

Abstract
In skeletal muscle, L-type calcium channels (DHPRs), localized to plasma membrane sarcoplasmic reticulum junctions, are tightly packed into groups of four termed tetrads. Here, we have used bimolecular fluorescence complementation (BiFC) and targeted biotinylation to probe the structure and organization of β1a subunits associated with native CaV1.1 in DHPRs of myotubes. The construct YN-β1a-YC, in which the non-fluorescent fragments of YFP (“YN” corresponding to YFP residues 1–158, and “YC” corresponding to YFP residues 159–238) were fused, respectively, to the N- and C-termini of β1a, was fully functional and displayed yellow fluorescence within DHPR tetrads after expression in β1-knockout (β1KO) myotubes; this yellow fluorescence demonstrated the occurrence of BiFC of YN and YC on the β1a N- and C-termini. In these experiments, we avoided overexpression because control experiments in non-muscle cells indicated that this could result in non-specific BiFC. BiFC of YN-β1a-YC in DHPR tetrads appeared to be intramolecular between N- and C-termini of individual β1a subunits rather than between adjacent DHPRs because BiFC (1) was observed for YN-β1a-YC co-expressed with CaV1.2 (which does not form tetrads) and (2) was not observed after co-expression of YN-β1a-YN plus YC-β1a-YC in β1KO myotubes. Thus, β1a function is compatible with N- and C-termini being close enough together to allow BiFC. However, both termini appeared to have positional freedom and not to be closely opposed by other junctional proteins since both were accessible to gold-streptavidin conjugates. Based on these results, a model is proposed for the arrangement of β1a subunits in DHPR tetrads.
Keywords: bimolecular fluorescence complementation, confocal microscopy, dihydropyridine receptor, electrophysiology, excitation-contraction coupling, skeletal muscle, targeted biotinylation, yellow fluorescent protein, β1a
Introduction
Two major proteins involved in skeletal muscle excitation-contraction (EC) coupling are the dihydropyridine receptor (DHPR) localized in the plasma membrane and the ryanodine receptor type 1 (RyR1) residing in the sarcoplasmic reticulum (SR) membrane. DHPRs in skeletal muscle are arrayed in groups of four, termed tetrads, positioned over the homotetrameric subunits of every other RyR1.1 The DHPR acts as an L-type Ca2+ channel and as the transducer that links electrical excitation to muscle contraction. Specifically, DHPR voltage sensing domains translocate in response to membrane depolarization causing the pore-forming regions of the channel to open and to flux Ca2+, although this Ca2+ current is not necessary for skeletal muscle EC coupling. Instead, it is thought that depolarization-induced conformational changes of the DHPR are somehow transmitted to RyR1, causing it to open and release Ca2+ from the SR, which in turn elicits muscle contraction.
The skeletal muscle DHPR is a hetero-pentamer comprised of CaV1.1, β1a, γ1 and α2-δ1 subunits. EC coupling is still functional following knockout of γ1 or suppression of α2-δ1 by siRNA.2-4 By contrast, mice null for either CaV1.1 or β1 die at birth as of consequence of failed EC coupling and respiratory paralysis.4-6 CaV1.1 is a typical, four-repeat, voltage-gated ion channel and houses both the voltage sensing structures and the ion conducting pore. The loop between domains II-III has been shown to be vital for skeletal muscle EC coupling,7 and the I-II cytoplasmic loop binds the channel auxiliary β subunit.8
CaVβ subunits are encoded by four distinct genes (β1, β2, β3 and β4) with multiple splice variants.8,9 Based on primary sequence, β subunits are comprised of five domains (D1–D5); two of these, D2 (or SH3) and D4 (or GK), are strongly conserved, and three (D1, D3 and D5) are highly divergent.10,11 Crystals of the β subunit core (D2-D4) reveal structural similarities to PSD-95, a membrane-associated guanylate kinase (MAGUK) protein.12-14 The β subunit D2 and D4 domains have sequence and structure similarity, respectively, to the Src homology 3 (SH3) and guanylate kinase (GK) domains of PSD-95.15-17 Because they are unstructured, the orientation of the D1, D3 and D5 domains, both in free β subunits and within assembled Ca2+ channels, remains unknown.
