

Abstract
AMPA receptor (AMPAR) plasticity at glutamatergic synapses in the mesostriatal dopaminergic pathway has been implicated in persistent cocaine-induced behavioral responses; however, the precise mechanism underlying these changes remains unknown. Utilizing cocaine psychomotor sensitization in mice we find that repeated cocaine results in a basal reduction of Ser 845 GluA1 and cell surface GluA1 levels in the dorsal striatum (dStr) following a protracted withdrawal period, an adaptation that is dependent on Cav1.3 channels but not those expressed in the VTA. We find that the basally-induced decrease in this phosphoprotein is the result of recruitment of the striatal dopamine D2 pathway, as evidenced by enhanced levels of D2 receptor (D2R) mRNA expression and D2R function as examined using the D2R antagonist, eticlopride, as well as alterations in the phosphorylation status of several downstream molecular targets of D2R’s, including CREB, DARPP-32, Akt and GSK3β. Taken together with our recently published findings examining similar phenomena in the nucleus accumbens (NAc), these results underscore the utilization of divergent molecular mechanisms in the dStr, in mediating cocaine-induced persistent behavioral changes.
Keywords: AKT, Cav1.3, cocaine, CREB, DARPP-32, dopamine D2, dorsal striatum, Erk, GluA1, GSK3β, VTA
Introduction
Repeated exposure to cocaine results in long-lasting changes in dopaminergic and glutamatergic signaling in brain reward regions including the dorsal striatum (dStr), which has been implicated in the habit-forming aspects of addiction1 and the nucleus accumbens (NAc), which has been implicated in mediating the rewarding properties of drugs of abuse.2,3 Such adaptations are believed, in part, to underlie persistent behavioral responses despite extended drug free periods that mimic aspects of addictive behavior.4,5 Recent studies suggest that alteration in the function of AMPA receptor (AMPAR) subunits GluA1 and GluA2 underlies cocaine craving and cocaine seeking behavior.3,6 In particular, attention has focused on trafficking of GluA1 and GluA2 by dopamine D1 receptor (D1R) and D2 receptor (D2R)-mediated mechanisms following withdrawal from cocaine exposure. Specifically, trafficking of the GluA1 subunit has been shown to occur via regulation of phosphorylation at Ser 831,7 a Ca2+/calmodulin kinase (CaMKII) site and Ser 845,8 a protein kinase A (PKA) site. Recent work from our laboratory has identified Cav1.2 and Cav1.3 L-type Ca2+ channels (LTCCs) as key mediators of cocaine-induced long-term behavioral and molecular plasticity,9-11 including phosphorylation and trafficking of AMPARs in the NAc.11 However, the role of these channels in regulating AMPARs in the dStr following cocaine exposure remains unexplored.
A useful rodent model for studying drug-induced long-term plasticity in the dStr and NAc is psychomotor sensitization, which involves a progressive increase in psychomotor activity occurring in response to repeated drug treatment, followed by a persistently elevated drug challenge-induced locomotor response following an extended withdrawal period.12,13 Adaptations in both D1R and D2R signaling have been found to underlie psychomotor sensitization.14 While acute cocaine activates D1R that stimulates PKA in the dStr and NAc which phosphorylates GluA1 at S845,15 we have recently reported that following extended withdrawal from a cocaine sensitization regimen, D1R activation increases phosphorylation of GluA1 at S831 in the NAc, via activation of Cav1.2 channels.11 In the present study, we further explore the role of Cav1.2 and Cav1.3 in regulating cocaine-induced GluA1 phosphorylation and cell surface levels in the dStr. We demonstrate that as opposed to a D1/Cav1.2 mechanism that regulates cocaine-induced phosphorylation and trafficking of GluA1 in the NAc,11 in the dStr Cav1.3 channels recruit the D2R pathway that regulates GluA1 phosphorylation and cell surface levels.
