

Abstract
Collapsin response mediator protein 2 (CRMP-2), traditionally viewed as an axon/dendrite specification and axonal growth protein, has emerged as nidus in regulation of both pre- and post-synaptic Ca2+ channels. Building on our discovery of the interaction and regulation of Ca2+ channels by CRMP-2, we recently identified a short sequence in CRMP-2 which, when appended to the transduction domain of HIV TAT protein, suppressed acute, inflammatory and neuropathic pain in vivo by functionally uncoupling CRMP-2 from the Ca2+ channel. Remarkably, we also found that this region attenuated Ca2+ influx via N-methylD-Aspartate receptors (NMDARs) and reduced neuronal death in a moderate controlled cortical impact model of traumatic brain injury (TBI). Here, we sought to extend these findings by examining additional neuroprotective effects of this peptide (TAT-CBD3) and exploring the biochemical mechanisms by which TAT-CBD3 targets NMDARs. We observed that an intraperitoneal injection of TAT-CBD3 peptide significantly reduced infarct volume in an animal model of focal cerebral ischemia. Neuroprotection was observed when TAT-CBD3 peptide was given either prior to or after occlusion but just prior to reperfusion. Surprisingly, a direct biochemical complex was not resolvable between the NMDAR subunit NR2B and CRMP-2. Intracellular application of TAT-CBD3 failed to inhibit NMDAR current. NR2B interactions with the post synaptic density protein 95 (PSD-95) remained intact and were not disrupted by TAT-CBD3. Peptide tiling of intracellular regions of NR2B revealed two 15-mer sequences, in the carboxyl-terminus of NR2B, that may confer binding between NR2B and CRMP-2 which supports CRMP-2's role in excitotoxicity and neuroprotection.
Keywords: N-methylD-aspartate receptor, CRMP-2, focal cerebral ischemia, MCAO model, peptide
Introduction
Neuronal insults such as TBI and stroke lead to a pronounced increase in extracellular glutamate surrounding the insult foci.1,2 The increased glutamate triggers neuronal death via hyperactivation of NMDARs which is also known as excitotoxicity. Based upon these findings, expansive efforts have been made to develop NMDAR antagonists to prevent death of neurons following an excitotoxic insult. While in vitro discoveries have been plenty, the majority of clinical trials using these antagonists have failed to yield treatment options.3-6 We reasoned that an alternative approach that may be more therapeutically beneficial is to modulate, rather than completely block, NMDARs to reduce excitotoxicity. This led to the identification and characterization of a novel modulator of NMDARs, a 15 amino acid peptide (CBD3) derived from the CRMP-2 protein and demonstrated its usefulness in preventing glutamate toxicity following neuronal injury.7
CRMP-2, initially believed to be a regulator of neuronal plasticity, neurite growth and extension8,9 has emerged as a modulator of presynaptic voltage-gated Ca2+ channels in our recent studies characterizing the interaction between CRMP-2 and N-type Ca2+ channels (CaV2.2).10-12 The CRMP-2 peptide CBD3 was identified as a disrupter of the CRMP-2-CaV2.2 interaction and resulted in reduced Ca2+ channel current density and CaV2.2 trafficking.13 Fusion of the CBD3 peptide to the trans-activating transcriptional activator (TAT) from human immunodeficiency virus conferred cell penetrating ability to CBD3 resulting in in vivo suppression of evoked pain in rat models of acute, inflammatory and neuropathic pain.13 Since TAT-CBD3 exhibited a favorable safety profile in rodents,13,14 and CRMPs have been linked to excitotoxicity,15-20 we asked if TAT-CBD3 could have beneficial effects against excitotoxicity. We found that TAT-CBD3 selectively reduced NMDAR surface expression in dendrites and attenuated Ca2+-influx without completely ablating NMDA/Glutamate responses, a strategy that could be especially useful for reducing the excitotoxic death of neurons without preventing the “normal” post-injury glutamate excitatory synaptic transmission—which are important for continued survival of glutamatergic neurons.21 In this addendum, we sought to further extend the neuroprotective scope of TAT-CBD3 by testing its efficacy in another excitotoxic injury model and to explore the biochemical mechanism(s) by which TAT-CBD3 targets NMDARs.
