fig. 8A). Our previous studies showed that chronic overexpression of AKAP79 in 0- to 12-d-old cultured neurons promoted spine maturation and increased mEPSC activity (Robertson et al., 2009). To reduce impacts on basal AMPAR activity that might occlude LTP, here we overexpressed 79WT or C36,129S for <48 h from day 10 to 12 in culture. Accordingly, acute expression of 79WT showed similar mean mEPSC frequencies (Fig. 8A,B) and amplitudes (Fig. 8A,C,D) as GFP controls. Surprisingly, C36,129S increased both mean mEPSC frequency (approximately fivefold; Fig. 8A,B) and amplitude (∼46%; Fig. 8A,C) compared with GFP. Increased mEPSC amplitudes for C36,129S were also seen as a large, uniform rightward shift on cumulative distribution plots (Fig. 8D). Interestingly, AKAP150 RNAi for <48 h also significantly increased mEPSC frequency compared with GFP (approximately twofold; Fig. 8E), and while it did not significantly increase mean mEPSC amplitude (Fig. 8F), it did produce a rightward shift toward larger mEPSC amplitudes >15 pA on cumulative plots (Fig. 8G). This 150RNAi effect on frequency and amplitude distribution was largely reversed by coexpression of 79WT but not C36,129S, which still strongly increased frequency (approximately fivefold; Fig. 8E) and amplitude (∼23%; Fig. 8F,G). Thus, the increase of mEPSC activity with C36,129S may in small part be a response to general reduction of AKAP functions similar to RNAi, but clearly involves additional dominant effects likely related to C36,129S mistargeting.
Figure 8.
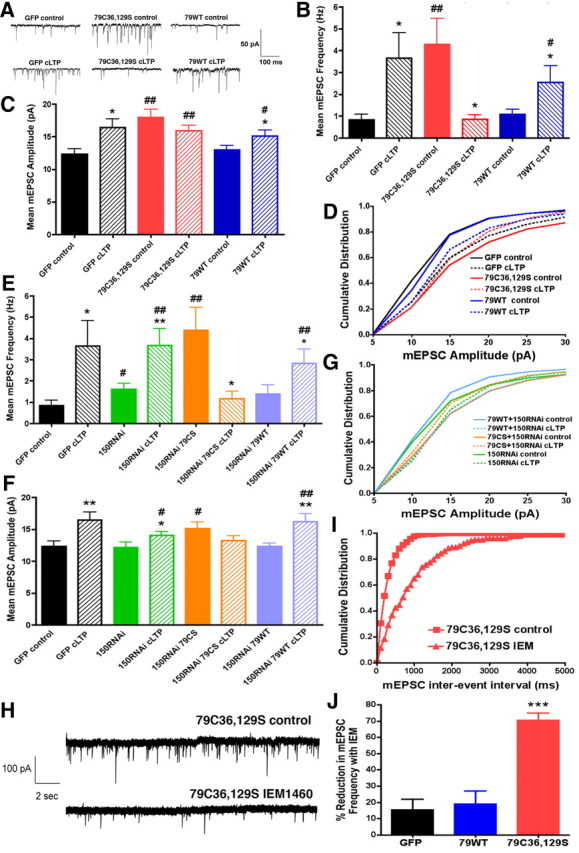
AKAP79 C36,129S increases basal synaptic activity through GluR2-lacking AMPA receptors but prevents maintenance of synaptic potentiation following cLTP. A, Representative mEPSC recordings for hippocampal neurons transfected (10–12 DIV) with GFP, 79WT-GFP, and C36,129S-GFP before (control) and 20–30 min after cLTP (100 μm glycine, 0 Mg2+, 10 min). B, C36,129S increases basal mean mEPSC frequency compared with GFP alone but after cLTP treatment shows synaptic depression instead of potentiation that is seen for 79WT and GFP: C36,129S (4.3 ± 1.2 Hz, n = 16, ##p < 0.01 to GFP control; cLTP, 0.9 ± 0.2 Hz, n = 9, *p < 0.05 to control); 79WT (control, 0.9 ± 0.2 Hz, n = 13; cLTP, 2.6 ± 0.8 Hz, n = 13, *p < 0.05 to control); GFP (control, 0.8 ± 0.2 Hz, n = 10; cLTP, 3.7 ± 1.2 Hz, n = 9, *p < 0.05 to control). Comparisons by t test. C, C36,129S increases basal mean mEPSC amplitude compared with GFP but fails to show additional potentiation after cLTP like 79WT and GFP: C36,129S (control, 17.9 ± 1.2 pA, n = 16, ##p < 0.01 to GFP control; cLTP, 16.0 ± 0.7 pA, n = 9); 79WT (control, 13.0 ± 0.7 pA, n = 13; cLTP, 15.1 ± 0.9 pA, n = 13, *p < 0.05 to control; #p < 0.05 to GFP control); GFP (control, 12.3 ± 0.8 pA, n = 10; cLTP, 16.5 ± 1.3 pA, n = 9, **p < 0.01 to control). Comparisons by t test. D, Cumulative distributions of mEPSC amplitudes (binned by 5 pA increments) showing that both GFP and 79WT have similar control distributions of mEPSC amplitudes and show rightward distribution shifts toward larger mEPSC amplitudes after cLTP; however, C36,129S starts with a control distribution containing larger mEPSC amplitudes than GFP or 79WT and then shows a leftward shift toward smaller mEPSC amplitudes after cLTP. E, RNAi suppression of AKAP150 increases basal mEPSC frequency (control, 1.6 ± 0.3 Hz; n = 16; #p < 0.05 to GFP control) but still allows cLTP stimulation to significantly increase it further (cLTP, 3.6 ± 0.8 Hz, n = 13, **p < 0.01 to control; ##p < 0.01 to GFP control). Basal and cLTP potentiation of mEPSC frequency are not significantly changed by 150RNAi+79WT rescue (control, 1.4 ± 0.4 Hz, n = 15; cLTP, 2.8 ± 0.7 Hz, n = 9; *p < 0.05 to control; ##p < 0.01 to GFP control). 