Results and Discussion
Repeated cocaine exposure results in lower levels of Ser 845 GluA1 phosphorylation and cell surface GluA1 levels in the dorsal striatum following 21 d of withdrawal, an adaptation dependent on Cav1.3 channels
To evaluate levels of Ser 831 and Ser 845 GluA1 phosphorylation (S831 P-GluA1 and S845 P-GluA1, respectively) and GluA1 and GluA2 cell surface levels in the dStr and the role of Cav1.3 channels therein, we utilized western blot analysis as previously described in Schierberl et al.11 Striatal tissue was isolated from saline (sal) and cocaine (coc) pre-exposed Cav1.3 wild-type (WT) and knockout (KO) mice (15 mg/kg i.p. cocaine, once a day for 5 d) challenged 21 d later with saline or cocaine. We found that cocaine pre-exposure resulted in significantly lower basal levels of S845 P-GluA1 (Fig. 1A, WT: coc-sal vs. sal-sal) and surface GluA1 (Fig. 1B, WT: coc-sal vs. sal-sal) in Cav1.3 WT mice when examined 21 d following the last cocaine treatment, a change that was absent in Cav1.3 KO mice (significant interaction, treatment group x genotype, S845: F1,70 = 67.43; p < 0.0001; surface GluA1: F3,60 = 37.83; p < 0.0001). Our finding of lower basal cell surface GluA1 is in contrast to that observed by Ferrario et al.,6 who report no change in GluA1 cell surface levels in the dStr of rats after 15 d of withdrawal from a sensitizing regimen of cocaine. We found that a cocaine challenge further lowered the levels of S845 P-GluA1 (Fig. 1A, WT: coc-coc vs. coc-sal) and surface GluA1 in WT mice (Fig. 1B, WT: coc-coc vs. coc-sal) compared with saline challenged mice; however, the differences did not reach significance. Both these changes were absent in Cav1.3 KO mice (Fig. 1A and B, KO: coc-coc vs. coc-sal). Our findings of a decrease in cell surface GluA1 levels following a cocaine challenge in WT mice are also in disagreement with those of Ferrario et al. (2010)6 in that they show that GluA1 cell surface levels in cocaine pre-exposed rats challenged with both saline and cocaine are indistinguishable from each other both 30 min and 24 h after challenge. However, their challenge injections were administered on withdrawal day 14 as opposed to day 21 and their studies were conducted in rats vs. mice. An acute cocaine injection increased S845 P-GluA1 levels in Cav1.3 WT mice, as has been previously demonstrated,15 a change that was also observed in KO mice (Fig. 1A, WT and KO: sal-coc vs. sal-sal). Acute cocaine treatment also increased cell surface levels of GluA1 in Cav1.3 WT and KO mice (Fig. 1B, WT and KO: sal-coc vs. sal-sal) demonstrating no role for Cav1.3 channels in regulating GluA1 following acute cocaine treatment. Ser 831 P-GluA1 was unaltered in the dStr following cocaine pre-exposure or cocaine challenge (data not shown) and no change in GluA2 cell surface levels was observed (data not shown), consistent with Ferrario et al.6 The above findings were further confirmed in Cav1.2 dihydropyridine (DHP)-insensitive mutant mice (Fig. 1C). In this mutant mouse line treatment with the LTCC blocker nifedipine specifically blocks only Cav1.3 channels.10,11,16 This circumvents any developmental compensatory mechanisms that may have occurred in constitutive Cav1.3 KO mice. Consistent with our Cav1.3 KO data, systemic injection of nifedipine (25 mg/kg i.p.), prior to each cocaine injection (15 mg/kg i.p., once a day for 5 d), blocked the decrease in S845 P-GluA1 (Fig. 1C; nif-coc vs. veh-coc) and surface GluA1 (Fig. 1D; nif-coc vs. veh-coc) in the dStr when examined 21 d later. To examine the role of Cav1.2 channels in GluA1 phosphorylation in the dStr we utilized CNS-specific Cav1.2 KO mice (Cav1.2CNSKO).11 We found no role of Cav1.2 channels in regulating S845 P-GluA1. Cocaine pre-exposed Cav1.2 WT (Cav1.2CNSWT) and Cav1.2CNSKO mice exhibited significantly lower levels of S845 P-GluA1 (Fig. 1E). These findings demonstrate that the decrease in S845 P-GluA1 and surface GluA1 in the dStr following extended withdrawal from cocaine treatment are dependent on Cav1.3 and not Cav1.2 channels.

