

Abstract
Spermidine and spermine, are endogenous polyamines (PAs) that regulate cell growth and modulate the activity of numerous ion channel proteins. In particular, intracellular PAs are potent blockers of many different cation channels and are responsible for strong suppression of outward K+ current, a phenomenon known as inward rectification characteristic of a major class of KIR K+ channels. We previously described block of heterologously expressed voltage-gated Na+ channels (NaV) of rat muscle by intracellular PAs and PAs have recently been found to modulate excitability of brain neocortical neurons by blocking neuronal NaV channels. In this study, we compared the sensitivity of four different cloned mammalian NaV isoforms to PAs to investigate whether PA block is a common feature of NaV channel pharmacology. We find that outward Na+ current of muscle (NaV1.4), heart (NaV1.5), and neuronal (NaV1.2, NaV1.7) NaV isoforms is blocked by PAs, suggesting that PA metabolism may be linked to modulation of action potential firing in numerous excitable tissues. Interestingly, the cardiac NaV1.5 channel is more sensitive to PA block than other isoforms. Our results also indicate that rapid binding of PAs to blocking sites in the NaV1.4 channel is restricted to access from the cytoplasmic side of the channel, but plasma membrane transport pathways for PA uptake may contribute to long-term NaV channel modulation. PAs may also play a role in drug interactions since spermine attenuates the use-dependent effect of the lidocaine, a typical local anesthetic and anti-arrhythmic drug.
Keywords: inward rectification, lidocaine, local anesthetics, Polyamines, sodium channels, spermidine, spermine, use-dependence, voltage-gated Na+ channels
Introduction
Putrescine [NH2(CH2)4NH2], spermidine [NH2(CH2)3NH(CH2)4NH2], and spermine [NH2(CH2)3NH(CH2)4NH(CH2)3NH2] are cationic polyamine (PA) molecules found in all bacterial, plant and animal cells. They are required for cell growth and bind electrostatically to a variety of anionic molecules such as nucleotides, nucleic acids, heparin sulfate groups of glycosaminoglycans, and phospholipids.1 In mammalian cells, putrescine (direct precursor to spermidine and spermine) is synthesized from ornithine by ornithine decarboxylase (ODC), an essential enzyme shown by the lethal phenotype of gene knockout in mice.2 Intracellular PA levels are highly regulated, consistent with the fact that PAs themselves regulate important cellular functions involving RNA including transcription, translation, cell cycle progression and apoptosis.3 Membrane transport of PAs involves both endocytotic uptake and solute carrier mechanisms although specific PA transport proteins have not been conclusively identified in mammals.3-6 In mammalian brains, PAs are taken up by glial cells and presynaptic terminals, accumulated in synaptic vesicles, and have been proposed to function as neuromodulators.4 Alteration of PA metabolism has been recently implicated in human Parkinson disease.7,8 In the field of cardiac physiology, it has long been known that agonist activation of the β-adrenergic receptor results in a rapid rise in ODC activity with a corresponding increase in the concentration of PAs in cardiac myocytes.9,10 The β-adrenergic agonist isoproterenol induces rapid hypertrophy of hearts of transgenic mice overexpressing ODC.11 Thus, PAs are recognized as ubiquitous metabolites with profound effects on cell and systems physiology and are molecules of current interest in various disease processes.
In 1994, PAs were found to function as endogenous blockers of K+ current through a class of plasma membrane K+ channels known as inward rectifiers or KIR channels.12-15 Strong block of outward current by spermidine and spermine is the principal mechanism underlying inward current rectification. KIR channels play a major role in setting the cell resting potential, enabling long-duration action potentials, preventing loss of cytoplasmic K+ and cell migration.16-18 Moreover, PAs have been found to modulate the function of many other ion channel proteins by acting as inhibitors and activators at various intracellular and extracellular sites of action.3,19-21 Known interactions of PAs with ion channels include: intracellular block of KIR channels;22-24 permeant block of AMPA, kainate, and NMDA subtypes of glutamate receptor channels (GluR) from both sides of the membrane;3,25-28 potentiation of NMDA GluR channels from the external side;3,27 intracellular block of neuronal nicotinic acetylcholine receptor channels;29 permeant block of olfactory and visual cyclic nucleotide-gated channels from both sides;30,31 permeant block of the human lymphocyte TRPM7 channel from the external side;32 activation of excitatory currents through the TRPV1 capsaicin receptor channel of sensory afferent nociceptive neurons from the external side;33 inhibition of L-type voltage-gated Ca2+ channels (CaV) of smooth muscle from both sides of the membrane;34,35 external block and positive voltage shift of activation gating of N-type CaV channels of superior cervical ganglion neurons.36
Studies of ion permeation through the voltage-gated Na+ channel (NaV) of rat skeletal muscle revealed that certain mutations of selectivity filter residues (i.e., K1237A) enhance the permeation of large organic cation molecules that normally block the channel.36–38 This line of research led to the observation that outward Na+ currents through the cloned rat muscle NaV channel expressed in human fibroblast HEK293 cells exhibit a slow time-dependent increase in the whole-cell recording mode, as if an endogenous intracellular blocking substance is slowly diluted by exchange with the pipette solution.39 When applied to the intracellular side of excised macropatches containing the skeletal muscle isoform of NaV channels, spermidine and spermine mimicked this behavior by producing strong voltage-dependent block of outward current at concentrations in the range of 1–100 μM.39 These observations suggested that PAs may function as endogenous modulators of NaV channels.