Results and Discussion
Neuroprotection by TAT-CBD3 against focal cerebral ischemic damage in the rat middle cerebral artery occlusion (MCAO) model
TAT-CBD3 administered systemically within minutes of a moderate controlled cortical injury protected ~50% of the neurons in the granular cell layer within the hippocampus against TBI and protected against an excitotoxic stimulation in vitro at 30 min post-challenge, supporting the notion that TAT-CBD3 works in the early phase of neuronal death observed following excitotoxic insults.22 To extend these findings, we asked if TAT-CBD3 would be effective in a transient model of focal cerebral ischemia (i.e., MCAO) and whether neuroprotection could be observed at a time after the onset of injury. The MCAO model was used here as (1) cell death in this model is dependent on excessive NMDAR activation,23-25 (2) NMDAR antagonists have been shown to be neuroprotective in animal models of ischemia,26 and (3) CRMP-2 levels are upregulated in the MCAO stroke penumbra.27 To confirm our hypothesis that TAT-CBD3 is effective in the early phase of neuronal death following excitotoxic insults, we first injected TAT-CBD3 (20 mg/kg) intraperitoneally (i.p.) 1 h prior to occlusion (Fig. 1A). The animals then underwent occlusion followed by a 2 h reperfusion and were sacrificed 48 h later. Cortices were sectioned and stained with 1% 2, 3 and 5-triphenyltetrazolium chloride monohydrate (TTC) to label viable tissue.28 TAT-CBD3 prior to MCAO caused an ~20% increase in surviving brain compared with saline injected MCAO animals (Fig. 1B and C). This finding is consistent with our previous report showing TAT-CBD3 reduces neuronal death if given immediately following TBI as well as with a recent report that demonstrated a reduction in brain infarct volume following intracerebroventricular injection of a CRMP-2 peptide, partially overlapping (i.e., 6 of 15 amino acids) with our CBD3 peptide, in mice just before MCAO.29
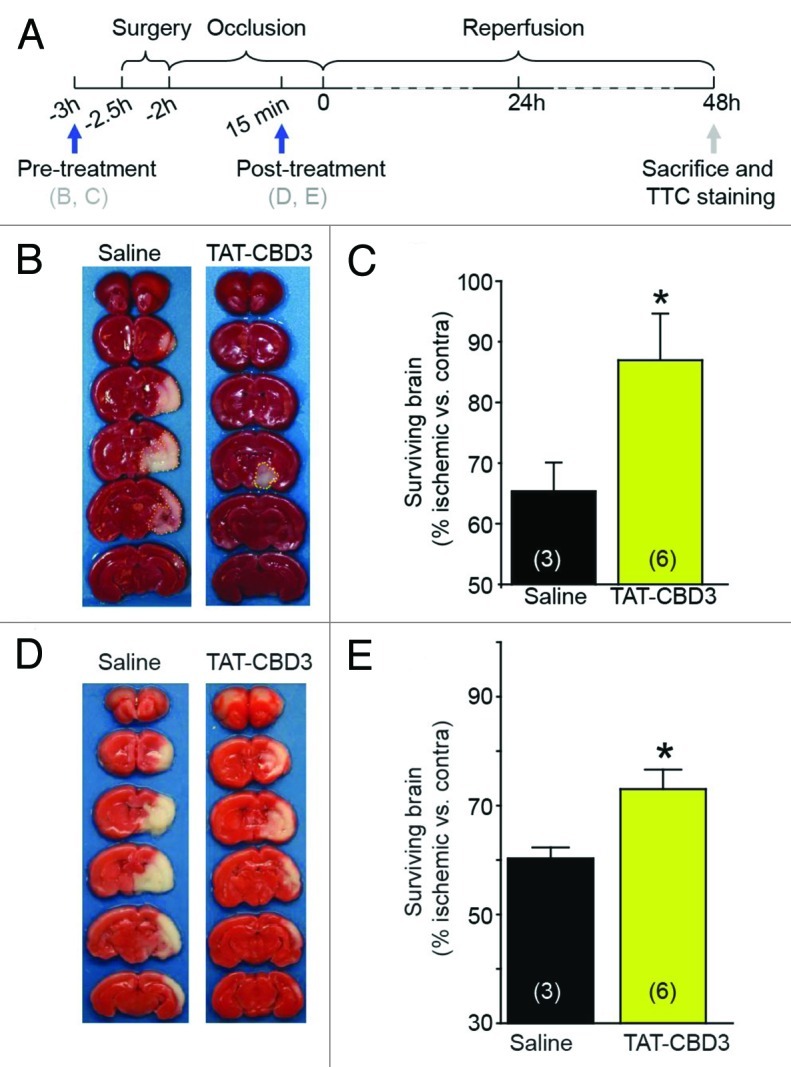
Figure 1. TAT-CBD3 reduces infarct volume following focal cerebral ischemia. (A) Ten day old Sprague-Dawley rats were challenged with focal cerebral ischemia by MCAO. The occlusion lasted for 2 h and infarct volume was assessed 48 h after the onset of reperfusion. (B) Animals were pre-treated with either 20 mg/kg TAT-CBD3 of saline 1 h prior to occlusion. Representative images of TTC stained cortices are shown. (C) Bar graph representation of surviving brain volume (% ischemic vs. contralateral) compared between saline and TAT-CBD3 pre-treated animals. (D) Animals were treated with either 20 mg/kg TAT-CBD3 or saline 15 min prior to reperfusion. (E) Bar graph representation of surviving brain volume (% ischemic vs. contralateral) compared between animals treated with TAT-CBD3 or saline 15 min prior to reperfusion.
To determine if TAT-CBD3 is effective if given hours after the occlusion, important since most stroke patients receive therapeutic intervention between a 3–8 h window, the peptide was injected i.p. 2 h after the onset of occlusion but 15 min prior to reperfusion. Two days after this procedure, the animals were sacrificed and cortices stained with TTC as above. A smaller (~10%) but significant sparing of surviving brain was observed in this paradigm compared with saline-injected animals (Fig. 1D and E). Collectively, these findings demonstrate that TAT-CBD3 acts as a neuroprotective agent both immediately and at least 2 h following an ischemic insult to the CNS.