Figure 1. Repeated cocaine decreases S845 P-GluA1 and GluA1 cell surface levels in the dorsal striatum following 21 d of withdrawal, an adaptation dependent on Cav1.3 channels. (A and B) Cocaine pre-exposed Cav1.3 WT but not Cav1.3 KO mice had significantly lower levels of S845 P-GluA1 (A) and surface (S)/intracellular (I) GluA1 (B) in the dorsal striatum (dStr) when examined 21 d following withdrawal from cocaine. Acute treatment with cocaine (sal-coc) significantly increased both S845 P-GluA1 and S/I GluA1 in Cav1.3 WT and KO mice. n = 8–12/treatment. (C and D) Vehicle pretreated Cav1.2DHP−/− mice had significantly lower levels of S845 P-GluA1 (C) and S/I GluA1 (D) compared with saline controls (sal-sal) when examined 21 d following withdrawal from cocaine. Nifedipine pretreatment (nif-coc) blocked this decrease in S845 P-GluA1. n = 8–12/treatment. (E) Knockout of Cav1.2 had no effect on S845 P-GluA1 levels. Cocaine pre-exposed Cav1.2CNSWT and Cav1.2CNSKO mice exhibited significantly lower levels of S845 P-GluA1 compared with saline control mice (dashed line). n = 5–7/group. (F and G) Intra-VTA stereotaxic delivery of Cav1.3 siRNA (F) or ERK2 siRNA (G) had no effect on the lower S845 P-GluA1 levels in the dStr. Both siRNA treatment groups (F), Cav1.3 and (G), ERK2) and their respective controls had significantly lower levels of S845 P-GluA1 compared with saline control mice (dashed line). n = 12–14/treatment. *p < 0.05, vs. sal-sal. sal, saline; coc, cocaine. Data represent the mean ± SEM.
We have recently shown that changes in GluA1 phosphorylation in the NAc following extended withdrawal are dependent on Cav1.3 channels in the VTA during the development of cocaine sensitization.11 Thus, we next evaluated if VTA Cav1.3 channels are also necessary for the decrease in Ser 845 P-GluA1 in the dStr (Fig. 1F). We additionally examined the role of ERK2 in the VTA (Fig. 1G), a downstream target of Cav1.3 channels.11 Recombinant adenoassociated viral vector (rAAV) expressing Cav1.3 siRNA or control siRNA was stereotaxically delivered bilaterally into the VTA three weeks before the start of the cocaine sensitization regimen [see detailed methods in Schierberl et al. (2011)].11 Twenty-one days following the last cocaine sensitization treatment, dStr tissue was examined for S845 P-GluA1 levels. We found that VTA Cav1.3 plays no role in regulating S845 P-GluA1 levels in the dStr. S845 P-GluA1 levels did not differ between control siRNA and Cav1.3 siRNA injected mice (Fig. 1F). Similarly, stereotaxic delivery of a rAAV-expressing ERK2 siRNA (described in Schierberl et al.11) had no effect on S845 P-GluA1 levels (Fig. 1G). This lack of a role for VTA Cav1.3 channels in mediating GluA1 adaptations observed in the dStr following extended withdrawal is not surprising as VTA neurons project primarily to the NAc,17 a pathway implicated in the initial stages of drug taking, whereas the substantia nigra to dStr pathway has been implicated in the more compulsive aspects of addictive behavior.18
Lower S845 GluA1 phosphorylation in the dorsal striatum following withdrawal from cocaine is a result of enhanced dopamine D2R signaling
As dopamine D2 receptors have been shown to regulate S845 P-GluA1 in the dStr14,19 and we have previously shown that repeated treatment with the psychostimulant amphetamine increases the D2R long-splice variant (D2L) mRNA in the dStr via a Cav1.3-dependent mechanism,9 we next tested the hypothesis that repeated cocaine enhances D2-mediated signaling that results in the lower S845 P-GluA1 we observe. We measured D2L mRNA levels (Fig. 2A), in addition to mRNA levels of dopamine D1, D2S, and D3 in the striatum of Cav1.3 WT and KO mice 21 d following repeated cocaine treatment using quantitative real time PCR (qPCR) and receptor-specific primers (Fig. 2A). We found significantly higher levels of D2L mRNA in the dStr of cocaine pre-exposed Cav1.3 WT but not Cav1.3 KO mice (Fig. 2A, significant interaction, treatment x genotype, F1,32 = 100.3; p < 0.0001). D2L levels were not altered in the NAc nor were D1 (WT, sal, 100 ± 4 vs. coc, 98 ± 6), D2S (WT, sal, 100 ± 7 vs. coc, 103 ± 9) or D3 (WT, sal, 100 ± 7 vs. coc, 95 ± 5) receptor mRNAs altered in the striatum or NAc (D1, WT, sal, 100 ± 5 vs. coc, 102 ± 4; D2S, WT, sal, 100 ± 4 vs. coc, 97 ± 5; D3, WT, sal, 100 ± 6 vs. coc, 105 ± 5). We additionally found that D2L mRNA levels positively correlated with expression of the cocaine sensitized response (Fig. 2B, r = 0.68, p < 0.01, Pearson correlation), in line with other studies that have found enhanced D2R agonist-induced psychomotor activity following withdrawal from repeated amphetamine20 and in rats self-administering cocaine21,22 and is in agreement with D2L receptors as important mediators of striatal plasticity.23 In line with our findings of a lack of a role of VTA Cav1.3 channels or ERK2 in mediating changes in S845 P-GluA1 in the dStr, we found no role of either molecule in the VTA on levels of D2L mRNA (Fig. 2C and D). SiRNA-mediated KD of Cav1.3 (Fig. 2C) or ERK2 (Fig. 2D) in the VTA did not alter levels of D2L mRNA in the dStr compared with control siRNA microinjected mice.