More recently Fleidervish et al.40 reported that depletion of intracellular PAs by an irreversible inhibitor of ODC increases the probability of persistent late opening of single NaV channels in mouse neocortical neurons as observed by cell-attached patch recording in brain slices. Depletion of intracellular PAs similarly increased the magnitude of whole-cell NaV current by 3-fold in these neurons. Both effects were reversed by exposure of brain slices to 500 μM spermine, suggesting that intracellular PAs block neuronal NaV channels. Action potential (AP) recordings showed that spermine produced an activity-dependent block and inhibition of rate-of-rise (dV/dt) of APs. These results, including the observation of spontaneous firing of PA-depleted pyramidal neurons, support the hypothesis that PAs may function as endogenous anti-epileptic molecules that stabilize NaV channel activity in mammalian brain.40 Considering that altered gating of NaV channels by various human genetic mutations is linked to pathological conditions of hyperexcitability in skeletal muscle, heart and neurons,41-44 it is possible that PA metabolism may generally affect NaV channel availability and electrical excitability in all tissues. Thus, the present study explores and compares various biophysical parameters of PA interaction with cloned mammalian NaV channel isoforms naturally expressed in skeletal muscle (NaV1.4), heart (NaV1.5) and neurons (NaV1.2, NaV1.7). The results demonstrate that block by intracellular spermidine and spermine is a well conserved pharmacological interaction for different NaV channel isoforms.
Results
As described previously,39 whole-cell Na+ current evoked by voltage-activation of rat NaV1.4 channels expressed in HEK293 cells exhibits time-dependent behavior when continuously monitored for 30 min after break-in to whole-cell mode. Observation of this phenomenon is facilitated by solutions containing symmetrical 140 mM Na+ in both the bath and pipette to enhance outward current. Both inward and outward current magnitude slowly increases with time of observation as shown by an overlay series of peak I-V data collected from ~12 sec to 30 min after break-in (Fig. 1A). The voltage-activated inward current stabilizes to a stable level relatively quickly within 10 min after break-in; but the outward current rectifies strongly in the positive voltage range at early times and progressively becomes more ohmic (linear) with time. Since the rectifying I-V behavior is closely mimicked by addition of spermidine or spermine to the cytoplasmic side of excised patches,39 it is likely that the time-dependent increase in current is at least partly due to slow dialysis of intracellular PAs by the PA-free solution in the pipette.

Figure 1. Time-dependent rectification of whole-cell I-V behavior of different NaV channel gene isoforms in the presence of symmetrical 140 mM Na+. (Left) Current traces are representative whole-cell current records from single cells expressing different NaV channel isoforms taken at 30 min after membrane break-in to whole-cell mode. I-V behavior was monitored by a consecutive series of step voltage pulses 15 ms in duration at intervals of 0.5 sec from a holding potential of -120 mV to membrane potentials ranging from -140 to +200 mV in 10 mV increments. (Right) Corresponding peak I-V data from NaV channel isoforms were collected from the same cell at various times noted by symbols ranging from 0.2–30 min after membrane break-in to whole-cell mode. I-V data are normalized to the expected peak current at +100 mV, assuming ohmic behavior of outward current in the low positive voltage range at 30 min as indicated by the dashed line. (A–C) correspond to NaV channel clones from rat NaV1.4, human NaV1.5 and human NaV1.7 respectively, expressed in HEK293 cell line. (D) rat NaV1.2 expressed in CHO cell line.
To examine whether such behavior is a common feature of different NaV channel gene isoforms, we compared the behavior of cloned rat NaV1.4 with that of cloned human NaV1.5 and NaV1.7 expressed in the human HEK293 cell line and cloned rat NaV1.2 expressed in the hamster CHO cell line. Comparison of whole-cell peak I-V data in Figure 1 shows similar time-dependent inward current rectification for all four NaV channel isoforms. The time course of outward current increase was compared by plotting the average peak current at +200 mV (Fig. 2). By fitting each time course to a single exponential function assuming a maximum current based on linear I-V behavior at infinite time, we obtained the following time constants for recovery from endogenous rectification: NaV1.4, 16 min; NaV1.7, 28 min; NaV1.2, 32 min; NaV1.5, 37 min. These results suggest that mammalian NaV channel isoforms normally expressed in skeletal muscle (NaV1.4), brain neurons (NaV1.2), peripheral nerve (NaV1.7), and heart (NaV1.5) are all sensitive to block by cytoplasmic PAs. The order of the recovery time constant for outward current (NaV1.4 < NaV1.7 < NaV1.2 < NaV1.5) may reflect intrinsic differences in blocking affinity for PAs or other cytosolic inhibitors and/or differences in the content or diffusion rate of cytosolic inhibitors among the various cell lines.
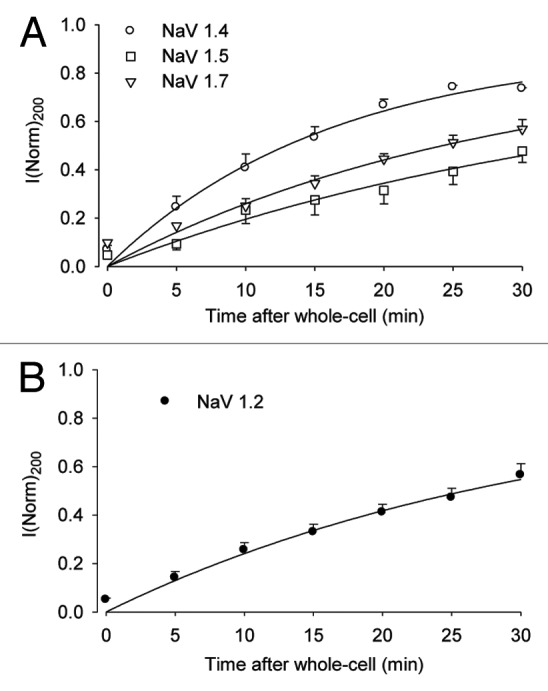
Figure 2. Time-dependence of outward current for different NaV channel gene isoforms. Whole-cell peak currents at +200 mV are plotted as a function of time after membrane break-in from experiments of Figure 1. Data points (mean ± SE of four to five cells) are normalized to the expected peak current at +200 mV, assuming ohmic behavior of outward current. (A) NaV gene isoforms 1.4, 1.5 and 1.7 expressed in HEK293 cells. (B) NaV1.2 expressed in CHO cells. Solid lines correspond to the best fit to a single exponential function with time constants: (A) NaV1.4, 16.0 min; NaV1.7, 28.0 min; NaV1.5, 37.0 min; (B) NaV1.2, 32.1 min.