Mechanistically, we have demonstrated that TAT-CBD3-mediated neuronal preservation in the face of an excitotoxic challenge likely occurs via attenuation of NMDAR-mediated Ca2+ influx. TAT-CBD3 slowed the onset of sustained [Ca2+]c elevation induced by excitotoxic glutamate and, consequently, decelerated mitochondrial depolarization (Fig. 2). Thus, TAT-CBD3 alleviated glutamate-induced Ca2+ dysregulation and protected mitochondria against Ca2+-dependent injury. These findings are entirely consistent with neuroprotection conferred by NMDAR antagonists in animal models of ischemia.26 One caveat however, is that as neuroprotection is also observed following treatment with CaV2.2 antagonists in animal models of stroke and TBI,30-34 the role of CaV2.2 block in neuroprotection in these models cannot be completely ruled out given our previous demonstration of TAT-CBD3 mediated reduction in Ca2+ influx via these channels. That NMDARs are involved is unequivocal, however the role of CaV2.2 as it relates to TAT-CBD3-mediated neuroprotection is unclear. CaV2.2 blockers work to reduce extracellular glutamate and subsequently lead to reduced activation of NMDARs. Thus, neuroprotection with TAT-CBD3 in vivo likely results from a summed action on both NMDARs and CaV2.2. While there are obvious benefits of polypharmacology of action of TAT-CBD3 on two targets, ongoing mutagenesis efforts in our laboratory are geared toward the development of peptide(s) that may allow discrimination of CaV2.2 or NMDARs. It is still possible that the mechanism for antagonism of CaV2.2 and NMDARs may rely on the same motif within CRMP-2. While the regions on CaV2.2 mediating binding to TAT-CBD3 have been identified,10,13 no such regions have been identified on NMDARs (but see Fig. 5).

Figure 2. Protective effect of TAT-CBD3 against glutamate-induced Ca2+ dysregulation and mitochondrial depolarization. Tat-CBD3 failed to affect the resting cytosolic Ca2+ and mitochondrial membrane potential (Δψ). In (A–D), the representative individual (thin, gray traces obtained from individual neurons) and average Fura-2FF (F340/F380, thick gray traces) (A) and (C) and Rhodamine 123 (Rh123, F/F0, thick black traces) fluorescence signals (B) and (D). Neurons were treated with 25 μM glutamate (plus 10μM glycine) as indicated. In (C) and (D), neurons were pre-incubated for 10 min with 10 μM TAT-CBD3.

Figure 5. Two NMDAR carboxyl-terminus peptides bind to CRMP-2. Peptides (15-mers), with a moving window of three amino acids, were generated for intracellular regions of the rat NMDAR. The SPOTS membranes were overlaid with rat brain lysates and then probed with polyclonal CRMP-2 and monoclonal phospho-CRMP-2 (3F4) antibodies. The intensity of each spot was measured using the Odyssey infrared imaging system utilizing the Li-Cor Biosciences one-dimensional software. Graphs represent normalized binding of poly (top) and mono (bottom) CRMP-2 antibodies to NMDAR peptides. Peptides were defined to bind if binding with both antibodies was observed above an arbitrary threshold of 0.6 of maximum. Using these criteria, two carboxyl-terminal peptides bound CRMP-2. The sequences of these peptides are highlighted in boldface font.
A biochemical interaction between CRMP-2 and NR2B?
TAT-CBD3 attenuates NMDAR-mediated Ca2+ influx as well as reduces NMDAR currents within 5 min, although the exact mechanism for this antagonism is unknown. The fast action of TAT-CBD3 may result from either a direct inhibition of the NMDAR or via a direct interaction with the NMDAR. To test the first possibility, we introduced TAT-CBD3 into neurons via the pipette and measured NMDAR currents. If TAT-CBD3 acts on the NMDAR exclusively at an extracellular site, then this method of peptide delivery is not expected to block currents. Conversely, if TAT-CBD3 acts on a NMDAR site intracellularly, inhibition of NMDAR currents would be observed.
We have previously shown that extracellular application of TAT-CBD3 is able to inhibit NMDAR Ca2+-currents.7 The mechanism of TAT-CBD3′s inhibition of NMDARs was proposed to be due to intracellular disruption of CRMP-2-NMDAR signaling. This conclusion was supported by the finding that application of TAT-CBD3 but not CBD3 sans TAT was neuroprotective. While these findings supported the conclusion that the TAT penetration peptide is required for neuroprotection by CBD3 it did not directly demonstrate that the target of TAT-CBD3 was intracellular. Therefore, we tested if intracellular application of TAT-CBD3 inhibits NMDARs. This was tested by measuring NMDA-induced Ca2+-currents in hippocampal neurons with TAT-CBD3 in the recording pipette. In experiments where TAT-CBD3 was applied intracellularly via the recording pipette, there was no observed decrease in NMDAR Ca2+-currents compared with controls (Fig. 3). This finding suggests that TAT-CBD3 does not inhibit NMDARs through an intracellular target, suggesting instead that the observed inhibition of NMDARs by TAT-CBD3 may occur through an extracellular mechanism, such as channel block. This also suggests that the conformation per se of the fused version of the TAT with CBD3 bestows inhibition on the NMDARs, rather than either sequence alone. At present, however, it is unclear whether the previously observed internalization of NMDARs in response to TAT-CBD3 occurs through the proposed extracellular mechanism or through a distinct intracellular signaling target. Ongoing studies in our laboratory are investigating these scenarios.