150RNAi+C36,129S still increases basal mEPSC frequency dramatically (control, 4.4 ± 1.1 Hz, n = 14; ##p < 0.01 to GFP control) and exhibits a strong reduction in response to cLTP (cLTP, 1.2 ± 0.3 Hz, n = 8; *p < 0.05 to 79C36,129S control by t test). GFP control and cLTP values are from B. Comparisons by t test. F, 150RNAi does not change basal mean mEPSC amplitude (control, 12.2 ± 0.8 pA; n = 16) but allows only a small increase with cLTP (cLTP, 14.0 ± 0.6 pA; n = 13; *p < 0.05 to control; #p < 0.05 to GFP control). 150RNAi+C36,129S enhances basal mean amplitude (control, 15.1 ± 1.0 pA; n = 13; #p < 0.05 to GFP control by t test) but shows no potentiation with cLTP (cLTP, 13.2 ± 0.8 pA; n = 8). 150RNAi+79WT rescue shows normal basal and cLTP regulation of mean mEPSC amplitudes (control, 12.3 ± 0.6 pA, n = 15; cLTP, 16.2 ± 1.3 pA, n = 8; **p < 0.01 to control; ##p < 0.01 to GFP control). GFP control and cLTP values are from C. Comparisons by t test. G, Cumulative distribution of mEPSC amplitudes showing that 150RNAi causes a rightward shift in basal mEPSC amplitude distribution compared with GFP (D) and 150RNAi+79WT rescue controls and allows only a small additional rightward shift with cLTP. 150RNAi+79WT shows normal basal and cLTP regulation of mEPSC amplitude distribution similar to GFP control (D). In contrast, 150RNAi+C36,129S exhibits a strong rightward shift in basal mEPSC amplitude distribution compared with GFP control and then a leftward shift toward smaller amplitudes with cLTP as seen above in D. H, Representative mEPSC recording traces for the same C36,129S-expressing neuron before (control) and after addition of the Ca2+-permeable AMPAR blocker IEM1460 (60 μm). I, Representative cumulative distribution plots of mEPSC interevent intervals (100 ms bins) for the C36,129S-expressing neuron in H before (control) and after addition of IEM1460 showing a large increase in interevent interval indicating a large decrease in mEPSC frequency. All C36,129S neurons recorded from (n = 8) showed significant increases by the Kolmogorov–Smirnov test (7 of 8 cells, p < 0.01; 1 of 8 cells, p < 0.05) in interevent interval distribution after IEM compared with before. GFP (n = 7) and 79WT (n = 5) (data not shown) showed no significant changes after IEM. J, Significant percentage reduction in mean mEPSC frequency after IEM1460 for C36,129S (n = 8; 71 ± 5%) but not 79WT (n = 5; 19 ± 8%) or GFP (n = 7; 15 ± 7%). ***p < 0.001 by one-sample t test compared with before (control) and by ANOVA compared with GFP and 79WT. IEM caused no significant changes in mean mEPSC amplitude for GFP (−0.3 ± 4%), 79WT (−0.8 ± 3%), or C36,129S (−2.4 ± 9%) by one-sample t test compared with before (control). Error bars indicate SEM.
We detected increases (∼30–45%) in total dendritic spine numbers for C36,129S (5.3 ± 0.3 spines/10 μm; **p < 0.01 by t test, n = 6) and 79WT (5.9 ± 0.3 spines/10 μm; ***p < 0.001 by t test, n = 7) compared with GFP controls (4.1 ± 0.3 spines/10 μm; n = 5). 150RNAi neurons showed similar spine densities as GFP controls (4.4 ± 0.2 spines/10 μm; n = 14). However, total synapse numbers identified by staining with the presynaptic marker bassoon increased only for C36,129S (∼23%) but not for 79WT (Fig. 9A–D). In addition, C36,129S, but not 79WT, increased the number (∼21%) and percentage (∼10%) of synaptic GluR1 puncta colocalized with bassoon (Fig. 9E,F). These GluR1/bassoon immunostaining results for C36,129S are in agreement with the increase in mEPSC amplitude in Figure 8, C and D, and indicate that the increase in mEPSC frequency is due to both new synapse formation and postsynaptic unsilencing by AMPARs.
Figure 9.