To directly test for sensitivity to block by intracellular PAs, we compared whole-cell peak I-V behavior for the four NaV channel isoforms recorded under conditions of 140 mM symmetrical Na+ and either 100 μM spermidine or spermine in the pipette solution. As described previously,39 whole-cell NaV current recorded under these conditions equilibrates to a steady-state inhibited level within 5 min after break-in. I-V data were quantitatively compared by normalizing each peak I-V relation to the largest magnitude of inward current observed in the negative voltage range. The results indicate that all four NaV channel isoforms are susceptible to strongly voltage-dependent block by intracellular spermidine and spermine as indicated by the steep negative-resistance region of outward current in the positive voltage range (Fig. 3). Both of the tested PAs produced similar blocking behavior with spermine being slightly more effective than spermidine for each NaV isoform. Comparision of the magnitude of the maximal level of outward current in this experiment indicates that the order of effectiveness of intrinsic block by intracellular PAs for the four NaV isoforms is: NaV1.2 < NaV1.4 < NaV1.7 < NaV1.5.

Figure 3. Comparison of stable whole-cell peak I-V behavior of different NaV channel gene isoforms under conditions of symmetrical 140 mM Na+ and 100 μM intracellular polyamine delivered via the pipette solution. Data points (mean ± SE of four to five cells) are normalized to the maximum peak inward current. (A–C) respectively correspond to NaV1.4, NaV1.5 and NaV1.7 expressed in HEK293 cells. (D) NaV1.2 in CHO cells. Closed symbols, 100 μM spermine; open symbols, 100 μM spermidine.
Previous studies of K+ channels and the present results for Na+ channels show that ion permeation through these channels is blocked by intracellular PAs. The intracellular free concentration of spermidine and spermine in a mammalian lymphocyte is estimated at 200 and 80 μM, respectively.21 Estimated levels of extracellular PAs in plasma and inflamed tissues range from μM to mM.33 Evidence for a functional role of extracellular PAs include: vesicular release of PAs by depolarization of brain slices,4 transport of exogenous PAs from dietary intake and gut bacteria sources into gastrointestinal cells,5 external block and activation of GluR channels,25-28 activation of the TRPV1 capsaicin receptor channel in peripheral sensory neurons,33 and gating shift of CaV channel current.34-36 Therefore, we investigated whether PAs may also affect NaV channels at an extracellular site of action. Figure 4 shows the results of an experiment where extracellular solution containing 1 mM spermine was perfused into the bath solution of an HEK293 cell expressing NaV1.4. Control records (Fig. 4A) show a family of voltage-activated currents collected under conditions of 140 mM symmetrical Na+ and recorded 30 min after break-in to whole-cell mode to allow for substantial recovery of endogenous block. After exposure of the cell for 1 min to extracellular solution containing 1 mM spermine, a sufficient time for complete solution exchange of the small recording chamber, there was virtually no effect on NaV1.4 channel currents as shown by overlay of peak I-V data before and after spermine (Fig. 4B). A similar lack of effect of 5 mM extracellular spermine on peak I-V behavior of voltage-activated Na+ current was reported for rat superior cervical ganglion neurons (Fig. 7 of ref. 36). These results suggest that PAs do not modulate NaV channels at an external site of action.

Figure 4. Lack of acute effect of 1 mM extracellular spermine on NaV1.4 channels. Whole-cell current of NaV1.4 was recorded under conditions of symmetrical 140 mM Na+ for 30 min to allow substantial recovery from outward current rectification. (A) Typical whole-cell current traces for assay of peak I-V behavior from the same cell before, during, and after 1 min exposure to 1 mM spermine in the bath solution. (B) Peak I-V relations of NaV1.4 current before and after exposure to 1 mM extracellular spermine in the bath solution.
However, it is possible that cellular uptake of extracellular PAs might affect NaV channel function. To examine this possibility with the present system we measured the time course of recovery of outward Na+ current from internal block by whole-cell recording in the presence of external PAs. Figure 5A and B show examples of peak I-V relations of NaV1.4 current recorded at various times from a single HEK293 cell maintained in whole-cell recording mode with continuous exposure to 1 mM spermine (Fig. 5A) or 1 mM spermidine (Fig. 5B) in the bath solution. The plot of Figure 5C compares the average time course of recovery from endogenous block in the absence and presence of 1 mM extracellular spermidine or spermine as monitored by the peak current at +200 mV. The results indicate that the time course of recovery from endogenous block is strongly suppressed (63% inhibition at 30 min) in the presence of 1 mM extracellular PAs. Since the tested PAs block NaV1.4 from the intracellular side, the results of this experiment suggests that cellular uptake of extracellular PAs on the time scale of minutes can contribute to suppression of outward NaV current.

Figure 5. Suppression of recovery rate of outward current in the presence of extracellular polyamine. (A) and (B) Examples of whole-cell peak I-V data collected from cells expressing NaV1.4 in the presence of 140 mM symmetrical Na+ and plotted as described for Figure 1 at various times ranging from 0.2–30 min after membrane break-in with continuous perfusion of 1 mM spermine (A) or spermidine (B) in the bath solution. (C) Average whole-cell peak currents at +200 mV (mean ± SE of four cells) as a function of time after membrane break-in, in the absence of polyamine (control) and presence of extracellular 1 mM spermine or 1 mM spermidine. Data points are normalized to the expected peak current at +200 mV (indicated by the dashed line) as in Figure 2. The control time course taken in the absence of extracellular polyamine is fitted to a single exponential function with a time constant of 16.0 min.