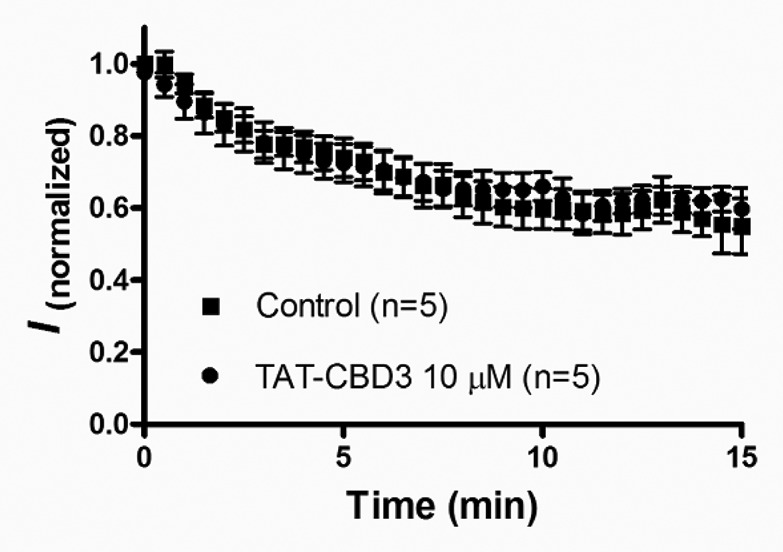
Figure 3. TAT-CBD3 does not intracellularly modulate NMDA current. The time course of NMDA current rundown in the condition that the pipette solution was supplemented with or without 10 μM TAT-CBD3. Peptide TAT-CBD3 was internally microdialyzed via the patch pipette. Current amplitude was normalized to the current evoked by the first application of NMDA (50 μM) at Time 0. No significant difference of current rundown between two conditions during the 15 min time course.
A previous study has reported CRMP-2 as a biochemical binding partner of NMDAR NR2A/B subunits,20 and hence it is possible that the TAT peptide may mediate neuroprotection by disrupting the interaction between CRMP-2 and the NMDAR, rather than via current inhibition. To explore this interaction and determine if TAT-CBD3 affected it, we performed a series of co-immunoprecipitation experiments. Lysates from embryonic day 19 cortical neurons grown for 8 d in culture were exposed to TAT-CBD3 (100 μM) or TAT-control (100 μM) peptides or vehicle (1% DMSO) for 1 h and immunoprecipitated with an anti-NR2B antibody (recognizes proximal carboxyl-terminus, BD Biosciences) (Fig. 3A). Immunoblotting of these immunoprecipitates with a CRMP-2 antibody (raised against amino acids 486–528, Immuno-Biological Laboratories) failed to yield a detectable interaction despite detection of PSD-95, a well-established interaction partner of NMDARs.35,36 Importantly, the interaction between PSD-95 and NR2B was not disrupted by TAT-CBD3, relevant here because disruption of this interaction has neuroprotective properties.37,38 The reciprocal co-immunoprecipitation with the anti-CRMP-2 (C4G) antibody on membrane enriched fractions from postnatal day 2 rat brains (Fig. 3B) also failed to pull down NR2B. It should be noted that the CRMP-2 antibody C4G was the same used in the previous study reporting an interaction between CRMP-2 and NMDARs. To rule out epitope specific or conformation specific NMDAR antibodies as the potential reasons for the lack of interaction in our cultures, we used two additional NR2B antibodies that have their epitopes in the N-terminus (NeuroMab) or in the C-terminus (Millipore) (Fig. 3C). These too failed to resolve an interaction between NR2B and CRMP-2. To exclude the possibility that lysis and immunoprecipitation buffers were too stringent for maintenance of the interaction, a range of RIPA immunoprecipitation buffers were used in which the concentration of detergents (0.25–1% NP-40 (nonyl phenoxypolyethoxylethanol), 0.1% sodium dodecylsulfate, 0.5–1% sodium deoxycholate, 0.25% Triton X-100, or n-Octyl-Glucopyranoside) or salt (50 mM HEPES or 50 mM TRIS-HCl) were varied. None of these ten conditions, performed on embryonic or early postnatal day rat brains, synaptosomes or cultures yielded a positive interaction between CRMP-2 and NMDAR, whereas the interaction between NMDAR and PSD-95 was routinely observed. Cross-linking agents formaldehyde, as well ortho-, and para-isomers of phenylenedimaleimide (PDM), which have different geometries and vary 2.5-fold in their cross-linking spans, were also used but failed to capture the NMDAR-CRMP-2 complex. Bacterially purified CRMP-2-GST fusion protein also failed to pull down NR2B from rat brain lysates or synaptosomes (Fig. 3D).