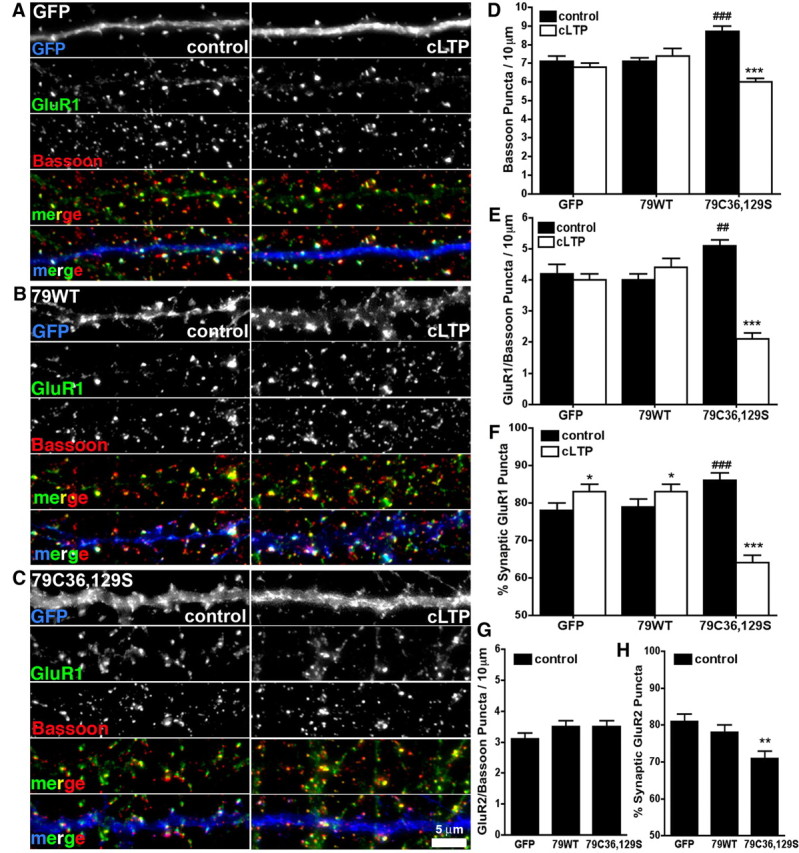
AKAP79 C36,129S increases basal synapse number and GluR1 synaptic localization, but these changes are reversed by cLTP stimulation. A–C, Hippocampal neurons dendrites expressing GFP (control) (A), 79WT-GFP (B), or C36,129S-GFP (all in blue) (C) immunostained for GluR1 (green), and Bassoon (red) before (control) or 20–30 min after cLTP stimulation. Colocalization of GluR1 and Bassoon is yellow in RG and RGB merge panels. Additional colocalization with GFP is white in RGB merge panels. D, Quantification of the numbers of bassoon-labeled presynaptic puncta/10 μm of dendrite showing that C36,129S (control, 8.7 ± 0.3), but not 79WT (control, 7.1 ± 0.2), significantly increases presynapse numbers compared with GFP (control, 7.1 ± 0.3; ###p < 0.001 by t test). cLTP reverses this increase in bassoon puncta for C36,129S (cLTP, 6.0 ± 0.2; ***p < 0.001). No changes are seen for GFP (cLTP, 6.8 ± 0.2) or 79WT (cLTP, 7.4 ± 0.4); n = 31–52 dendrites from 8–16 neurons. E, Quantification of the number of GluR1 puncta colocalized with bassoon/10 μm of dendrite showing that C36,129S (control, 5.1 ± 0.2), but not 79WT (control, 4.0 ± 0.2), increases AMPAR-GluR1 synaptic localization compared with GFP control (control, 4.2 ± 0.3; ##p < 0.01 by t test). cLTP reverses this increase in GluR1/bassoon puncta for 79C36,129S (cLTP, 2.1 ± 0.2; ***p < 0.001). No changes are seen for GFP (cLTP, 4.0 ± 0.2) or 79WT (cLTP, 4.4 ± 0.4); n = 31–52 dendrites from 8–16 neurons. F, Quantification of the percentage of synaptic GluR1 puncta (number of GluR1 colocalized with bassoon/total number of GluR1 puncta) showing that C36,129S (control, 86 ± 2%), but not 79WT (control, 79 ± 2%), significantly increases the number of synaptic GluR1 puncta compared with GFP control (control, 78 ± 2%; ###p < 0.01 by t test). cLTP reverses this increase in percentage synaptic GluR1 puncta for C36,129S (cLTP, 64 ± 2%; ***p < 0.001) but increases this parameter for GFP (cLTP, 83 ± 2%; *p < 0.05) and 79WT (cLTP, 83 ± 2%; *p < 0.05); n = 31–52 dendrites from 8–16 neurons. G, Quantification of the number of GluR2 puncta colocalized with bassoon puncta/10 μm of dendrite (images not show) showing that C36,129S (control, 3.5 ± 0.2) and 79WT (control, 3.5 ± 0.2) do not change the number of synaptic GluR2 puncta compared with GFP control (control, 3.1 ± 0.2); n = 29–50 dendrites from 9–13 neurons. H, Quantification of the percentage of synaptic GluR2 puncta (number of GluR2 colocalized with bassoon/total number of GluR2 puncta) showing that C36,129S (71 ± 2%), but not 79WT (control, 78 ± 2%), decreases GluR2 synaptic localization compared with GFP control (control, 81 ± 2%; **p < 0.01 by ANOVA); n = 29–50 dendrites from 9–13 neurons. Error bars indicate SEM.