Since comparison of four different NaV isoforms in the experiments of Figures 1 and 3 indicates that the human cardiac NaV1.5 channel is most sensitive to PAs, we characterized the effect of spermidine and spermine on the cytoplasmic side of NaV1.5 as recorded in macropatches excised from transfected HEK293 cells. The effect of PAs on the peak I-V relation for NaV1.5 current in macropatches is similar to that previously reported for NaV1.4.39 However the negative slope inflection of the I-V relation at positive voltage and intermediate PA concentration is less pronounced for NaV1.5 than NaV1.4 (compare Fig. 6 of this paper with Fig. 6 of ref. 41), suggesting that the voltage-dependence of block or permeation characteristics of PAs may differ for the two NaV channel isoforms. Best-fit titration curves (Fig. 7) for block of outward current at +100 mV are described by KD values of 9.7 μM (NaV1.5), 16.5 μM (NaV1.4) for spermidine; and, 3.2 μM (NaV1.5), 7.8 μM (NaV1.4) for spermine. By this comparison NaV1.5 channels are inhibited by these PAs with ~2-fold higher affinity than NaV1.4.

Figure 6. Effect of polyamines on NaV1.5 cardiac Na+ channel recorded in inside-out macropatches in the presence of symmetrical 140 mM Na+. (A) Representative families of current traces from macropatch assay of peak I-V behavior of NaV1.5 before, during, and after exposure to 100 μM spermine in the bath solution (cytoplasmic side). (B) and (C) Peak I-V data for macropatch current of NaV1.5 in the absence and presence of 1, 5, 10, 50 and 100 μM spermine (B) or spermidine (C) in the bath solution. Data points are normalized to the peak current at +100 mV and plotted as the mean ± SE of three to seven patches.

Figure 7. Concentration-dependence of inhibition of steady-state outward current of (A) NaV1.4 and (B) NaV1.5 by spermine and spermidine. Outward NaV current was evoked by 15 ms voltage pulses from -120 mV to +100 mV delivered once every 20 sec under conditions of 140 mM symmetrical Na+ in inside-out macropatches in the absence and presence of various concentrations of spermidine and spermine in the bath solution (cytoplasmic side). Data are plotted as the ratio (I/I0) of peak current at +100 mV in the presence (I) vs. the absence (I0) of polyamine for the same patch. Data points are the mean ± SE for three to five cells. Titrations are fit to I/I0 = KD/[(PA) + KD] using best fit KD values of: (A) 16.5 μM spermidine, 7.8 μM spermine; (B) 9.7 μM spermidine, 3.2 μM spermine.
By mutual binding within the permeation pathway, endogenous intracellular PAs may interact with antiarrhythmic or local anesthetic drugs that act on NaV channels. To evaluate this possibility, we studied use-dependent inhibition of NaV1.5 expressed in HEK293 cells by lidocaine in the absence and presence of 100 μM spermine added to the pipette solution. Figure 8A compares normalized inward currents recorded on the first, second and thirtieth consecutive pulses (100 ms voltage step to +20 mV from -120 mV) stimulated at 5 Hz pulse frequency in the presence of 20 μM lidocaine without or with 100 μM spermine in 135 K+ pipette solution. The comparison shows that accumulation of inhibition due to the use-dependence action of lidocaine45,46 on human heart NaV channels is somewhat less pronounced in the presence of 100 μM spermine. Analysis of the time course of development of use-dependence shows that 100 μM spermine does not affect the slight decrease in control current magnitude induced by stimulation at 5 Hz in the absence of lidocaine (Fig. 8B). However, the extra block measured at the thirtieth pulse (~58% inhibition from the first pulse) induced by 5 Hz stimulation in the presence of 20 mM lidocaine is significantly reduced (to ~42% inhibition from the first pulse) by 100 μM spermine in the pipette solution (Fig. 8B and C). Significant reduction of use-dependent accumulation of lidocaine block in the presence of added spermine is observed for stimulation frequencies tested in the range of 1–5 Hz (Fig. 8C). These results suggest that intracellular PAs may attenuate the use-dependent action of drugs that target NaV channels.

Figure 8. Effect of intracellular spermine on use-dependent block of NaV1.5 channels expressed in HEK293 cells. Whole-cell current behavior with 135 K+ internal pipette solution was collected and analyzed starting at 20 min after break-in. Use-dependence was assayed by delivering consecutive voltage pulses to +20 mV for 100 ms from a holding potential of -120 mV at a pulse frequency of 1, 2 and 5 Hz. (A) Representative currents recorded during the first, second and thirtieth pulses at 5 Hz in the presence of 20 μM lidocaine and in the absence or presence or 100 μM spermine in pipette solution. (B) Time course of development of use-dependence at 5 Hz in the absence (Control) or presence of 20 μM lidocaine without or with 100 μM spermine in pipette solution. Peak Na+ currents (I) were normalized to the first pulse (I1) and plotted vs. the pulse number. (C) Steady-state use dependence at the 30th pulse (I30/I1) of NaV 1.5 channels stimulated at 1, 2 and 5 Hz in the presence of 20 μM lidocaine without or with 100 μM spermine in pipette solution. Data points in (B) and (C) are plotted as the mean ± SE for four cells. *p < 0.05, **p < 0.01.