Figure 2. Repeated cocaine exposure recruits the dopamine D2L pathway in the dorsal striatum following extended withdrawal. (A) Repeated cocaine significantly increased D2L mRNA levels in the dStr of Cav1.3 WT but not Cav1.3 KO mice. No change was seen in the NAc. **p < 0.01. n = 8–9/treatment. (B) DStr D2L mRNA levels correlated with expression of sensitization. n = 9. r = 0.68, p < 0.01, Pearson correlation. (C and D) Knockdown of VTA Cav1.3 (C) or VTA ERK2 (D) had no effect on the increase in D2L mRNA in the dStr. Both VTA Cav1.3 siRNA and ERK2 siRNA injected mice had significantly higher levels of D2L mRNA compared with saline controls (dashed line). **p < 0.01. n = 12–14/treatment. (E) Eticlopride (etic; 0.2 mg/kg i.p.) significantly increased S845 P-GluA1 in the dStr of cocaine pre-exposed mice compared with saline pre-exposed mice. **p < 0.05, coc-etic vs. sal-etic. *p < 0.05 vs. sal-sal. (F) Eticlopride increased S845 P-GluA1 in the NAc to the same extent in saline- and cocaine-pre-exposed mice (coc-etic vs. sal-etic). *p < 0.05 vs. sal-sal. n = 10–14/treatment. Data represent the mean ± SEM.
We next utilized the D2R antagonist, eticlopride, to examine the functional relevance of higher D2L mRNA on S845 P-GluA1 levels. Blocking D2R’s with eticlopride has been shown to increase S845 P-GluA1 levels;19 thus, we hypothesized that eticlopride treatment should result in higher levels of S845 P-GluA1 in the dStr of cocaine pre-exposed mice compared with saline pre-exposed mice, reflective of higher D2LR levels. Saline and cocaine pre-exposed C57BL/6 mice were treated with eticlopride (0.2 mg/kg, i.p.) 21 d following the last cocaine injection (Fig. 2E). Consistent with our hypothesis eticlopride treatment resulted in significantly higher levels of S845 P-GluA1 in the dStr of cocaine pre-exposed mice compared with saline pre-exposed mice (Fig. 2E, coc-etic vs. sal-etic; significant interaction, cocaine pre-exposure x eticlopride treatment, F1,46 = 70.11; p < 0.0001), an effect that was not seen in the NAc of the same mice (Fig. 2F, coc-etic vs. sal-etic), demonstrating increased functional D2L receptors in the dStr.
We also examined phosphorylation changes of downstream D2R targets including CREB, DARPP-32, Akt and GSK3β24,25 that have been implicated in cocaine’s actions.24,26-28 Ser 133 P-CREB (Fig. 3A), Thr 34 P-DARPP-32 (Fig. 3B) and Thr 308 P-Akt (Fig. 3C) were significantly lower in the striatum of cocaine pre-exposed Cav1.3 WT mice but not Cav1.3 KO mice (significant interaction, treatment x genotype, Ser 133 P-CREB: F1, 34 = 12.48; p < 0.001; Thr 34 P-DARPP-32: F1, 46 = 30.49; p < 0.0001; Thr 308 P-Akt: F1, 36 = 15.31; p < 0.001) while Thr 75 P-DARPP-32 (Fig. 3D) and Ser 9 P-GSK3β (Fig. 3E), a molecule inhibited by