In response to cLTP, GFP neurons increased both mean mEPSC frequency (approximately fourfold; Fig. 8A,B) and amplitude (∼35%; Fig. 8A,C) as expected from previous studies of this form of synaptic potentiation (Lu et al., 2001; Fortin et al., 2010). 79WT neurons also increased mean mEPSC frequency (approximately threefold) and amplitude (∼16%; Fig. 8A–C) with cLTP. Accordingly, both GFP and 79WT showed uniform rightward distribution shifts toward larger mEPSC amplitudes on cumulative plots with cLTP (Fig. 8D). In contrast, C36,129S neurons, rather than simply occluding cLTP due to already elevated basal AMPAR activity, responded to cLTP with a dramatic reduction in mEPSC frequency (approximately fivefold; Fig. 8A,B) with the active synapses remaining showing a trend toward a reduction in mean mEPSC amplitude (∼11%; Fig. 8C; p = 0.1) and a clear reduction in the number of high-amplitude mEPSCs >15 pA on a cumulative plot (Fig. 8D). For 150RNAi, cLTP stimulation induced some additional potentiation of mEPSC frequency (approximately twofold; Fig. 8E) and amplitude (∼15%; Fig. 8F) but with only a small rightward shift in amplitude distribution primarily for events <20 pA (Fig. 8G). Rescue of 150RNAi with 79WT restored the ability of cLTP to induce a more robust increase in mean amplitude (∼32%; Fig. 8F) with a corresponding larger, uniform rightward shift in mEPSC amplitude distribution (Fig. 8G) while maintaining a modest increase in mean mEPSC frequency (approximately twofold; Fig. 8E). In contrast, 150RNAi+C36,129S exhibited a large decrease in mEPSC frequency with cLTP (approximately fourfold; Fig. 8E), a trend toward decreased mean amplitude (∼13%; Fig. 8F; p = 0.1) and a leftward shift in the cumulative distribution of larger mEPSC amplitudes (Fig. 8G) similar to when C36,129S was expressed alone (Fig. 8B–D).
The pronounced decrease in mEPSC frequency seen with C36,129S is consistent with the silencing of active synapses and is more characteristic of LTD in dissociated cultures (Beattie et al., 2000; Lu et al., 2001). Thus, C36,129S neurons cannot maintain elevated levels of AMPAR activity when challenged with a stimulus that should promote potentiation and instead inappropriately respond by inducing depression. Consistent with loss of synaptic AMPARs, C36,129S neurons exhibited large decreases after cLTP in the number (∼58%; Fig. 9E) and percentage of synaptic GluR1 puncta (∼26%; Fig. 9F). In contrast, GFP and 79WT both increased (∼7%) the percentage of synaptic GluR1 puncta after cLTP (Fig. 9F). C36,129S also showed decreases in bassoon puncta (∼31%; Fig. 9D) and spine numbers following cLTP (∼20%; **p < 0.01 by t test; 4.2 ± 0.2 spines/10 μm; n = 5) compared with untreated controls above, indicating some synapse elimination. No significant changes in bassoon puncta (Fig. 9D) or spine numbers were seen for GFP (4.0 ± 0.3 spines/10 μm; n = 5), 79WT (5.3 ± 0.3 spines/10 μm; n = 6), or 150RNAi (4.5 ± 0.2 spines/10 μm; n = 15) following cLTP.
These mEPSC recordings and GluR1 staining results indicate that C36,129S increases basal AMPAR synaptic localization and function but prevents maintenance of synaptic GluR1 and potentiation following cLTP. To address this paradox, we first investigated the large effects of C36,129S on basal mEPSC frequency. One explanation for this large enhancement would be if C36,129S favored increased synaptic accumulation of Ca2+-permeable, GluR1 homomeric receptors that lack the GluR2 subunit; recruitment of even a small number of GluR1 homomers, which have high single-channel conductance, would have a large impact on synapse unsilencing. Indeed, glycine-cLTP is known to induce potentiation and spine enlargement through recruitment of Ca2+-permeable AMPARs to synapses (Fortin et al., 2010). Consistent with this idea, immunostaining for GluR2 and bassoon reveled that C36,129S did not change the number of synaptic GluR2 puncta and actually decreased (∼12%) the percentage of GluR2 puncta localized to synapses compared with GFP and 79WT (Fig. 9G,H). To explore this possibility further, we recorded mEPSCs and then added N,N,H,-trimethyl-5-[(tricyclo[3.3.1.13,7]dec-1-ylmethyl)amino]-1-pentanaminiumbromide hydrobromide (IEM1460), a polyamine blocker of GluR2-lacking AMPARs (Fig. 8H) (Fortin et al., 2010). After 5 min, IEM dramatically increased interevent interval in C36,129S neurons (Fig. 8I), resulting in a reduction in mean mEPSC frequency of ∼70% (Fig. 8J). In contrast, IEM had no significant impact on mEPSC frequency in GFP and 79WT neurons. Thus, consistent with increased synaptic GluR1, but not GluR2, the large increase in mEPSC frequency for C36,129S can be accounted for by postsynaptic unsilencing by increased synaptic localization and function of GluR2-lacking AMPARs.
AKAP79 palmitoylation is required for GluR1 surface delivery and increased exocytosis from recycling endosomes associated with cLTP
These alterations in basal and cLTP regulation of GluR1 synaptic localization and activity for C36,129S could be due to a changes in local recycling and exocytosis that maintain synaptic strength by controlling lateral exchange of perisynaptic receptors in and out of the synapse (Petrini et al., 2009). In addition to its absence from endosomes, C36,129S is almost completely TX-100 soluble (Fig. 2), consistent with its likely exclusion from the PSD and presence only at perisynaptic and extrasynaptic plasma membrane locations in dendrites. Importantly, PKA phosphorylation of GluR1 promotes recycling, exocytosis, and stabilization of Ca2+-permeable GluR1 homomers at perisynaptic/extrasynaptic locations where they can subsequently exchange with synaptic receptors during plasticity in response to additional signals (Ehlers, 2000; Esteban et al., 2003; Oh et al., 2006; He et al., 2009; Fortin et al., 2010). In particular, a failure to increase dendritic AMPAR and recycling endosome exocytosis in response to cLTP (Park et al., 2004; Kennedy et al., 2010) could result in postsynaptic silencing through redistribution of GluR1 from synapses back to extrasynaptic sites in C36,129S-expressing neurons. To test the hypothesis that AKAP79 palmitoylation regulates AMPAR exocytosis, we measured GluR1 surface expression in dendrites before and after cLTP by labeling under nonpermeabilized conditions with an antibody to the receptor extracellular domain (Fig. 10A,B). Consistent with previous studies showing that AKAP150 overexpression somewhat increases basal AMPAR surface expression (Bhattacharyya et al., 2009), neurons expressing 79WT-GFP with 150RNAi showed elevated basal levels of GluR1 surface labeling (∼75%) when normalized to GFP controls (Fig. 10C). However, cLTP still led to a robust increase (approximately threefold) in GluR1 surface labeling in 79WT neurons (Fig. 10A,C). In contrast, C36,129S also showed some increased basal GluR1 surface labeling, but cLTP failed to increase GluR1 dendritic surface expression further (Fig. 10B,C). The interference of C36,129S with AMPAR trafficking was specific for cLTP-induced exocytosis and not due to a more general endosomal trafficking alterations, as NMDA-cLTD induced GluR1 internalization in dendrites was comparable for 79WT and C36,129S (Fig. 10D). This result indicates that the depalmitoylated C36,129S mutant supports normal LTD-associated AMPAR trafficking and is consistent with our observations above that NMDA-cLTD induces depalmitoylation of AKAP79/150 (Fig. 2D).