Discussion
The human genome contains nine functional NaV channel genes located on four different chromosomes.47 Activation of these channels by membrane depolarization, is the basis for initiation and propagation of action potentials in excitable cells. Although all nine human NaV channels exhibit considerable sequence identity (~50% over ~2,000 amino acid residues), they differ markedly in affinity for various toxins and drugs such as tetrodotoxin, μ-conotoxin, huwenotoxin-IV, and lidocaine.47,48 In this work we compared PA sensitivity of four mammalian NaV channels expressed in skeletal muscle (NaV1.4, human gene SCN4A), heart (NaV1.5, SCN5A), brain (NaV1.2, SCN2A) and peripheral nerve (NaV1.7, SCN9A). All four tested NaV channels are subject to voltage-dependent block of outward Na+ current by 100 μM intracellular spermidine and spermine as indicated negative resistance I-V behavior in the presence of PAs (Fig. 3). Since the tested isoforms include members from each of the three major phylogenetically-related sub-branches of NaV genes, it is likely that all mammalian NaV channels are sensitive to block by intracellular PAs. Additionally, the phenomenon of slow recovery of outward current in whole-cell recordings with symmetrical Na+ is exhibited by all four tested Na+ channels (Fig. 1). This observation implies that all NaV channels are modulated by endogenous PAs or molecules that resemble PAs in their mode of action. Our results also suggest that cardiac NaV1.5 is somewhat more sensitive to PAs than other NaV isoforms. This evidence includes a ~2-fold slower time course of recovery from endogenous rectification (Fig. 2) and ~2-fold higher affinity of NaV1.5 compared with NaV1.4 for block by cytoplasmic-side application of spermidine and spermine to macropatches (Fig. 7).
Based on lack of effect of acute exposure of NaV1.4 to 1 mM extracellular spermine (Fig. 4), it is likely that PAs modulate NaV channels solely from cytoplasmic site(s) of action. Similar insensitivity of NaV current to 5 mM extracellular spermidine was observed in experiments using dissociated rat superior cervical ganglion neurons; however, high concentrations of external PAs appear to reduce sensitivity to block by tetrodotoxin in these neurons.36 A prolonged 30 min exposure of transfected HEK293 cells to 1 mM extracellular spermidine or spermine suppresses the time course of recovery from inward rectification in the whole-cell mode (Fig. 5), suggesting that slow cellular uptake of PAs by endocytosis or transport proteins5,6 may be a physiological mechanism of NaV channel modulation. The fact that cytoplasmic PAs block outward NaV current in a voltage-dependent manner39 indicates that PAs are likely to bind within the Na+ conduction pathway accessible from the intracellular side of the channel. Mutational analysis previously identified the intracellular vestibule formed by S6 transmembrane helices from each of four homologous I-IV pseudosubunit domains of NaV channels as the site of action of local anesthetics such as lidocaine.46 The possibility that PAs and local anesthetics have overlapping sites of action in the internal vestibule is further supported by use-dependent accumulation of extra block at high frequency rates of channel activation for both classes of molecules39,46 and attenuation of lidocaine use-dependence by spermine for NaV1.5 as described here (Fig. 8).
The approach we used to characterize the interaction of different NaV channel isoforms with PAs takes advantage of non-physiological conditions (i.e., symmetrical Na+ concentration, transfected cells) to favor observation of blocking interactions that affect outward current. This approach is useful in screening for channel interactions with cytoplasmic PAs but does not readily predict how PAs may affect NaV channel activity underlying action potential firing in native cells. Since NaV channels typically function in the rapid upstroke of membrane potential from negative to positive voltage polarity, any factor that inhibits this process is likely to affect excitability. Very small changes in inward current magnitude are sufficient to change firing patterns near threshold. Similarly, cardiac action potential duration and hence cardiac rhythm, can be modulated by small changes in persistent sodium current during the plateau phase when overall membrane conductance is low.
Using a more physiological approach, Fleidervish et al.40 demonstrated that intracellular PAs act to stabilize and suppress AP firing rate of mouse neocortical neurons. Their work highlighted the possibility that intracellular PAs eliminate so-called late openings of NaV channels that persist for hundreds of milliseconds after a voltage-activation pulse. Such late openings or “persistent” current have been attributed to modal gating properties of NaV channels which include kinetic pathways that preclude inactivation or give rise to atypical slow inactivation.49,50 Persistent current is also a prominent feature of mammalian cardiac NaV channels and may account for as much as 1% of the peak transient Na+ current.51 Persistent cardiac NaV current is thought to contribute to the onset of arrhythmia under conditions of ischemia and heart failure.51,52 Drugs that target persistent NaV current are a potential therapeutic strategy in treatment of cardiac arrhythmia as well as epilepsy and pain.40,41,52 Thus, manipulation of PA levels is likely to have an impact on electrical excitability in various tissues. In conclusion, sensitivity of multiple NaV channel isoforms to block by intracellular PAs documented by our work warrants further investigation of the physiological role of spermine and spermidine in the regulation of NaV channel activity and pathological conditions of hyperexcitation.
Materials and Methods
Channel expression and cell culture
Rat skeletal muscle NaV channel (NaV1.4, μ1), human heart NaV channel (NaV1.5, hH1) and human neuronal NaV channel (NaV1.7, hNE-Na) were stably expressed in HEK293 cells as previously described.41,51 Rat brain NaV channel (NaV1.2, rat II) was stably expressed in the Chinese hamster ovary (CHO) cell line. HEK293 cells were grown at 37°C using Dulbecco’s modified essential medium (Gibco BRL) supplemented with 10% fetal bovine serum plus 600 μg/ml (NaV1.4 and NaV1.7) or 200 μg/ml G-418 (Geneticin, Gibco BRL). CHO cells were grown at 37°C in Dulbecco’s modified essential medium and Ham F12 medium (Invitrogen) with 10% fetal bovine serum plus 600 μg/ml G-418. Stably transfected cells were seeded for growth on small polylysine-coated coverslips and used for electrophysiological recording after 2–3 d in culture.