Finally, we utilized a peptide array13 where we synthesized 15 amino acid peptides (overlapping by 12 amino acids) spanning the N-terminus (92 peptides), intracellular loop 1 (2 peptides), intracellular loop 2 (1 peptide), intracellular loop 3 (26 peptides) and C-terminus (106 peptides) of NR2B which was then overlaid with embryonic rat brain lysate for 1 h and probed in a far-Western simultaneously with a polyclonal CRMP-2 antibody and a monoclonal phospho-CRMP-2 (3F4) antibody that recognizes the cyclin dependent kinase 5 (cdk5) and glycogen synthase kinase 3β (GSK-3β) phosphorylation sites. Two peptides within the NMDAR C-terminus bound CRMP-2: peptide KPGMVFSISRGIYSC (residues 857–871 of the rat NR2B sequence) and DWEDRSGGNFCRSCP (residues 1205–1219 of the rat NR2B sequence) (Fig. 4). These peptides demonstrated relatively high (> 0.6 of normalized maximal) binding to both unphosphorylated and phosphorylated CRMP-2. The carboxyl-terminus of NR2 is believed to be inherently unstructured39 and may “switch” between folded and unfolded states and this may explain the lack of an interaction between CRMP-2 and NR2B in our experiments. Following binding to adaptor proteins, the NR2B may embrace a tertiary confirmation that allows for binding of other proteins or peptides. In the peptide array here, while structural integrity is lost given the short lengths of the peptides, it is nevertheless possible that these “open” unstructured peptides facilitate binding to CRMP-2. While further work is necessary to definitively prove that these two peptides indeed bind CRMP-2 in other biochemical/biophysical experiments, it is perhaps not surprising that the carboxyl-terminus is the likely site of CRMP-2 interaction as this region is a proven nucleation point for NMDARs with demonstrated binding of > 75 proteins including receptor, adaptor, signaling, cytoskeletal and novel proteins.40

Figure 4. CRMP-2 is not found in a complex with NR2B. The biochemical interaction between CRMP-2 and NR2B was investigated using in vitro binding assays. (A) Lysates from E19 DIV 8 cortical neurons were pre-incubated with vehicle (0.5% DMSO), TAT-Control (100 μM), or TAT-CBD3 (100 μM) prior to performing co-immunoprecipitations (IP w/) using an anti-NR2B antibody and immunoblotted for NR2B, PSD-95, and CRMP-2 as indicated. The commercial suppliers of the antibodies are indicated in parentheses. (B) Postnatal day 2 rat brain fractions were used to perform co-immunoprecipitations using anti-CRMP-2 C4G followed by western blot (WB) analyses. (C) Cortical neuron lysates were used to coimmunoprecipitate NR2B using two additional NR2B antibodies antibody and immunoblotted as in (A). (D) Bacterially expressed purified GST (control) or CRMP-2 GST proteins were utilized in pull downs (PD) from cortical neuron lysates along with either a monoclonal IgG (control) or NR2B antibody, and then the immune-captured complexes were blotted with NR2B and CRMP-2 antibodies. Both GST tagged and endogenous CRMP-2 are indicated. Molecular weight markers are indicated in kilodaltons (kDa).
Conclusions
We have advanced the usefulness of TAT-CBD3 as a new therapeutic that may prove useful in managing chronic pain13,14 as it has been shown to alleviate inflammatory and neuropathic hypersensitivity by preventing CRMP2-mediated enhancement of CaV2.2 function. We also demonstrated that TAT-CBD3 could mitigate neuronal death in the face of excitotoxic damage such as that resulting from a traumatic brain injury7 or focal cerebral ischemia. This latter effect involved block and/or internalization of NR2B. As it has been recently proposed that neurodegeneration/neuronal cell death may be an underlying cause of neuropathic pain,41 TAT-CBD3′s proven preservation of neurons and success at amelioration of neuropathic pain in rodent models puts it at the interface of these important and widespread medical problems. TAT-CBD3 may be a first-in-class therapeutic for treatment of neuroprotection as well as neuropathic pain.
Materials and Methods
All methods except those listed below were described by us previously.10,22
Materials and reagents
The antibodies (sources listed in parentheses) used were: whole mouse IgG (Jackson Immunoresearch), whole rabbit IgG (Jackson Immunoresearch), mouse anti-CRMP-2 C4G (Immuno-Biological Laboratories), rabbit anti-CRMP-2 (Sigma-Aldrich), mouse anti-NR2B (UC Davis/NIH NeuroMab Facility), mouse anti-NR2B (BD Biosciences), rabbit anti-NR2B (Millipore), and mouse anti-PSD-95 (UC Davis/NIH NeuroMab Facility). All chemicals, unless stated otherwise, were purchased from Sigma-Aldrich.
Animals and surgery
All experiments using animals were approved by the Animal Care Committee of the Indiana University School of Medicine and followed IASP ethical guidelines. All rats (Harlan Laboratories) were housed under a 12 h light and 12 h dark cycle in individual cages with access to water and standard laboratory chow ad libitum. All surgical procedures were performed on 10-d-old Sprague-Dawley rats as the rat brain at this stage approximates that observed in a human brain at term. Transient focal cerebral ischemia was produced using the MCAO method with some modifications.28,42 Briefly, each rat pup was weighed and anesthetized with 30% isoflurane in a mixture of 70% N2O and 30% O2. Following induction of anesthesia, 1.5% isoflurane was maintained and rectal temperature was monitored and maintained at 36–37°C with a heating blanket. With the animal supine, the left common carotid artery (CCA), external carotid artery (ECA) and internal carotid artery (ICA) were exposed. The left ECA was ligated. A noose was placed around the ICA to control backflow while another noose was placed on the CCA 1 cm proximal to the bifurcation. Then, the ECA close to the ligature was cut off and a 5–0 nylon monofilament suture, that had been blunted at the tip and coated with silicone, was introduced into the ECA lumen and gently advanced through the ICA up to the middle cerebral artery until a slight resistance was felt. The noose on the CCA was loosened. Similar procedures were performed on sham controls except that the suture was not advanced. After awakening, a brief behavioral assessment was performed on rats. The rats that were unable to exhibit circling to the right were regarded to have unsuccessful MCAO, and these animals were excluded from further study. Two hours later, the animals were re-anesthetized under the same conditions and the filament was removed to allow reperfusion.