The β subunit core plays an important role in membrane trafficking of high-voltage activated Ca2+ channels, including CaV1.1.18,19 However, studies of β subunit expression in myotubes obtained from mice with a knocked-out β1 gene (β1KO myotubes) indicate that β1a also has a more direct role in skeletal-type EC coupling. In particular, skeletal-type EC coupling, but not membrane trafficking of CaV1.1, is suppressed by truncation or alteration of the C-terminal (D5) domain of β1a.20-22 Given the importance of the β1a C-terminal, and the lack of information about its orientation, as well as that of the N-terminal, the goal of the studies described here was to probe this orientation by means of bimolecular fluorescence complementation (BiFC) and targeted biotinylation.
BiFC23,24 employs two non-fluorescent fragments of yellow fluorescent protein (YFP), YFP-N (YN: residues 1–158) and YFP-C (YC: residues 159–238). When expressed together as individual fragments in living cells, complementation, and subsequent yellow fluorescence, is infrequent and dim. This complementation, or BiFC, is greatly enhanced when the two fragments are attached to proteins or protein domains that interact with each other.23 An advantage of this approach, compared with fluorescent resonance energy transfer (FRET), is that BiFC can provide information from a single excitation/emission image without the need for extensive analysis.
In the current study, we have attached YN and YC to the N- and C-termini of the β1a subunit to test whether fluorescence complementation would occur for the resulting construct (YN-β1a-YC) either as a free protein in the cytoplasm of tsA201 cells or in β1KO myotubes co-assembled with native CaV1.1 into groups of functional DHPRs at plasma membrane/SR junctions. We found that complementation occurred both in tsA201 cells (indicating intramolecular BiFC between the N- and C-termini of free β1a), and in groups of DHPRs in β1KO myotubes. These data suggest the existence of intramolecular BiFC of N- and C-termini of individual β1a subunits and/or intermolecular BiFC between β1a subunits of DHPRs neighboring one another in tetrads. Arguing against the latter of these two possibilities, we failed to observe intermolecular BiFC after co-expression of YN-β1a-YN and YC-β1a-YC in β1KO myotubes. Furthermore, BiFC was observed in dysgenic (CaV1.1-null) myotubes when YN-β1a-YC was co-expressed with CaV1.2, which is closely related to CaV1.1 but does not group into tetrad arrays.25 Thus, when β1a is incorporated into DHPRs at plasma membrane/SR junctions, the N- and C-termini have the ability to come into close apposition.
To further probe the positional restraints on the β1a N- and C-termini, we tested for the binding of gold-streptavidin conjugates applied to myotubes expressing β1a constructs with a biotin acceptor domain (BAD) fused to either the N- or C-terminus.26 Large (> 5 nm) gold-streptavidin conjugates could bind to either terminus, suggesting that the N- and C-termini are not constrained to be in tight apposition to one another. The results with BiFC and gold-streptavidin binding provide the basis for a proposed model of the arrangement of β1a subunits within DHPR tetrads.
Results
Functional equivalence of YN-β1a-YC and YFP-β1a
As one approach for probing the spatial relationships between the N- and C-termini of the β1a subunit of the skeletal muscle L-type Ca2+ channel (DHPR), we tested whether BiFC would occur for YFP amino acids 1–158 (“YN”) and 159–238 (“YC”) fused to the β1a N- and C-terminus, respectively. As a first step, we tested whether this construct (YN-β1a-YC) was functional after expression in β1KO myotubes, which are null for endogenous β1. In particular, we compared it to YFP-β1a because previous work has shown that attachment of intact fluorescent proteins to the N- and/or C-termini has no obvious effect on function of β1a.27,28 Both YFP-β1a and YN-β1a-YC constructs produced robust Ca2+ currents, which were not present in non-transfected β1KO myotubes (Fig. 1A). Furthermore, the currents resulting from expression of the two constructs were similar in both magnitude and voltage-dependence as reflected in the peak current-voltage (I-V) relationships (Fig. 1B). Because both channel function and membrane trafficking are affected by β subunits, we assessed membrane expression by measuring intramembrane charge movements after block of L-type ionic current by addition of 0.5 mM CdCl2 and 0.1 mM LaCl3 to the external solution. Representative charge movements for a depolarization to +30 mV are shown in Figure 1C and charge-voltage (Q-V) relationships, obtained by integrating the ON charge movements at each test potential, are illustrated in Figure 1D. The β1KO myotubes produced very modest charge movements, which presumably arose, at least in part, from voltage-gated sodium and potassium channels that would be expected to be present in myotubes whether or not L-type Ca2+ channels are also present. The charge movements in myotubes expressing either YFP-β1a or YN-β1a-YC were roughly 3-fold larger than those in non-transfected β1KO myotubes. Additionally, the charge movements for YFP-β1a and YN-β1a-YC had similar voltage dependence (Fig. 1D).