Solutions and electrophysiology
For whole-cell recording, standard 140 Na+ bath (extracellular) solution was 140 mM NaCl, 2 mM KCl, 2 mM CaCl2, 1mM MgCl2, 10 mM glucose, 10 mM Hepes, pH 7.3 with NaOH. The whole-cell 140 Na+ pipette (intracellular) solution was 120 mM NaF, 20 mM NaCl, 5 mM EGTA, 10 mM glucose, 10 mM Hepes, pH 7.3 with NaOH. The whole-cell 135 K+ pipette (intracellular) solution used for the lidocaine experiments of Figure 8 was 120 mM KF, 15 mM KCl, 3 mM NaCl, 1 mM MgCl2, 5 mM EGTA, 10 mM glucose, 10 mM Hepes, pH 7.3 with KOH. For inside-out macropatch recording, the standard pipette (extracellular) solution was the same Na+ bath solution as described above for whole-cell recording. The macropatch 140 Na+ bath (intracellular) solution was 120 mM NaF, 20 mM NaCl, 1.5 mM EGTA, 10 mM glucose, 10 mM Hepes, pH 7.3 with NaOH.
Spermine and spermidine (Sigma) were prepared as 0.1 M stock solutions and added to the above recording solutions at the desired final concentration. Lidocaine hydrochloride (ICN Biomedicals) was prepared as 0.2 M stock solution and diluted to 20 μM by whole-cell recording bath solution.
Patch clamp recording was performed at room temperature (23°C) using an EPC-9 amplifier (HEKA Electronic) with Pulse and Pulse-fit software (Instrutech). The resistance of whole-cell patch pipettes fabricated from Kimax-51 glass capillaries (Fisher Scientific) was 1–2 MΩ filled with 140 Na+ or 135 K+ intracellular solution. The resistance of macropatch pipettes was 0.5–0.7 MΩ filled with extracellular Na+ solution. Current records were acquired by filtering at 10 kHz and sampling at 50 kHz except in use-dependence experiments which sampled at 10 kHz. Current data was subsequently filtered at 3 kHz for analysis and presentation. For whole-cell and macropatch recordings (except in the use-dependent experiments), capacitance transients and leak currents were subtracted by using a negative P/5 pulse protocol delivered at -140 mV. For whole-cell recording, series resistance was electronically compensated to at least 70%. A KCl-agar bridge was used to connect the bath solution to the Ag/AgCl ground electrode.
Current-voltage behavior of NaV channels was typically monitored by recording the response to a consecutive series of step voltage pulses 15 ms in duration to membrane potentials ranging between -140 and +200 mV in increments of 10 mV. The holding potential and pulse interval were normally -120 mV and 0.5 sec, respectively. Whole-cell data was collected beginning at 10 min from the time of break-in when investigating the intracellular effect of polyamine added to the pipette solution. The acute effect of extracellular spermine on whole-cell currents was tested beginning at 30 min after break-in to allow for recovery from endogenous block. The use-dependent action of lidocaine in NaV1.5 without or with spermine was explored 20 min after whole-cell break-in. Recording from excised macropatches was initiated after outward current exhibited a stable response during continuous gravity perfusion of control bath solution.
Concentration-dependent block of PAs was monitored with 15 ms pulses to +100 mV from a holding potential of -120 mV, delivered at 20 sec intervals. Use-dependence of lidocaine was studied by stimulating with a consecutive train of 100 ms test pulse to +20 mV delivered at frequencies of 1, 2 and 5 Hz from a holding potential of -120 mV.
Data analysis
Summary data are the mean ± SE. Except where behavior of a typical cell is illustrated, most results are plotted as average normalized peak I-V relations using two methods of normalization. In the first method, denoted on ordinate axes by I(Norm), each peak current value collected from a given cell was divided by the absolute value of the maximum inward peak current from the same cell (e.g., Fig. 3). For the second method, denoted on ordinate axes by I(Norm)100, each peak current value collected from a given cell/patch was divided by the actual or expected peak current value in the absence of PA at +100 mV (e.g., Figs. 1, 5A–B and 6B–C). The expected peak current value in the absence of PA was obtained by fit the current-voltage data to the following transform of a Boltzmann function: INa = Gmax(V-Vrev)/{1+exp[(V-V0.5)/k]}, where INa is the peak Na+ current, V is the test potential, Vrev is the reversal potential, Gmax is the maximal conductance, V0.5 is the voltage activation midpoint, and k corresponds to a slope factor.
The extent of steady-state, use-dependent block was assessed as the fraction of block at the 30th pulse relative to the 1st pulse (i.e., 1 – (I 30/I 1), where I 1 and I 30 represent the peak currents measured during the 1st and 30th pulses, respectively]. Tonic block was assessed as the fraction of block of the first pulse. Statistical significance was determined using Student’s t-test, with p < 0.05 representing significance.
Acknowledgments
This work was supported by a Grant-in-Aid from the American Heart Association (0150058N) and an Early Career LDRD award from Sandia National Laboratories to EGM. Sandia National Laboratories is a multi-program laboratory managed and operated by Sandia Corporation, a wholly owned subsidiary of Lockheed Martin Corporation, for the US Department of Energy's National Nuclear Security Administration under contract DE-AC04-94AL85000. TRC was funded in part by the National Institutes of Health National Institute of Neurological Disorders and Stroke (Grant R01NS053422).