NMDA current “rundown” assay in hippocampal neuron cultures
The methods for whole-cell voltage clamp recordings were described previously.10,22 For the NMDA current rundown assay, the pipette solution was supplemented with or without 10 μM TAT-CBD3. In both conditions, once the whole-cell configuration was achieved, a 30–40 sec period of time elapsed before collecting the first trace. For statistical evaluation of rundown, the peak currents were normalized to the current amplitude of first traces and expressed as means ± SEM.
TTC Staining
To assess infarct volume, 48 h after the reperfusion, rat brains were removed and sectioned coronally into six 2 mm slices in a brain matrix (Rodent Brain Matrix, Adult Rat, Coronal Sections, RBM-4000C). Slices were incubated in 1% 2, 3, 5-triphenyltetrazolium chloride monohydrate (TTC; Sigma) at 37°C for 10 min and fixed in 10% buffered formalin. Photographs of the sections were taken with a digital camera, and the infarct area was measured using Image J. The tissue survival for each rat was calculated as the ratio of the nonischemic (i.e., TTC-stained ipsilateral region) to ischemic areas of (i.e., TTC-stained contralateral region).
Culturing embryonic neurons
Cortical or hippocampal neurons were dissociated from embryonic day 19 rat and cultured as previously described.22
Calcium and mitochondrial membrane potential (Δψ) imaging
Neurons incubated in the growth medium were loaded at 37°C simultaneously with 2.6 μM Fura-2FF-AM (Molecular Probes) to follow changes in cytosolic Ca2+ and with 1.7 μM Rhodamine 123 (Molecular Probes) to monitor changes in mitochondrial membrane potential.43 During fluorescence recordings, neurons were incubated in the standard bath solution containing 139 mM NaCl, 3 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 10 mM NaHEPES, pH 7.4, 5 mM glucose, and 45 mM sucrose. Fura-2FF and Rhodamine 123 fluorescence signals were followed with an inverted microscope Nikon Eclipse TE2000 using Nikon objective Super Fluor 20• 0.75 NA and a Photometrics CCD camera CoolSNAPHQ (Roper Scientific) controlled by MetaFluor software 6.1 (Universal Imaging Corporation). The excitation light was delivered by a Lambda-LS system (Sutter Instruments). The excitation filters (340 ± 5, 380 ± 7, and 480 ± 20) were controlled by a Lambda 10-2 optical filter changer (Sutter Instruments). Fluorescence was recorded through a 505 nm dichroic mirror at 535 ± 25 nm. The changes in [Ca2+]c were monitored by following F340/F380 calculated after subtracting the background from both channels. The changes in Δψ were monitored by following changes in fluorescence of Rhodamine 123 expressed as F/F0. The Rhodamine 123 fluorescence traces were constructed after subtracting background fluorescence.
Cortical neuron lysates
Embryonic day 19 cortical neurons were grown for 10 d in culture before being washed in PBS (pH 8.0) and lysed in a modified RIPA buffer (1 mM EDTA, 50 mM Tris-HCL pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% Sodium deoxycholate, and freshly supplemented with protease inhibitors) by gentle rotation. Lysates were then clarified by centrifugation at 15,000 x g for 20 min at 4°C.
Coimmunoprecipitations and Immunoblotting
Coimmunoprecipitations were performed on freshly prepared or flash frozen lysates. For coimmunoprecipitations in the presence of TAT peptides, lysates were incubated with 100 μM TAT-Control or TAT-CBD3 or an equal volume of vehicle (DMSO) for 1 h at 37°C prior to addition of the primary antibody. Lysates were incubated with the primary antibody of interest under gentle agitation for 2 h or overnight at 4°C. Samples were then clarified to remove any additional precipitate and incubated with Protein A/G Plus for rabbit primary antibodies or Protein G Plus agarose for mouse primary antibodies (Santa Cruz Biotechnology) for 2 h at 4°C. The immune-captured complexes were then washed three times with lysis buffer before being boiled in equal volumes of SDS loading dye (Invitrogen). Samples were then processed by immunoblotting as previously described.13,22 Blots were probed with 1:1000 anti-NR2B (BD bioscience), 1:1000 anti-CRMP-2 (Sigma-Aldrich), or 1:1000 anti-PSD-95 (UC Davis/NIH NeuroMab Facility). All blots are representative of at least three experiments.
Preparation of postnatal day 2 (P2) rat brain membrane fraction
P2 rat brain membrane fractions were generated as previously described.38
Bacterial expression of CRMP-2 GST fusion protein
CRMP-2 GST was expressed in BL21 Escherichia coli as previously described with minor modifications.10 CRMP-2 GST was purified using a glutathione-cellulose column and eluted using a stepwise gradient of glutathione. The eluted protein was then dialyzed against PBS with 10% glycerol and stocks were flash frozen and stored at -80°C prior to use.