Akt, were significantly higher in Cav1.3 WT but not KO mice (significant interaction, treatment x genotype, Thr 75 P-DARPP-32: F1, 34 = 27.39; p < 0.001; Ser 9 P-GSK3β: F1, 36 = 11.60; p < 0.01). Our findings of lower levels of Ser 133 P-CREB and Thr 34 P-DARPP-32, both PKA sites, in the dStr of cocaine pre-exposed mice that have higher levels of D2R’s is consistent with D2 inhibiting PKA.29 Furthermore, the presence of higher levels of Thr 75 P-DARPP-32, a form that converts DARPP-32 into a PKA inhibitor,30 additionally supports our finding of lower levels of Ser 133 P-CREB, Thr 34 P-DARPP-32 and Ser 845 P-GluA1. Our finding of lower levels of Thr 308 P-Akt and higher levels of Ser 9 P-GSK3β are in line with D2 receptors inactivating Akt via dephosphorylation at this particular threonine residue, which would increase Ser 9 GSK-3 phosphorylation31 and the involvement of the D2/Akt/GSK pathway in several DA-associated behaviors32 and cocaine sensitization.33,34
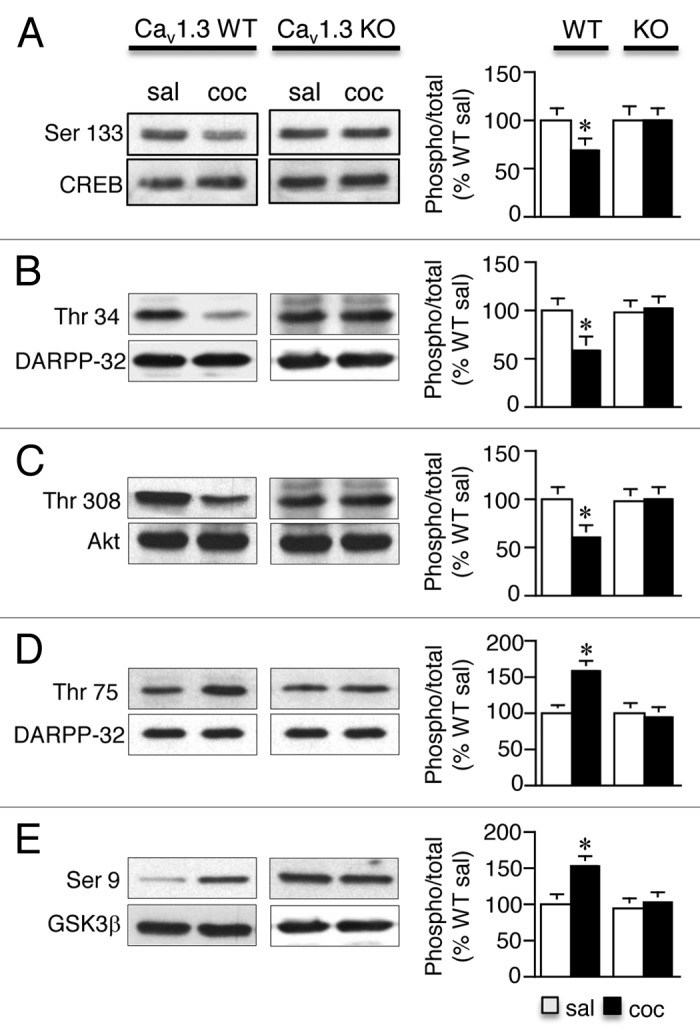
Figure 3. Repeated cocaine-induced change in phosphorylation of downstream D2 receptor targets are mediated by Cav1.3 channels. (A–E) Ser 133 P-CREB (A), Thr 34 P-DARPP-32 (B) and Thr 308 P-Akt (C) were significantly lower whereas Thr 75 P-DARPP-32 (D) and Ser 9 P-GSK3β (E) were significantly higher in the striatum of cocaine pre-exposed Cav1.3 WT mice compared with Cav1.3 KO mice when examined 21 d following cocaine withdrawal. *p < 0.05 vs. sal. n = 8–12/treatment. Data represent the mean ± SEM.
Conclusion
Here we demonstrate that repeated cocaine causes a non-VTA Cav1.3-dependent recruitment of the D2L signaling pathway in the striatum following extended withdrawal, which decreases basal S845 P-GluA1 and cell surface GluA1 levels. This is in contrast to the VTA Cav1.3-dependent recruitment of the Cav1.2 signaling pathway in the NAc that has no effect on the basal increase in S845 P-GluA1 and cell surface GluA1 levels that we have recently identified, further elucidating the divergent molecular mechanisms utilized by interconnected regions within addiction circuitry to mediate reward-related behavior.11
Materials and Methods
Animals
Male C57BL/6 mice (Charles River Laboratories), Cav1.3 wild-type (WT) and knockout mice (KO),35 Cav1.2 dihydropyridine (DHP)-insensitive mice16 and CNS-specific Cav1.2 WT and KO mice11,36 generated on the C57BL/6 background were 9–10 weeks of age at the start of all experiments. Mice were provided food and water ad libitum. Animals were maintained on a 12 h light/dark cycle (7:00 a.m. to 7:00 p.m.). All procedures were conducted in accordance with the Massachusetts General Hospital Subcommittee on Research Animal Care and the Weill Cornell Medical College Institutional Animal Care and Use Committee rules.