Glossary
Abbreviations:
- NaV
voltage-gated Na+ channel
- PA
polyamine
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/19001
References
- 1.Cohen SS. A guide to the polyamines. 1998. Oxford University Press, New York, NY. [Google Scholar]
- 2.Pendeville H, Carpino N, Marine JC, Takahashi Y, Muller M, Martial JA, et al. The ornithine decarboxylase gene is essential for cell survival during early murine development. Mol Cell Biol. 2001;21:6549–58. doi: 10.1128/MCB.21.19.6549-6558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Igarashi K, Kashiwagi K. Modulation of cellular function by polyamines. Int J Biochem Cell Biol. 2010;42:39–51. doi: 10.1016/j.biocel.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Masuko T, Kusama-Eguchi K, Sakata K, Kusama T, Chaka S, Okuyama S, et al. Polyamine transport, accumulation, and release in brain. J Neurochem. 2003;84:610–7. doi: 10.1046/j.1471-4159.2003.01558.x. [DOI] [PubMed] [Google Scholar]
- 5.Uemura T, Stringer DE, Blohm-Manone KA, Gerner EW. Polyamine transport is mediated by both endocytotic and solute carrier transport mechanisms in the gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol. 2010;299:G517–22. doi: 10.1152/ajpgi.00169.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poulin R, Casero RA, Soulet D. Recent advances in the molecular biology of metazoan polyamine transport. Amino Acids. 2011;2011:1–13. doi: 10.1007/s00726-011-0987-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antony T, Hoyer W, Cherny D, Heim G, Jovin TM, Subramaniam V. Cellular polyamines promote the aggregation of α-synuclein. J Biol Chem. 2003;278:3235–40. doi: 10.1074/jbc.M208249200. [DOI] [PubMed] [Google Scholar]
- 8.Lewandowski NM, Ju S, Verbitsky M, Ross B, Geddie ML, Rockenstein E, et al. Polyamine pathway contributes to the pathogenesis of Parkinson disease. Proc Natl Acad Sci USA. 2010;107:16970–5. doi: 10.1073/pnas.1011751107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flamigni F, Rossoni C, Stefanelli C, Caldarera CM. Polyamine metabolism and function in the heart. J Mol Cell Cardiol. 1986;18:3–11. doi: 10.1016/S0022-2828(86)80977-4. [DOI] [PubMed] [Google Scholar]
- 10.Fan CC, Koenig H. The role of polyamines in β-adrenergic stimulation of calcium influx and membrane transport in rat heart. J Mol Cell Cardiol. 1988;20:789–99. doi: 10.1016/S0022-2828(88)80004-X. [DOI] [PubMed] [Google Scholar]
- 11.Shantz LM, Feith DJ, Pegg AE. Targeted overexpression of ornithine decarboxylase enhances β-adrenergic agonist-induced cardiac hypertrophy. Biochem J. 2001;358:25–32. doi: 10.1042/0264-6021:3580025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ficker E, Tagliatela M, Wible BA, Henley CM, Brown AM. Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science. 1994;266:1068–72. doi: 10.1126/science.7973666. [DOI] [PubMed] [Google Scholar]
- 13.Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994;372:366–9. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- 14.Fakler B, Brändle U, Glowatzki E, Weidemann S, Zenner HP, Ruppersberg JP. Strong voltage-dependent inward rectification of inward rectifier K+ channels is caused by intracellular spermine. Cell. 1995;80:149–54. doi: 10.1016/0092-8674(95)90459-X. [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Jan YN, Jan LY. Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+ channel. Neuron. 1995;14:1047–54. doi: 10.1016/0896-6273(95)90343-7. [DOI] [PubMed] [Google Scholar]
- 16.Hille B. Ion channels of excitable membranes. 3rd ed. 2001. Sinauer Associates, Inc. Sunderland MA. [Google Scholar]
- 17.deHart GW, Jin T, McCloskey DE, Pegg AE, Sheppard D. The α9β1 integrin enhances cell migration by polyamine-mediated modulation of an inward rectifier potassium channel. Proc Natl Acad Sci USA. 2008;105:7188–93. doi: 10.1073/pnas.0708044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vandenberg CA. Integrins step up the pace of cell migration through polyamines and potassium channels. Proc Natl Acad Sci USA. 2008;105:7109–10. doi: 10.1073/pnas.0803231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forsythe ID. A physiological function for polyamines? Curr Biol. 1995;5:1248–51. doi: 10.1016/S0960-9822(95)00249-1. [DOI] [PubMed] [Google Scholar]
- 20.Williams K. Interactions of polyamines with ion channels. Biochem J. 1997;325:289–97. doi: 10.1042/bj3250289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Igarashi K, Kashiwagi K. Polyamines: Mysterious modulators of cellular functions. Biochem Biophys Res Commun. 2000;271:559–64. doi: 10.1006/bbrc.2000.2601. [DOI] [PubMed] [Google Scholar]
- 22.Lopatin AN, Makhina EN, Nichols CG. The mechanism of inward rectification of potassium channels: “long-pore plugging” by cytoplasmic polyamines. J Gen Physiol. 1995;106:923–55. doi: 10.1085/jgp.106.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo D, Lu Z. Mechanism of IRK1 channel block by intracellular polyamines. J Gen Physiol. 2000;115:799–814. doi: 10.1085/jgp.115.6.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu Z. Mechanism of rectification in inward-rectifier K+ channels. Annu Rev Physiol. 2004;66:103–29. doi: 10.1146/annurev.physiol.66.032102.150822. [DOI] [PubMed] [Google Scholar]