Acknowledgments
The authors would like to acknowledge Sarah Wilson, Dr May Khanna and colleagues at the Stark Neurosciences Research Institute (SNRI) for comments on the manuscript. The peptide blot was synthesized at the SNRI core facility by Dr Andy Hudmon with assistance from Nicole Ashpole. This work was supported, in part, by grants from the Indiana Clinical and Translational Sciences Institute funded, in part by a Project Development Team Grant Number (RR025761) from the National Institutes of Health, National Center for Research Resources, Clinical and Translational Sciences Award, the Indiana State Department of Health—Spinal Cord and Brain Injury Fund (A70-9-079138 to R.K.), the Indiana University Biomedical Committee—Research Support Funds (2286501 to R.K.), a National Scientist Development from the American Heart Association (SDG5280023 to R.K.), and the Elwert Award in Medicine to R.K. J.M.B. is the recipient of a Larry Kays Medical Neuroscience fellowship. R.K. is a majority shareholder of Sophia Therapeutics L.L.C.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/18919
References
- 1.Benveniste H, Drejer J, Schousboe A, Diemer NH. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem. 1984;43:1369–74. doi: 10.1111/j.1471-4159.1984.tb05396.x. [DOI] [PubMed] [Google Scholar]
- 2.Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 3.Villmann C, Becker CM. On the hypes and falls in neuroprotection: Targeting the NMDA receptor. Neuroscientist. 2007;13:594–615. doi: 10.1177/1073858406296259. [DOI] [PubMed] [Google Scholar]
- 4.Muir KW. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol. 2006;6:53–60. doi: 10.1016/j.coph.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Orgogozo JM, Rigaud AS, Stoffler A, Mobius HJ, Forette F. Efficacy and safety of memantine in patients with mild to moderate vascular dementia: a randomized, placebo-controlled trial (MMM 300) Stroke. 2002;33:1834–9. doi: 10.1161/01.STR.0000020094.08790.49. [DOI] [PubMed] [Google Scholar]
- 6.Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348:1333–41. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 7.Brittain JM, Chen L, Wilson SM, Brustovetsky T, Gao X, Ashpole NM, et al. Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2) J Biol Chem. 2011;286:37778–92. doi: 10.1074/jbc.M111.255455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt EF, Strittmatter SM. The CRMP family of proteins and their role in Sema3A signaling. Adv Exp Med Biol. 2007;600:1–11. doi: 10.1007/978-0-387-70956-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hensley K, Venkova K, Christov A, Gunning W, Park J. Collapsin response mediator protein-2: an emerging pathologic feature and therapeutic target for neurodisease indications. Mol Neurobiol. 2011;43:180–91. doi: 10.1007/s12035-011-8166-4. [DOI] [PubMed] [Google Scholar]
- 10.Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem. 2009;284:31375–90. doi: 10.1074/jbc.M109.009951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chi XX, Schmutzler BS, Brittain JM, Wang Y, Hingtgen CM, Nicol GD, et al. Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J Cell Sci. 2009;122:4351–62. doi: 10.1242/jcs.053280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Brittain JM, Wilson SM, Khanna R. Emerging roles of collapsin response mediator proteins (CRMPs) as regulators of voltage-gated calcium channels and synaptic transmission. Commun Integr Biol. 2010;3:172–5. doi: 10.4161/cib.3.2.10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca(2) channel complex. Nat Med. 2011;17:822–9. doi: 10.1038/nm.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson SM, Brittain JM, Piekarz AD, Ballard CJ, Ripsch MS, Cummins TR, et al. Further insights into the antinociceptive potential of a peptide disrupting the N-type calcium channel-CRMP-2 signaling complex. Channels. 2011;5:449–56. doi: 10.4161/chan.5.5.17363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou ST, Jiang SX, Aylsworth A, Ferguson G, Slinn J, Hu H, Leung T, Kappler J, Kaibuchi K. CaMKII phosphorylates collapsin response mediator protein 2 and modulates axonal damage during glutamate excitotoxicity. J Neurochem. 2009;111:870–81. doi: 10.1111/j.1471-4159.2009.06375.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z, Ottens AK, Sadasivan S, Kobeissy FH, Fang T, Hayes RL, et al. Calpain-mediated collapsin response mediator protein-1, -2, and -4 proteolysis after neurotoxic and traumatic brain injury. J Neurotrauma. 2007;24:460–72. doi: 10.1089/neu.2006.0078. [DOI] [PubMed] [Google Scholar]
- 17.Touma E, Kato S, Fukui K, Koike T. Calpain-mediated cleavage of collapsin response mediator protein(CRMP)-2 during neurite degeneration in mice. Eur J Neurosci. 2007;26:3368–81. doi: 10.1111/j.1460-9568.2007.05943.x. [DOI] [PubMed] [Google Scholar]
- 18.Bretin S, Rogemond V, Marin P, Maus M, Torrens Y, Honnorat J, et al. Calpain product of WT-CRMP2 reduces the amount of surface NR2B NMDA receptor subunit. J Neurochem. 2006;98:1252–65. doi: 10.1111/j.1471-4159.2006.03969.x. [DOI] [PubMed] [Google Scholar]
- 19.Chung MA, Lee JE, Lee JY, Ko MJ, Lee ST, Kim HJ. Alteration of collapsin response mediator protein-2 expression in focal ischemic rat brain. Neuroreport. 2005;16:1647–53. doi: 10.1097/01.wnr.0000176520.49841.e6. [DOI] [PubMed] [Google Scholar]