Figure 1. YFP-β1a and YN-β1a-YC are functionally equivalent in membrane-targeted L-type Ca2+ channels. (A) Representative Ca2+ currents for test potentials of 0 and +30 mV applied to a β1KO myotube, and β1KO myotubes expressing YFP-β1 or YN-β1a-YC. Scale bars: vertical 2 pA/pF; horizontal, 50 ms. (B) Average peak Ca2+ current density vs. test potential for β1KO myotubes (open squares, n = 10), and β1KO myotubes expressing YFP-β1a (filled squares, n = 16) or YN-β1a-YC (gray squares, n = 7). Fits of the averaged peak current-voltage data with Equations 1 and 2 yielded values of Gmax (210 and 235 nS/nF), V1/2 (18.5 and 19.0 mV) and k (5.4 and 6.0 mV) for YFP-β1a and YN-β1a-YC, respectively. (C) Representative, membrane-bound charge movements for a step from the holding potential (-50 mV) to a test potential of +30 mV applied to a β1KO myotube, and β1KO myotubes expressing YFP-β1 or YN-β1a-YC. Scale bars: vertical 1 pA/pF; horizontal, 5 ms. (D) Average integral of the ON component of charge movement (Qon) vs. voltage for β1KO myotubes (open squares, n = 4), and β1KO myotubes expressing YFP-β1a (filled squares, n = 2) or YN-β1a-YC (gray squares, n = 3). Averaged data points were fit with Equation 2 yielding values of Qmax (6.2, 7.0 and 1.8 nC/μF) V1/2 (0.0, -2.8 and -12.1 mV) and k (12.2, 13.2 and 10.2 mV) for YFP-β1a, YN-β1a-YC and β1KO, respectively. Error bars represent ± SEM (in some cases smaller than the symbol).
To test for EC coupling, myotubes were depolarized by a 5 sec application of 80 mM KCl. As shown in Figure 2, left, this depolarization did not elicit a visible response from a β1KO myotube, but caused pronounced, qualitatively similar contractions of β1KO myotubes expressing either YFP-β1a or YN-β1a-YC. Moreover the fraction of myotubes contracting was similar for these two β1a constructs (Fig. 2, right). These data, together with those in Figure 1 indicate that YFP-β1a and YN-β1a-YC were equivalent in regard to membrane expression of CaV1.1, and its function as both L-type Ca2+ channel and as voltage-sensor for EC coupling.

Figure 2. YFP-β1a and YN-β1a-YC are functional for EC coupling. (Left) β1KO myotubes did not contract in response to 80 mM KCl membrane depolarization whereas expression of either YFP-β1a or YN-β1a-YC in β1KO myotubes rescued EC coupling. Horizontal scale bar: 5 sec. (Right) Percentage of non-transfected β1KO (n = 0/4 cells), YFP-β1a (n = 8/14 cells) or YN-β1a-YC (n = 8/13 cells) expressing myotubes that produced visible contractions in response to 80 mM KCl.
BiFC occurs for YN-β1a-YC
Having established the functional equivalency of YFP-β1a and YN-β1a-YC, we next compared the two constructs in terms of fluorescence intensity and subcellular distribution after expression in β1KO myotubes. A comparison of low-magnification (10X) images, which were obtained with identical confocal settings from fields having comparable coverage by myotubes, revealed that the fraction of detectably yellow cells was similar in four dishes of β1KO myotubes transfected with YFP-β1a (38/310 cells) and four dishes transfected with YN-β1a-YC (33/272 cells). Examined at higher power (63X), myotubes transfected with either construct displayed large numbers of yellow fluorescent puncta superimposed on a diffuse, yellow background (Fig. 3A). These puncta represent presumptive sites of plasma membrane junctions with the sarcoplasmic reticulum at which DHPRs containing CaV1.1 and β1a interact with RyRs,27,29 whereas the diffuse background likely represents cytoplasmic β1a not associated with CaV1.1.19,27 Thus, it appears that BiFC occurred for YN-β1a-YC located either cytoplasmically or incorporated into junctionally inserted DHPRs. This result is consistent with earlier work showing that FRET occurred between CFP and YFP attached to the N- and C-termini of β1a both in the cytoplasm and in junctional DHPRs.27