Reagents
Cocaine HCl, nifedipine and eticlopride were obtained from Sigma-Aldrich. Cocaine and eticlopride were dissolved in 0.9% saline and nifedipine was dissolved in 0.9% saline containing 1.5% DMSO and 1.5% Tween-80. Anti-rabbit Ser 831 P-GluA1 and Ser 845 P-GluA1 were obtained from Abcam. Anti-rabbit GluA1, GluA2, Ser 133 P-CREB, and CREB were obtained from Millipore. Anti-rabbit Thr 34 and Thr 75 P-DARPP-32, DARPP-32, Thr 308 P-Akt, Akt, Ser 9 P-GSK3β and GSK3β antibodies were obtained from Cell Signaling. Goat anti-rabbit and horse anti-mouse secondary antibodies were obtained from Vector Laboratories.
Psychomotor sensitization protocol
Cocaine psychomotor sensitization was performed as previously described in Giordano et al. (2010).10 Briefly, mice were habituated to open-field locomotor activity chambers (Med Associates Inc.) for 30 min. Mice were then administered saline or cocaine (15 mg/kg, i.p.) once a day for five days (Day 1–5) and locomotor activity was measured for 30 min on each testing day. Following a 21 d drug-free period, mice were challenged with saline or 15 mg/kg i.p. cocaine and locomotor activity measured for 30 min.
Immunoblot analysis
Immunoblotting was performed as previously described in Schierberl et al.11 Bilateral striatal tissue punches spanning approximately 1.7–1.2 mm A/P relative to Bregma (Paxinos and Franklin, 2004),37 were obtained with a 17-gauge stainless steel stylet in a cryostat. Twenty to 40 μg of protein were separated on a 10% (GluA1 and GluA2) or 12% (CREB, DARPP-32, Akt and GSK3β) SDS-polyacrylamide gels. Blots were probed with anti-rabbit (1:850 Ser 831 P-GluA1, Ser 845 P-GluA1, GluA1, GluA2; 1:1000 Ser 133 P-CREB, CREB, Thr 34 P-DARPP-32, Thr 75 P-DARPP-32, Thr308 P-Akt, Akt, Ser 9 P-GSK3β, and GSK3β) primary antibodies overnight at 4°C. Blots were then incubated with goat anti-rabbit (1:5000 for all antibodies) horseradish peroxidase-linked IgG.
Surface GluA1 and GluA2 detection using BS3 cross-linking
Experiments were performed as described in Schierberl et al. (2011).11 Briefly, a 0.5 mm fresh brain coronal section (spanning 1.7–1.2 mm relative to Bregma, Paxinos and Franklin, 2004) was obtained using a mouse brain matrix and placed on an ice-cold surface. Bilateral striatum tissue punches were rapidly dissected using a stainless steel stylet (15 gauge). Tissue was processed as described in Schierberl et al.11 and 15 μg protein was loaded on a 4–15% gradient TRIS-HCl gel (BioRad) and run at 100 V constant voltage. Gels were processed for GluA1 (1:850) and GluA2 (1:1000) immunoblot analysis.
Quantitative real time PCR (qPCR)
For quantitation of mRNA levels, RNA was isolated from striatum tissue punches as described in Schierberl et al.11 Amplification was performed for 40 cycles (95°C for 15 sec, 60°C for 30 sec, 72°C for 30 sec, extension 72°C for 10 min). For D2L and D2S mRNA detection, D2L- and D2S-specific primers (D2L-specific forward primer, 5′-AACTGTACCCACCCTGAGGA-3′; D2S-specific forward primer, 5′-CACCACTCAAGGATGCTGCCCG-3′ and reverse primer common to D2L and D2S, 5′-GTTGCTATGTAGACCGTG-3′) as published in Giordano et al.,9 were used. D1 and D3 mRNA was detected using D1- and D3-specific primers (QuantiTect Primer assay D1, QT00263396 and D3 QT00170527; Qiagen). Cycle threshold (Ct) values for all genes were normalized to the housekeeping gene β-actin. Each experiment was performed in triplicate and values were averaged. For mRNA data analysis, the ΔCt method was used as previously described in Schierberl et al.11 To obtain nanogram (ng) values of D2L mRNA levels used for correlation analysis in Figure 2B, the standard curve method was used as published in Giordano et al.10
Cav1.3 and ERK2 knockdown in the ventral tegmental area
Adenoassociated viral vector (AAV) expressing Cav1.3 siRNA or ERK2 siRNA was utilized to knockdown Cav1.3 or ERK2 in the VTA (−3.4 and −3.5 mm posterior to Bregma and +/0.53 mm lateral to the sagittal suture based on Paxinos and Franklin,37 and as described in Schierberl et al.11 A 26 s-gauge Hamilton syringe was used to deliver 0.5 μL of virus expressing either Cav1.3 siRNA, ERK2 siRNA or scrambled control siRNA into each hemisphere of the VTA at a rate of 0.1 μL/min. Mice were allowed a 3 week period for maximal Cav1.3 and ERK2 knockdown. Injection placement was confirmed by detection of green fluorescent protein (GFP) by fluorescent immunohistochemistry.