- 25.Bähring R, Bowie D, Benveniste M, Mayer ML. Permeation and block of rat GluR6 glutamate receptor channels by internal and external polyamines. J Physiol. 1997;502:575–89. doi: 10.1111/j.1469-7793.1997.575bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Araneda RC, Lan JY, Zheng R, Zukin S, Bennett MVL. Spermine and arcaine block and permeate N-methyl-D-aspartate receptor channels. Biophys J. 1999;76:2899–911. doi: 10.1016/S0006-3495(99)77445-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mony L, Kew JNC, Bunthorpe MJ, Paoletti P. Allosteric modulators of NR2B-containing NMDA receptors: Molecular mechanisms and therapeutic potential. Br J Pharmacol. 2009;157:1301–17. doi: 10.1111/j.1476-5381.2009.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perrais D, Veran J, Mulle C. Gating and permeation of kainite receptors: Differences unveiled. Trends Pharmacol Sci. 2010;31:516–22. doi: 10.1016/j.tips.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 29.Haghighi AP, Cooper E. A molecular link between inward rectification and calcium permeability of neuronal nicotinic α3β4 and α4β2 receptors. J Neurosci. 2000;20:529–41. doi: 10.1523/JNEUROSCI.20-02-00529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lynch JW. Rectification of the olfactory cyclic nucleotide-gated channel by intracellular polyamines. J Membr Biol. 1999;170:213–27. doi: 10.1007/s002329900551. [DOI] [PubMed] [Google Scholar]
- 31.Lu Z, Ding L. Blockade of a retinal cGMP-gated channel by polyamines. J Gen Physiol. 1999;113:35–43. doi: 10.1085/jgp.113.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kerschbaum HH, Kozak JA, Cahalan MD. Polyvalent cations as permeant probes of MIC and TRPM7 pores. Biophys J. 2003;84:2293–305. doi: 10.1016/S0006-3495(03)75035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahern GP, Wang X, Miyares RL. Polyamines are potent ligands for the capsaicin receptor TRPV1. J Biol Chem. 2006;281:8991–5. doi: 10.1074/jbc.M513429200. [DOI] [PubMed] [Google Scholar]
- 34.Gomez M, Hellstrand P. Effects of polyamines on voltage-activated calcium channels in guinea-pig intestinal smooth muscle. Pflugers. 1995;430:501–7. doi: 10.1007/BF00373886. [DOI] [PubMed] [Google Scholar]
- 35.Gomez M, Hellstrand P. Endogenous polyamines modulate Ca2+ channel activity in guinea-pig intestinal smooth muscle. Pflugers Arch. 1999;438:445–51. doi: 10.1007/s004240051060. [DOI] [PubMed] [Google Scholar]
- 36.Chen W, Harnett MT, Smith SM. Modulation of neuronal voltage-activated calcium and sodium channels by polyamines and pH. Channels. 2007;1:281–90. doi: 10.4161/chan.4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun YM, Favre I, Schild L, Moczydlowski E. On the structural basis for size-selective permeation of organic cations through the voltage-gated sodium channel: effect of alanine mutations at the DEKA locus on selectivity, inhibition by Ca2+ and H+, and molecular sieving. J Gen Physiol. 1997;110:693–715. doi: 10.1085/jgp.110.6.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang CJ, Favre I, Moczydlowski E. Permeation of large tetra-alkyammonium cations through mutant and wild-type voltage-gated sodium channels as revealed by relief of block at high voltage. J Gen Physiol. 2000;115:435–454. doi: 10.1085/jgp.115.4.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang CJ, Moczydlowski E. Cytoplasmic polyamines as permeant blockers and modulators of the voltage-gated sodium channel. Biophys J. 2001;80:1262–79. doi: 10.1016/S0006-3495(01)76102-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fleidervish IA, Libman L, Katz E, Gutnick MJ. Endogenous polyamines regulate cortical neuronal excitability by blocking voltage-gated Na+ channels. Proc Natl Acad Sci USA. 2008;105:18994–9. doi: 10.1073/pnas.0803464105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–47. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- 42.Stafstrom CE. Severe epilepsy syndromes of early childhood: the link between genetics and pathophysiology with a focus on SCN1A mutations. J Child Neurol. 2009;24:15S–23S. doi: 10.1177/0883073809338152. [DOI] [PubMed] [Google Scholar]
- 43.Zimmer T, Surber R. SCN5A channelopathies—an update on mutations and mechanisms. Prog Biophys Mol Biol. 2008;98:120–36. doi: 10.1016/j.pbiomolbio.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 44.Cannon SC. Spectrum of sodium channel disturbances in the nondystrophic myotonias and periodic paralyses. Kidney Int. 2000;57:772–9. doi: 10.1046/j.1523-1755.2000.00914.x. [DOI] [PubMed] [Google Scholar]
- 45.Li RA, Ennis IL, Tomaselli GF, Marban E. Structural basis of differences in isoform-specific gating and lidocaine block between cardiac and skeletal muscle sodium channels. Mol Pharmacol. 2002;61:136–41. doi: 10.1124/mol.61.1.136. [DOI] [PubMed] [Google Scholar]
- 46.Nau C, Wang GK. Interactions of local anesthetics with voltage-gated Na+ channels. J Membr Biol. 2004;201:1–8. doi: 10.1007/s00232-004-0702-y. [DOI] [PubMed] [Google Scholar]
- 47.Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 48.Xiao Y, Bingham JP, Zhu W, Moczydlowski E, Liang S, Cummins TR. Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain II voltage sensor in the closed configuration. J Biol Chem. 2008;283:27300–13. doi: 10.1074/jbc.M708447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patlak JB, Ortiz M. Slow currents through single sodium channels of the adult rat heart. J Gen Physiol. 1985;86:89–104. doi: 10.1085/jgp.86.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alzheimer C, Schwindt PC, Crill WE. Modal gating of Na+ channels as a mechanism of persistent Na+ current in pyramidal neurons from rat and cat sensorimotor cortex. J Neurosci. 1993;13:660–73. doi: 10.1523/JNEUROSCI.13-02-00660.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maltsev VA, Undrovinas A. Late sodium current in failing heart: Friend or foe? Prog Biophys Mol Biol. 2008;96:421–51. doi: 10.1016/j.pbiomolbio.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saint DA. The cardiac persistent sodium current: an appealing therapueutic target. Br J Pharmacol. 2008;153:1133–42. doi: 10.1038/sj.bjp.0707492. [DOI] [PMC free article] [PubMed] [Google Scholar]