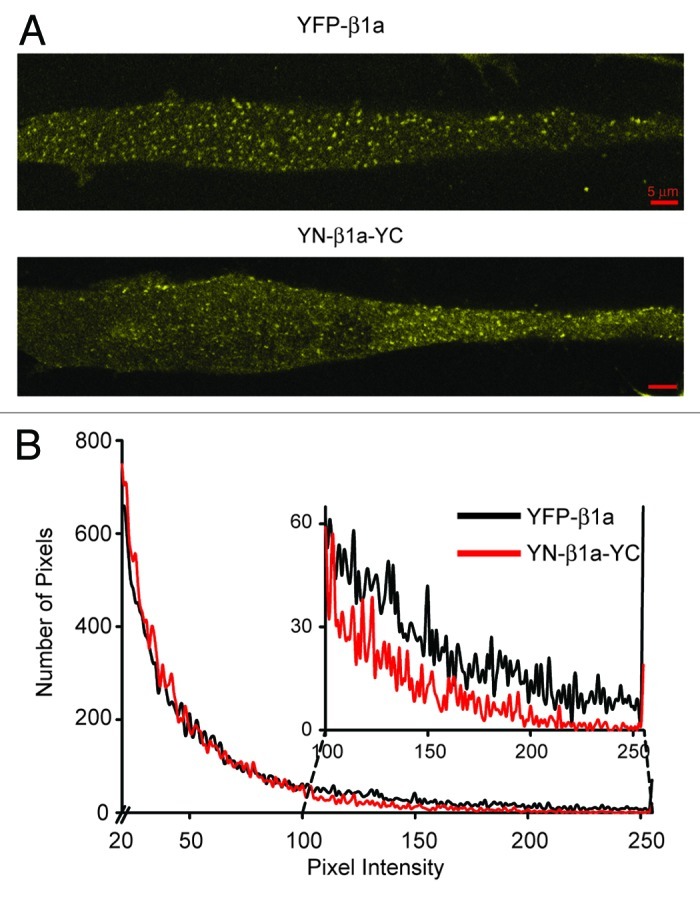
Figure 3. BiFC occurred for YN-β1a-YC incorporated with CaV1.1 into junctional DHPRs. (A) Confocal sections near the glass coverslip: surface membrane interface of β1KO myotubes expressing YFP-β1a or YN-β1a-YC. Both constructs produced yellow fluorescent foci. (B) Histogram of numbers of pixels vs. intensity for YFP-β1a (black line) and YN-β1a-YC cells (red line); pixel values < 20 were excluded. Inset shows a magnification of pixel intensities 100–255. The lower values for YN-β1a-YC than for YFP-β1a indicated that fluorescence complementation was incomplete. Red scale bars represent 5 μm in all images.
To compare the intensity resulting from fluorescence complementation with that resulting from intact YFP, four 512 × 512 pixel images were obtained at 63X of β1KO myotubes transfected with YFP-β1a or YN-β1a-YC. In similar images of non-transfected β1KO myotubes, we observed that 99.6% of all pixels had intensities < 20, which was thus used as a lower threshold for the analysis of the transfected myotubes. After exclusion of pixel intensities < 20, the average fluorescence intensity for YN-β1a-YC was 52 ± 35 (mean ± S.D.; n = 18,503) compared with 64 ± 50 (n = 19,518) for YFP-β1a (different at p < 0.0001, unpaired Student’s t-test). A comparison of intensity histograms (Fig. 3B) revealed that this difference in average intensity was a consequence of the fact that there were more “dim” pixels and fewer bright pixels for YN-β1a-YC (red line) than for YFP-β1a (black line). In particular, intensities of 20–99 accounted for 16,781 and 15,918 pixels, and intensities of 100–255 for 1,722 and 3,600 pixels in the images obtained from YN-β1a-YC and YFP-β1a, respectively. The lower fluorescence intensity for YN-β1a-YC is typical of the BiFC assay and is likely because complementation is incomplete even when the YN and YC moieties are closely apposed to one another.24
The L134P mutation eliminates BiFC between the N- and C-termini of β1a
To test whether the fluorescence complementation observed for YN-β1a-YC would be disrupted by alteration of the “MAGUK” core structure, we introduced the L134P mutation into the SH3 domain, which is structurally conserved between CaVβ subunits.16 Previous work had shown that the L134P mutation within β1a, and the corresponding mutation (L93P) within β2a, altered inactivation of co-expressed CaV2.116 and that the L93P mutation greatly reduced (although did not eliminate) the ability of β2a to produce membrane trafficking of CaV1.2.17 Furthermore, the homologous mutation (L460P) interferes with the interaction of the SH3 and GK domains of the MAGUK protein PSD-95.30 Thus, we constructed YN-β1aL134P-YC, as well as YFP-β1aL134P to serve as a control. Figure 4A demonstrates that the subcellular distribution of YFP-β1aL134P expressed in β1KO myotubes was much different than the dispersed, small puncta characteristic of YFP-β1a (Fig. 3A, top panel). In particular, 2–3 d post-transfection, YFP-β1aL134P was present in some cells as large juxta-nuclear clusters (Fig. 4A, top panel), which may represent an accumulation of misfolded protein in aggresomes,31 whereas in other cells it was more uniformly distributed and produced a faint, diffuse yellow fluorescence; this diffuse yellow fluorescence was sometimes also present in cells with aggregates but is not visible in the top panel of Figure 4A to avoid extreme over-saturation of the aggregates. By 4–5 d post-transfection, however, the aggregates had largely disappeared and YFP-β1aL134P was diffusely distributed in most cells at a level that was easily distinguishable from background (Fig. 4A, bottom panel).