We next wanted to determine how C36,129S could alter dendritic endosomal recycling in a manner that could enhance basal synaptic localization of GluR1 yet prevent additional delivery of GluR1 to maintain potentiation following cLTP. To this end, we used spinning-disc confocal microscopy to monitor exocytosis from dendritic recycling endosomes using a TfR reporter tagged on its extracellular domain with the pH-sensitive GFP variant SEP (SEP-TfR) (Park et al., 2006; Kennedy et al., 2010). The fluorescence of SEP-TfR is quenched when it resides in the acidic lumen of endosomes but rapidly becomes fluorescent when it is exocytosed to the plasma membrane and higher extracellular pH. Discrete flashes of SEP-TfR exocytosis were detected in dendrites of 79WT (Fig. 10E,G) or C36,129S-mCherry neurons (Fig. 10F,H) during 0–4 min of continuous imaging. C36,129S exhibited a ∼3.5-fold increase in basal exocytosis rate over 79WT (Fig. 10I). The elevated recycling exocytosis for C36,129S could promote the increases in basal GluR1 synaptic localization we observe and prevent decreases in overall GluR1 surface levels (Fig. 10B,C) even when receptors are lost from synapses with cLTP stimulation (Fig. 9C,E,F). During a subsequent 4–8 min of cLTP stimulation, the rate of SEP-TfR exocytosis increased ∼2.5-fold above baseline for 79WT (Fig. 10E,G,J) similar to previous studies (Park et al., 2006; Kennedy et al., 2010). However, cLTP was not able increase SEP-TfR exocytosis in C36,129S-expressing neurons (Fig. 10F,H,J), consistent with the impaired GluR1 surface delivery that we observed above (Fig. 10A,B). Overall, our findings indicate that preventing AKAP79 palmitoylation by the C36,129S double mutation interferes with normal regulation of endosomal recycling that is required for control of AMPAR synaptic localization, spine size, and synaptic strength (Fig. 10K).
Discussion
Using a combination of biochemical and fluorescence imaging methods, we characterized a novel role for S-palmitoylation in endosomal targeting of AKAP79/150. Importantly, using additional imaging and electrophysiological recordings, we showed that this dynamic posttranslational modification modulates activity-dependent dendritic spine targeting of AKAP79/150, endosome recycling, spine enlargement, and AMPAR potentiation (Fig. 10K).
Palmitoylation and mechanisms of endosome targeting
A striking observation we made was that C36,129S was absent from endosomes but not the plasma membrane in both COS7 cells and hippocampal neuron dendrites. Unpalmitoylated recombinant AKAP79 N-terminal fragments can bind directly to vesicles containing PI4,5P2 with weaker binding to PI4P (Dell'Acqua et al., 1998); thus, it is likely that the substantial negative charge provided by PI4,5P2 enriched at the plasma membrane allows even unpalmitoylated AKAP basic domains to engage in strong electrostatic interactions. This AKAP plasma membrane targeting may also be aided by binding to cortical F-actin at sites enriched in PI4,5P2 (Gomez et al., 2002; Horne and Dell'Acqua, 2007). Accordingly, a recent study found that PI4,5P2 depletion removed depalmitoylated, but not palmitoylated, AKAP79 from HEK-293 cell plasma membranes (Delint-Ramirez et al., 2011). While late endosomes contain PI3,5P2 and some late recycling endosomes contain PI4,5P2, early endosomes that precede trafficking to these compartments primarily contain PI3P (Nicot and Laporte, 2008). Thus, unpalmitoylated AKAP may be lost from early endosomes due to insufficient electrostatic interactions. In addition, lipid raft sorting of palmitoylated AKAP may be necessary for its initial endocytosis from the plasma membrane.