Statistical analyses
For immunoblot analysis, optical density values were used to calculate percentage fold change compared with control group (set to 100%). For qPCR, percentage fold change in mRNA levels was used. Data was analyzed by a one-way ANOVA followed by Bonferroni-Dunn post hoc test. Statview 4.5 software (SAS Institute Inc.) was used for all statistics.
Acknowledgments
This work was supported by NIDA Grants K01 DA14057 (A.M.R.), R21 DA023686 (A.M.R.) and DA007274-19 (K.S.), Austrian Science Fund P20670 (J.S.) and Deutsche Forschungsgemeinschaft (F.H. and S.M.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
These authors contributed equally to this work
Previously published online: www.landesbioscience.com/journals/channels/article/19324
References
- 1.Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–9. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- 2.Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–98. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt HD, Pierce RC. Cocaine-induced neuroadaptations in glutamate transmission: potential therapeutic targets for craving and addiction. Ann N Y Acad Sci. 2010;1187:35–75. doi: 10.1111/j.1749-6632.2009.05144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berke JD, Hyman SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–32. doi: 10.1016/S0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- 5.Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–28. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- 6.Ferrario CR, Li X, Wang X, Reimers JM, Uejima JL, Wolf ME. The role of glutamate receptor redistribution in locomotor sensitization to cocaine. Neuropsychopharmacology. 2010;35:818–33. doi: 10.1038/npp.2009.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson SM, Famous KR, Sadri-Vakili G, Kumaresan V, Schmidt HD, Bass CE, et al. CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat Neurosci. 2008;11:344–53. doi: 10.1038/nn2054. [DOI] [PubMed] [Google Scholar]
- 8.Ferrario CR, Loweth JA, Milovanovic M, Ford KA, Galiñanes GL, Heng LJ, et al. Alterations in AMPA receptor subunits and TARPs in the rat nucleus accumbens related to the formation of Ca2+-permeable AMPA receptors during the incubation of cocaine craving. Neuropharmacology. 2011;61:1141–51. doi: 10.1016/j.neuropharm.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giordano TP, III, Satpute SS, Striessnig J, Kosofsky BE, Rajadhyaksha AM. Upregulation of Dopamine D2L mRNA levels in the ventral tegmental area and dorsal striatum of amphetamine-sensitized C57BL/6 mice: role of Cav1.3 L-type Ca2+ channels. J Neurochem. 2006;99:1191–206. doi: 10.1111/j.1471-4159.2006.04186.x. [DOI] [PubMed] [Google Scholar]
- 10.Giordano TP, Tropea TF, Satpute SS, Sinnegger-Brauns MJ, Striessnig J, Kosofsky BE, et al. Molecular switch from L-type Ca v 1.3 to Ca v 1.2 Ca2+ channel signaling underlies long-term psychostimulant-induced behavioral and molecular plasticity. J Neurosci. 2010;30:17051–62. doi: 10.1523/JNEUROSCI.2255-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schierberl K, Hao J, Tropea TF, Ra S, Giordano TP, Xu Q, et al. Cav1.2 L-type Ca2+ channels mediate cocaine-induced GluA1 trafficking in the nucleus accumbens, a long-term adaptation dependent on ventral tegmental area Ca(v)1.3 channels. J Neurosci. 2011;31:13562–75. doi: 10.1523/JNEUROSCI.2315-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Brain Res Rev. 1991;16:223–44. doi: 10.1016/0165-0173(91)90007-U. [DOI] [PubMed] [Google Scholar]
- 13.Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–91. doi: 10.1016/0165-0173(93)90013-P. [DOI] [PubMed] [Google Scholar]
- 14.Borgkvist A, Fisone G. Psychoactive drugs and regulation of the cAMP/PKA/DARPP-32 cascade in striatal medium spiny neurons. Neurosci Biobehav Rev. 2007;31:79–88. doi: 10.1016/j.neubiorev.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Snyder GL, Allen PB, Fienberg AA, Valle CG, Huganir RL, Nairn AC, et al. Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J Neurosci. 2000;20:4480–8. doi: 10.1523/JNEUROSCI.20-12-04480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renström E, Wietzorrek G, Berjukov S, et al. Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca 2+ channels. J Clin Invest. 2004;113:1430–9. doi: 10.1172/JCI20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nestler EJ. Molecular neurobiology of addiction. Am J Addict. 2001;10:201–17. doi: 10.1080/105504901750532094. [DOI] [PubMed] [Google Scholar]