Figure 4. The point mutation L134P in β1a impaired trafficking and L-type channel function, and altered β1a folding as indicated by loss of BiFC. (A) Mid-level confocal sections of β1KO myotubes expressing YFP-β1aL134P at days 3 (top) and 5 (bottom) post-transfection. (B) Distribution of peak Ca2+ current densities at +30 mV of β1KO myotubes expressing YFP-β1aL134P at day 3 (black bars, n = 9) and days 4–5 (gray bars, n = 11); cells with either small outward currents or the smallest inward currents were grouped together. Mean peak current densities were 0.41 ± 0.43 and -0.14 ± 0.89 pA/pF at 3 and 4–5 d post-transfection, respectively. (C) Average integral of the ON component of the charge movement vs. voltage relationships in β1KO myotubes expressing YFP-β1aL134P 3 d (black circles, n = 3) and 4–5 d (gray circles, n = 5) post-transfection. Error bars represent ± SEM. Averaged data points were fit with Equation 1, with values of Qmax (2.7, 3.4) V1/2 (-1.4, -1.4 mV) and k (10.1, 16.9 mV) for YFP-β1aL134P day 3 and day 4–5, respectively. The fit of the averaged β1KO data from Figure 2E is shown as a reference (dashed line). (D) CaV1.1 did not co-localize with the juxtanuclear aggregates of β1aL134P. Two to three days after co-expression in β1KO myotubes, YFP-β1aL134P was typically present in presumed aggresomes whereas CaV1.1(726-CFP-727) was present in a reticular pattern. (E) Representative cell 5 d after transfection of β1KO myotubes with YN-β1aL134P-YC. No significant yellow fluorescence was seen with the same confocal settings used to acquire the image shown in A for YFP-β1aL134P at day 5 post-transfection. Similarly, no significant yellow fluorescence was seen for YN-β1aL134P-YC at day 3 post-transfection (not shown). In (A), (D) and (E), the red ellipses indicate approximate outlines of nuclei. Red scale bars represent 5 μm.
Although the L134P mutation caused a significant alteration in the subcellular distribution of β1a, it appeared not to entirely eliminate its ability to direct membrane expression of DHPRs. Thus, measureable Ca2+ current was present in one cell examined 3 d post-transfection with YFP-β1aL134P, and in half of all cells examined 4–5 d post-transfection (Fig. 4B), whereas no Ca2+ currents were detected in non-transfected β1KO myotubes (Fig. 1A and B). Figure 4C shows that the presence of L-type Ca2+ current in myotubes expressing YFP-β1aL134P can likely be attributed to increased DHPR surface expression as indicated by a small augmentation of charge movements at both 3 and 4–5 d post-transfection. Perhaps because the membrane targeting of DHPRs was very low, cells expressing YFP-β1aL134P did not contract when depolarized with 80 mM KCl (day 3, n = 4; day 4–5, n = 8; data not shown).
Whether or not the low level of DHPR expression in β1aL134P-transfected myotubes depended on association between β1aL134P and CaV1.1 was difficult to test directly. Figure 4D illustrates an experiment in which we co-transfected β1KO myotubes with YFP-β1aL134P and CaV1.1(726-CFP-727), in which CFP had been inserted between residues 726 and 727 of the II-III loop. As shown in Figure 4D at 2 d post-transfection, the YFP-β1aL134P was aggregated around nuclei. This pattern was not present for CaV1.1(726-CFP-727), which indicates that it does not bind strongly to the aggregated β1aL134P but also does not exclude the possibility that it binds, perhaps weakly, to β1aL134P that is more diffusely distributed in the cytoplasm.