Regulation of palmitoylation by neuronal activity
AKAP150 palmitoylation was regulated by seizure activity in vivo and by plasticity-inducing stimuli in cultured neurons. We observed that kainate seizures rapidly increased AKAP150 palmitoylation and localization to lipid rafts with PSD-95. This AKAP150 movement into postsynaptic rafts could involve palmitoylation-dependent delivery via endosomes. Interestingly, distribution of the nonraft TfR, which would be co-delivered from endosomes with AKAP150, did not change with kainate, consistent with studies showing that TfR is exocytosed with GluR1 in spines but is not retained there (Kennedy et al., 2010). In cultured neurons, NMDA-cLTD that moved AKAP from spines rapidly decreased palmityolation, whereas cLTP that recruited AKAP to spines rapidly increased palmitoylation. Several other neuronal proteins, including PSD-95, exhibit activity-dependent palmityolation (El-Husseini et al., 2002). A number of DHHC-PAT family members including DHHCs 2, 3, 5, 7, 8, and 15 catalyze PSD-95 palmitoylation (Fukata and Fukata, 2010). Many PATs are Golgi-localized proteins that palmitoylate newly synthesized membrane proteins; however, DHHC5/8 are plasma membrane localized (Greaves et al., 2010), present in dendrites (Thomas et al., 2012), and have C-terminal PDZ ligands that bind PSD-95 and GRIP1 (Li et al., 2010; Thomas et al., 2012). Interestingly, DHHC5 knock-out mice have learning and memory impairments (Li et al., 2010), and DHHC8 mutations in mice and humans are linked to schizophrenia (Mukai et al., 2004, 2008). Last, DHHC2 is localized to recycling endosomes in neuroendocrine cells (Greaves et al., 2011) and is recruited to dendritic spines to palmitoylate PSD-95 when activity decreases (Noritake et al., 2009). It will be important to investigate whether any of these PATs regulate AKAP79/150 palmityolation and how it is regulated through control of PAT enzymatic activity or proximity.
Potential roles for AKAP79/150 in regulation of dendritic endosome trafficking
The dominant interfering effects of C36,129S on cLTP-induced spine enlargement, GluR1 regulation, and recycling exocytosis indicate that exclusion of AKAP79 from endosomes interferes with trafficking regulation required for potentiation. Our observations of C36,129S impacts on TfR and GluR1 trafficking indicate that the AKAP signaling complex could function in both general control of endosome trafficking and specific control of AMPARs. CaN triggers AMPAR endocytosis during LTD (Beattie et al., 2000), PKC regulates AMPAR synaptic incorporation in LTP (Boehm et al., 2006), and PKA promotes AMPAR recycling and exocytosis to favor LTP (Ehlers, 2000; Esteban et al., 2003; Oh et al., 2006). The effects of CaN, PKA, PKC, and other kinases on AMPAR localization are in part due to regulation of GluR1 phosphorylation, although additional mechanisms such as phosphorylation of the AMPAR-associated protein Stargazin are involved (Esteban et al., 2003; Tomita et al., 2005; Boehm et al., 2006; Man et al., 2007; He et al., 2009; Lee et al., 2010; Opazo et al., 2010). More directly, AKAP79/150-CaN anchoring is required for LTD regulation of AMPAR currents and endocytosis in cultured neurons and slices (Tavalin et al., 2002; Hoshi et al., 2005; Bhattacharyya et al., 2009; Jurado et al., 2010), and AKAP binding to PSD-95 is required for LTD in cultured slices (Xu et al., 2008) and enhancement of spine maturation in developing cultured neurons (Robertson et al., 2009). Importantly, characterization of acute hippocampal slices from PKA anchoring-deficient AKAP150D36 knock-in mice revealed deficits in LTD and LTP involving PKA regulation of GluR2-lacking receptors (Lu et al., 2007, 2008). Thus, expression of C36,129S likely delocalizes one or more of these proteins away from endosomes (as seen in Fig. 6) to alter trafficking regulation. AKAP150RNAi also effected regulation of spine size and AMPAR activity, but not to the same extent as C36,129S, which was unable to increase or maintain AMPAR activity with cLTP. This greater effect of mutant expression is not surprising considering that RNAi suppression is likely incomplete after <48 h and AKAP150 knock-out mice have normal LTP under the same conditions that AKAP150D36 mice show impairments (Lu et al., 2007; Weisenhaus et al., 2010). Thus, there are clearly different impacts of reducing all AKAP functions versus eliminating anchoring of one signaling partner or eliminating palmityolation to alter localization.
AKAP79/150 palmitoylation and control of basal synaptic activity and potentiation
Another prominent feature of C36,129S was the large increase in basal mEPSC activity through a combination of new synapse formation and postsynaptic unsilencing by Ca2+-permeable GluR1-containing AMPARs. Acute AKAP150RNAi also increased basal mEPSC activity, in accordance with increases in basal AMPAR transmission previously seen with AKAP150RNAi in cultured rat hippocampal slices and with AKAP150 knock-out in mouse hippocampal slices (Jurado et al., 2010; Lu et al., 2011). However, these previous studies found differential involvement of anchored CaN versus PKA in basal transmission regulation, but as mentioned above, both the acute and chronic effects of AKAP150 suppression or knock-out are complicated because multiple and opposing activities are eliminated at the same time including those that relieve inhibitory constraints on basal transmission, such as CaN (Jurado et al., 2010), and those that remove positive modulators of intrinsic excitability, such as PKA downregulation of A-type K+ currents (Lin et al., 2011), which could lead to homeostatic increases in basal transmission. As mentioned above, the failure of C36,129S to associate with lipids rafts likely excludes it not only from endosomes but also from the PSD, leading to localization only in the extrasynaptic plasma membrane where it may increase basal GluR1 activity through PKA and PKC phosphorylation pathways that control surface delivery and synaptic incorporation (Boehm et al., 2006; Oh et al., 2006; He et al., 2009; Lin et al., 2009). In any case, the positive effects of C36,129S on basal AMPAR activity are not maintained after cLTP, as shown by mEPSC silencing, impaired GluR1 and SEP-TfR exocytosis, and GluR1 synaptic loss. Overall, it appears that palmitoylated AKAP79 normally applies brakes to endosome recycling and GluR1 synaptic incorporation that are then removed by cLTP stimulation (Fig. 10K); C36,129S is not able to apply these brakes but consequently cannot respond to stimulation. In addition, it is possible that interactions of AKAP79/150 that acutely stabilize it and AMPARs in the PSD during cLTP, such as linkage to PSD-95, require AKAP palmityolation and lipid raft association. In any case, our overall findings add significantly to recent studies that highlight the emerging role of palmitoylation of synaptic proteins in controlling neuronal functions that are central to synaptic plasticity mechanisms and neurological disease processes.