- 18.Belin D, Everitt BJ. Cocaine seeking habits depend upon dopamine-dependent serial connectivity linking the ventral with the dorsal striatum. Neuron. 2008;57:432–41. doi: 10.1016/j.neuron.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 19.Håkansson K, Galdi S, Hendrick J, Snyder G, Greengard P, Fisone G. Regulation of phosphorylation of the GluR1 AMPA receptor by dopamine D2 receptors. J Neurochem. 2006;96:482–8. doi: 10.1111/j.1471-4159.2005.03558.x. [DOI] [PubMed] [Google Scholar]
- 20.Vanderschuren LJ, Schoffelmeer AN, Mulder AH, De Vries TJ. Dopaminergic mechanisms mediating the long-term expression of locomotor sensitization following pre-exposure to morphine or amphetamine. Psychopharmacology (Berl) 1999;143:244–53. doi: 10.1007/s002130050943. [DOI] [PubMed] [Google Scholar]
- 21.De Vries TJ, Schoffelmeer AN, Binnekade R, Raasø H, Vanderschuren LJ. Relapse to cocaine- and heroin-seeking behavior mediated by dopamine D2 receptors is time-dependent and associated with behavioral sensitization. Neuropsychopharmacology. 2002;26:18–26. doi: 10.1016/S0893-133X(01)00293-7. [DOI] [PubMed] [Google Scholar]
- 22.Dias C, Lachize S, Boilet V, Huitelec E, Cador M. Differential effects of dopaminergic agents on locomotor sensitisation and on the reinstatement of cocaine-seeking and food-seeking behaviour. Psychopharmacology (Berl) 2004;175:414–27. doi: 10.1007/s00213-004-1839-1. [DOI] [PubMed] [Google Scholar]
- 23.Centonze D, Usiello A, Costa C, Picconi B, Erbs E, Bernardi G, et al. Chronic haloperidol promotes corticostriatal long-term potentiation by targeting dopamine D2L receptors. J Neurosci. 2004;24:8214–22. doi: 10.1523/JNEUROSCI.1274-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beaulieu JM, Gainetdinov RR, Caron MG. The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol Sci. 2007;28:166–72. doi: 10.1016/j.tips.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–96. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- 26.Rajadhyaksha AM, Kosofsky BE. Psychostimulants, L-type calcium channels, kinases, and phosphatases. Neuroscientist. 2005;11:494–502. doi: 10.1177/1073858405278236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nairn AC, Svenningsson P, Nishi A, Fisone G, Girault JA, Greengard P. The role of DARPP-32 in the actions of drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):14–23. doi: 10.1016/j.neuropharm.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 28.McGinty JF, Shi XD, Schwendt M, Saylor A, Toda S. Regulation of psychostimulant-induced signaling and gene expression in the striatum. J Neurochem. 2008;104:1440–9. doi: 10.1111/j.1471-4159.2008.05240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoof JC, Kebabian JW. Opposing roles for D-1 and D-2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature. 1981;294:366–8. doi: 10.1038/294366a0. [DOI] [PubMed] [Google Scholar]
- 30.Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, et al. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402:669–71. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- 31.Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36:131–7. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- 32.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–73. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 33.Miller JS, Tallarida RJ, Unterwald EM. Cocaine-induced hyperactivity and sensitization are dependent on GSK3. Neuropharmacology. 2009;56:1116–23. doi: 10.1016/j.neuropharm.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu CM, Wang J, Wu P, Zhu WL, Li QQ, Xue YX, et al. Glycogen synthase kinase 3beta in the nucleus accumbens core mediates cocaine-induced behavioral sensitization. J Neurochem. 2009;111:1357–68. doi: 10.1111/j.1471-4159.2009.06414.x. [DOI] [PubMed] [Google Scholar]
- 35.Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, et al. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/S0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 36.Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Müller J, et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–92. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates Second Edition ed2004: Academic Press. [Google Scholar]