- 20.Al-Hallaq RA, Conrads TP, Veenstra TD, Wenthold RJ. NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J Neurosci. 2007;27:8334–43. doi: 10.1523/JNEUROSCI.2155-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kloda A, Martinac B, Adams DJ. Polymodal regulation of NMDA receptor channels. Channels (Austin) 2007;1:334–43. doi: 10.4161/chan.5044. [DOI] [PubMed] [Google Scholar]
- 22.Brittain JM, Chen L, Wilson SM, Brustovetsky T, Gao X, Ashpole NM, et al. Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2) J Biol Chem. 2011;286:37778–92. doi: 10.1074/jbc.M111.255455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bordi F, Pietra C, Ziviani L, Reggiani A. The glycine antagonist GV150526 protects somatosensory evoked potentials and reduces the infarct area in the MCAo model of focal ischemia in the rat. Exp Neurol. 1997;145:425–33. doi: 10.1006/exnr.1997.6442. [DOI] [PubMed] [Google Scholar]
- 24.Dawson DA, Graham DI, McCulloch J, Macrae IM. Anti-ischaemic efficacy of a nitric oxide synthase inhibitor and a N-methyl-D-aspartate receptor antagonist in models of transient and permanent focal cerebral ischaemia. Br J Pharmacol. 1994;113:247–53. doi: 10.1111/j.1476-5381.1994.tb16201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sauter A, Rudin M. Strain-dependent drug effects in rat middle cerebral artery occlusion model of stroke. J Pharmacol Exp Ther. 1995;274:1008–13. [PubMed] [Google Scholar]
- 26.Park CK, Nehls DG, Graham DI, Teasdale GM, McCulloch J. The glutamate antagonist MK-801 reduces focal ischemic brain damage in the rat. Ann Neurol. 1988;24:543–51. doi: 10.1002/ana.410240411. [DOI] [PubMed] [Google Scholar]
- 27.Indraswari F, Wong PT, Yap E, Ng YK, Dheen ST. Upregulation of Dpysl2 and Spna2 gene expression in the rat brain after ischemic stroke. Neurochem Int. 2009;55:235–42. doi: 10.1016/j.neuint.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 28.Derugin N, Ferriero DM, Vexler ZS. Neonatal reversible focal cerebral ischemia: A new model. Neurosci Res. 1998;32:349–53. doi: 10.1016/S0168-0102(98)00096-0. [DOI] [PubMed] [Google Scholar]
- 29.Bu X, Zhang N, Yang X, Liu Y, Du J, Liang J, et al. Proteomic analysis of cPKCbetaII-interacting proteins involved in HPC-induced neuroprotection against cerebral ischemia of mice. J Neurochem. 2011;117:346–56. doi: 10.1111/j.1471-4159.2011.07209.x. [DOI] [PubMed] [Google Scholar]
- 30.Takahara A, Konda T, Enomoto A, Kondo N. Neuroprotective effects of a dual L/N-type Ca(2+) channel blocker cilnidipine in the rat focal brain ischemia model. Biol Pharm Bull. 2004;27:1388–91. doi: 10.1248/bpb.27.1388. [DOI] [PubMed] [Google Scholar]
- 31.Lee LL, Galo E, Lyeth BG, Muizelaar JP, Berman RF. Neuroprotection in the rat lateral fluid percussion model of traumatic brain injury by SNX-185, an N-type voltage-gated calcium channel blocker. Exp Neurol. 2004;190:70–8. doi: 10.1016/j.expneurol.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 32.Perez-Pinzon MA, Yenari MA, Sun GH, Kunis DM, Steinberg GK. SNX-111, a novel, presynaptic N-type calcium channel antagonist, is neuroprotective against focal cerebral ischemia in rabbits. J Neurol Sci. 1997;153:25–31. doi: 10.1016/S0022-510X(97)00196-2. [DOI] [PubMed] [Google Scholar]
- 33.Bowersox SS, Singh T, Luther RR. Selective blockade of N-type voltage-sensitive calcium channels protects against brain injury after transient focal cerebral ischemia in rats. Brain Res. 1997;747:343–7. doi: 10.1016/S0006-8993(96)01325-X. [DOI] [PubMed] [Google Scholar]
- 34.Valentino K, Newcomb R, Gadbois T, Singh T, Bowersox S, Bitner S, et al. A selective N-type calcium channel antagonist protects against neuronal loss after global cerebral ischemia. Proc Natl Acad Sci USA. 1993;90:7894–7. doi: 10.1073/pnas.90.16.7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–40. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- 36.Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16:2157–63. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martel MA, Soriano FX, Baxter P, Rickman C, Duncan R, Wyllie DJ, et al. Inhibiting pro-death NMDA receptor signaling dependent on the NR2 PDZ ligand may not affect synaptic function or synaptic NMDA receptor signaling to gene expression. Channels (Austin) 2009;3:12–5. doi: 10.4161/chan.3.1.7864. [DOI] [PubMed] [Google Scholar]
- 38.Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, et al. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science. 2002;298:846–50. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- 39.Ryan TJ, Emes RD, Grant SG, Komiyama NH. Evolution of NMDA receptor cytoplasmic interaction domains: implications for organisation of synaptic signalling complexes. BMC Neurosci. 2008;9:6. doi: 10.1186/1471-2202-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–9. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- 41.Bordet T, Pruss RM. Targeting neuroprotection as an alternative approach to preventing and treating neuropathic pain. Neurotherapeutics. 2009;6:648–62. doi: 10.1016/j.nurt.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonzalez FF, McQuillen P, Mu D, Chang Y, Wendland M, Vexler Z, et al. Erythropoietin enhances long-term neuroprotection and neurogenesis in neonatal stroke. Dev Neurosci. 2007;29:321–30. doi: 10.1159/000105473. [DOI] [PubMed] [Google Scholar]
- 43.Brustovetsky T, Li V, Brustovetsky N. Stimulation of glutamate receptors in cultured hippocampal neurons causes Ca2+-dependent mitochondrial contraction. Cell Calcium. 2009;46:18–29. doi: 10.1016/j.ceca.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]