Footnotes
This work was supported by NIH Grant R01NS040701 (M.L.D.), Canadian Institutes of Health Research Grants 20R90479, 20R91909, MOP44052, and MOP81144 (A.E.-H.), and The Michael Smith Foundation for Health Research (A.E.-H.). D.J.K. was supported by NIH Grant T32AT002688. J.L.S. was supported by NIH Grant T32AA007464 and an American Heart Association postdoctoral fellowship. H.R.R. and K.O. were supported by NIH Grant T32GM007635 and H.R.R. by an American Heart Association predoctoral fellowship. K.W. was supported by NIH Grant T32NS007083 and an American Heart Association postdoctoral fellowship. Spinning disc confocal imaging was performed in the University of Colorado Anschutz Medical Campus Advanced Light Microscopy Core supported by NIH–NINDS Rocky Mountain Neurological Disorders Core Center Grant P30NS048154 and NIH–NCRR Colorado CTSI Grant UL1RR025780. We thank Drs. Michael D. Ehlers, John D. Scott, and Alexander Sorkin for providing plasmids. We thank Dr. Eric A. Horne for assistance with generation of the 79CS mutants. We thank Drs. K. Ulrich Bayer and Mathew Kennedy for critical reading of this manuscript. Contents are the authors' sole responsibility and do not necessarily represent official NIH views.
This article is dedicated to the late Dr. Alaa El-Husseini, a wonderful scientific colleague, mentor, and friend.
References
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schlüter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Brandao KE, Dell'Acqua ML, Levinson SR. A-kinase anchoring protein 150 expression in a specific subset of TRPV1- and CaV 1.2-positive nociceptive rat dorsal root ganglion neurons. J Comp Neurol. 2012;520:81–99. doi: 10.1002/cne.22692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Delint-Ramirez I, Willoughby D, Hammond GV, Ayling LJ, Cooper DM. Palmitoylation targets AKAP79 to lipid rafts and promotes its regulation of the calcium sensitive adenylyl cyclase type 8. J Biol Chem. 2011;286:32962–32975. doi: 10.1074/jbc.M111.243899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua ML, Faux MC, Thorburn J, Thorburn A, Scott JD. Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4,5-bisphosphate. EMBO J. 1998;17:2246–2260. doi: 10.1093/emboj/17.8.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- El-Husseini Ael-D, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS. Synaptic strength regulated by palmitate cycling on PSD-95. Cell. 2002;108:849–863. doi: 10.1016/s0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Faux MC, Rollins EN, Edwards AS, Langeberg LK, Newton AC, Scott JD. Mechanism of A-kinase-anchoring protein 79 (AKAP79) and protein kinase C interaction. Biochem J. 1999;343:443–452. [PMC free article] [PubMed] [Google Scholar]
- Fortin DA, Davare MA, Srivastava T, Brady JD, Nygaard S, Derkach VA, Soderling TR. Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J Neurosci. 2010;30:11565–11575. doi: 10.1523/JNEUROSCI.1746-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci. 2002;22:7027–7044. doi: 10.1523/JNEUROSCI.22-16-07027.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Gomez LL, Scott JD, Dell'Acqua ML. Association of an A-kinase-anchoring protein signaling scaffold with cadherin adhesion molecules in neurons and epithelial cells. Mol Biol Cell. 2005;16:3574–3590. doi: 10.1091/mbc.E05-02-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Gorleku OA, Salaun C, Chamberlain LH. Palmitoylation of the SNAP25 protein family: specificity and regulation by DHHC palmitoyl transferases. J Biol Chem. 2010;285:24629–24638. doi: 10.1074/jbc.M110.119289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Carmichael JA, Chamberlain LH. The palmitoyl transferase DHHC2 targets a dynamic membrane cycling pathway: regulation by a C-terminal domain. Mol Biol Cell. 2011;22:1887–1895. doi: 10.1091/mbc.E10-11-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Song L, Cummings LW, Goldman J, Huganir RL, Lee HK. Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1–S845 phosphorylation. Proc Natl Acad Sci U S A. 2009;106:20033–20038. doi: 10.1073/pnas.0910338106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne EA, Dell'Acqua ML. Phospholipase C is required for changes in postsynaptic structure and function associated with NMDA receptor-dependent long-term depression. J Neurosci. 2007;27:3523–3534. doi: 10.1523/JNEUROSCI.4340-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, Scott JD. Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat Cell Biol. 2005;7:1066–1073. doi: 10.1038/ncb1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Biou V, Malenka RC. A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat Neurosci. 2010;13:1053–1055. doi: 10.1038/nn.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Swayze R, Lise MF, Gerrow K, Mullard A, Honer WG, El-Husseini A. Presynaptic trafficking of synaptotagmin I is regulated by protein palmitoylation. J Biol Chem. 2004;279:50524–50536. doi: 10.1074/jbc.M404981200. [DOI] [PubMed] [Google Scholar]
- Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, Thompson JX, Roth AF, Drisdel RC, Mastro R, Green WN, Yates JR, 3rd, Davis NG, El-Husseini A. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 2008;456:904–909. doi: 10.1038/nature07605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010;33:121–129. doi: 10.1016/j.tins.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141:524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK. Synaptic plasticity and phosphorylation. Pharmacol Ther. 2006;112:810–832. doi: 10.1016/j.pharmthera.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol. 2010;103:479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hu J, Höfer K, Wong AM, Cooper JD, Birnbaum SG, Hammer RE, Hofmann SL. DHHC5 interacts with PDZ domain 3 of post-synaptic density-95 (PSD-95) protein and plays a role in learning and memory. J Biol Chem. 2010;285:13022–13031. doi: 10.1074/jbc.M109.079426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Makino Y, Sharma K, Hayashi T, Neve R, Takamiya K, Huganir RL. Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat Neurosci. 2009;12:879–887. doi: 10.1038/nn.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Sun W, Kung F, Dell'Acqua ML, Hoffman DA. AKAP79/150 impacts intrinsic excitability of hippocampal neurons through phospho-regulation of A-type K+ channel trafficking. J Neurosci. 2011;31:1323–1332. doi: 10.1523/JNEUROSCI.5383-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 2007;26:4879–4890. doi: 10.1038/sj.emboj.7601884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhang M, Lim IA, Hall DD, Allen M, Medvedeva Y, McKnight GS, Usachev YM, Hell JW. AKAP150-anchored PKA activity is important for LTD during its induction phase. J Physiol. 2008;586:4155–4164. doi: 10.1113/jphysiol.2008.151662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zha XM, Kim EY, Schachtele S, Dailey ME, Hall DD, Strack S, Green SH, Hoffman DA, Hell JW. A-kinase anchor protein 150 (AKAP150)-associated protein kinase A limits dendritic spine density. J Biol Chem. 2011;286:26496–26506. doi: 10.1074/jbc.M111.254912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Man HY, Sekine-Aizawa Y, Huganir RL. Regulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci U S A. 2007;104:3579–3584. doi: 10.1073/pnas.0611698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai J, Liu H, Burt RA, Swor DE, Lai WS, Karayiorgou M, Gogos JA. Evidence that the gene encoding ZDHHC8 contributes to the risk of schizophrenia. Nat Genet. 2004;36:725–731. doi: 10.1038/ng1375. [DOI] [PubMed] [Google Scholar]
- Mukai J, Dhilla A, Drew LJ, Stark KL, Cao L, MacDermott AB, Karayiorgou M, Gogos JA. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nat Neurosci. 2008;11:1302–1310. doi: 10.1038/nn.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newpher TM, Ehlers MD. Spine microdomains for postsynaptic signaling and plasticity. Trends Cell Biol. 2009;19:218–227. doi: 10.1016/j.tcb.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Nicot AS, Laporte J. Endosomal phosphoinositides and human diseases. Traffic. 2008;9:1240–1249. doi: 10.1111/j.1600-0854.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noritake J, Fukata Y, Iwanaga T, Hosomi N, Tsutsumi R, Matsuda N, Tani H, Iwanari H, Mochizuki Y, Kodama T, Matsuura Y, Bredt DS, Hamakubo T, Fukata M. Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J Cell Biol. 2009;186:147–160. doi: 10.1083/jcb.200903101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Oliveria SF, Gomez LL, Dell'Acqua ML. Imaging kinase–AKAP79–phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol. 2003;160:101–112. doi: 10.1083/jcb.200209127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dell'Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo P, Choquet D. A three-step model for the synaptic recruitment of AMPA receptors. Mol Cell Neurosci. 2011;46:1–8. doi: 10.1016/j.mcn.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of Stargazin. Neuron. 2010;67:239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD, Choquet D. Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron. 2009;63:92–105. doi: 10.1016/j.neuron.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Robertson HR, Gibson ES, Benke TA, Dell'Acqua ML. Regulation of postsynaptic structure and function by an A-kinase anchoring protein-membrane-associated guanylate kinase scaffolding complex. J Neurosci. 2009;29:7929–7943. doi: 10.1523/JNEUROSCI.6093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Dell'Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist. 2011;17:321–336. doi: 10.1177/1073858410384740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, Dell'Acqua ML. cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J Neurosci. 2006;26:2391–2402. doi: 10.1523/JNEUROSCI.3092-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin SJ. AKAP79 selectively enhances protein kinase C regulation of GluR1 at a Ca2+-calmodulin-dependent protein kinase II/protein kinase C site. J Biol Chem. 2008;283:11445–11452. doi: 10.1074/jbc.M709253200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD. Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J Neurosci. 2002;22:3044–3051. doi: 10.1523/JNEUROSCI.22-08-03044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Hayashi T, Chiu SL, Chen CM, Huganir RL. Palmitoylation by DHHC5/8 targets GRIP1 to dendritic endosomes to regulate AMPA-R trafficking. Neuron. 2012;73:482–496. doi: 10.1016/j.neuron.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS. Bidirectional synaptic plasticity regulated by phosphorylation of Stargazin-like TARPs. Neuron. 2005;45:269–277. doi: 10.1016/j.neuron.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, Scott JD. Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci U S A. 2008;105:12557–12562. doi: 10.1073/pnas.0805922105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenhaus M, Allen ML, Yang L, Lu Y, Nichols CB, Su T, Hell JW, McKnight GS. Mutations in AKAP5 disrupt dendritic signaling complexes and lead to electrophysiological and behavioral phenotypes in mice. PLoS One. 2010;5:e10325. doi: 10.1371/journal.pone.0010325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Schlüter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudowski GA, Puthenveedu MA, Leonoudakis D, Panicker S, Thorn KS, Beattie EC, von Zastrow M. Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J Neurosci. 2007;27:11112–11121. doi: 10.1523/JNEUROSCI.2465-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]