Taken together, the fluorescence and electrophysiological data indicate that the L134P mutation altered the conformation of β1a such that its trafficking and function were affected. Importantly, this altered conformation could be directly detected by the absence of fluorescence complementation for the construct YN-β1aL134P-YC. In particular, we failed to detect yellow fluorescence above background in a total of eight dishes of β1KO myotubes from two separate primary cell cultures 3–5 d after transfection with YN-β1aL134P-YC (YFP-β1a and YFP-β1aL134P were transfected in parallel experiments as qualitative positive controls). A representative example of a myotube from a β1KO culture 5 d after transfection with YN-β1aL134P-YC is shown in Figure 4E, with the image obtained using the same confocal microscope settings as used for the YFP-β1aL134P image shown in the lower panel of Figure 4A. If it is assumed that subcellular trafficking of YN-β1aL134P-YC is similar to that of YFP-β1aL134P, these data support the idea that YN-β1aL134P-YC is present at both 3 and 5 d post-transfection, but that BiFC does not occur for the misfolded β1aL134P mutant.
Absence of BiFC in myotubes after co-expression of YN-β1a-YN and YC-β1a-YC
Because DHPRs in junctions of the plasma membrane with the sarcoplasmic reticulum are closely packed in arrays of tetrads,32 the punctate fluorescence observed in β1KO myotubes expressing YN-β1a-YC (Fig. 3A) could have arisen intramolecularly between N- and C-termini of individual β1a subunits and/or intermolecularly between N- and C-termini of subunits adjacent to one another in tetrads. Thus, we constructed YN-β1a-YN and YC-β1a-YC which after co-expression could only produce intermolecular fluorescence complementation. Specifically, if YN-β1a-YN and YC-β1a-YC assembled randomly into tetrads and produced intermolecular BiFC with the same efficiency as occurs for YN-β1a-YC in tetrads, then the total number of fluorescence complementation events per tetrad would be 50% of those occurring for YN-β1a-YC. Before testing this prediction, we performed control experiments in tsA201 cells that lack Ca2+ channel CaV1 and CaV2 subunits and would thus yield cytoplasmic localization of expressed β1a constructs.19
After transfection of tsA201 cells with YN-β1a-YC, yellow fluorescent cells were present, consistent with the notion that N- and C-termini of cytoplasmic β1a are closely apposed to one another, as well as with previous FRET measurements on β1a subunits in the cytosol of myotubes lacking CaV1.1.27 Surprisingly, however, we also observed fluorescent tsA201 cells after co-transfection with YN-β1a-YN plus YC-β1a-YC. Thus, intermolecular BiFC appeared to have occurred for YN-β1a-YN plus YC-β1a-YC perhaps due to the oligomerization of the CaVβ subunit that has been recently reported for heterologous expression systems that lack the CaV1 subunit.33,34 Another possibility is that the expression levels were sufficiently high in the tsA201 cells to allow non-specific YN–YC complementation. Indeed, complementation also occurred in tsA201 cells after expression of YN and YC fragments not attached to β1a, with the fraction of yellow cells (6/266) being about 14% of that observed (41/262) after transfection with intact YFP (data not shown). Such non-specific complementation appeared to occur with a lower incidence in β1KO myotubes, where co-transfection of YN and YC fragments yielded yellow fluorescence in 40 cells in five dishes that each contained hundreds of myotubes (two separate cultures) which was only about 4% of that observed after expression of YFP (338 cells in two dishes from two separate cultures).
Because the control experiments in tsA201 cells indicated that overexpression might lead to intermolecular BiFC between non-interacting proteins, we compared β1KO myotubes after transfection either with YN-β1a-YC (2 μl LT-1 and 1.5 μg cDNA per 35 mm dish) or with YN-β1a-YN plus YC-β1a-YC (2 μl LT-1 and 1.5 μg of each cDNA per 35 mm dish). In the myotubes transfected with YN-β1a-YC, yellow fluorescence was observed (Fig. 5, bottom left panel) in 12% of cells, whereas detectable yellow fluorescence was not observed (Fig. 5, bottom right panel) with the same confocal settings in eight dishes co-transfected with YN-β1a-YN plus YC-β1a-YC.
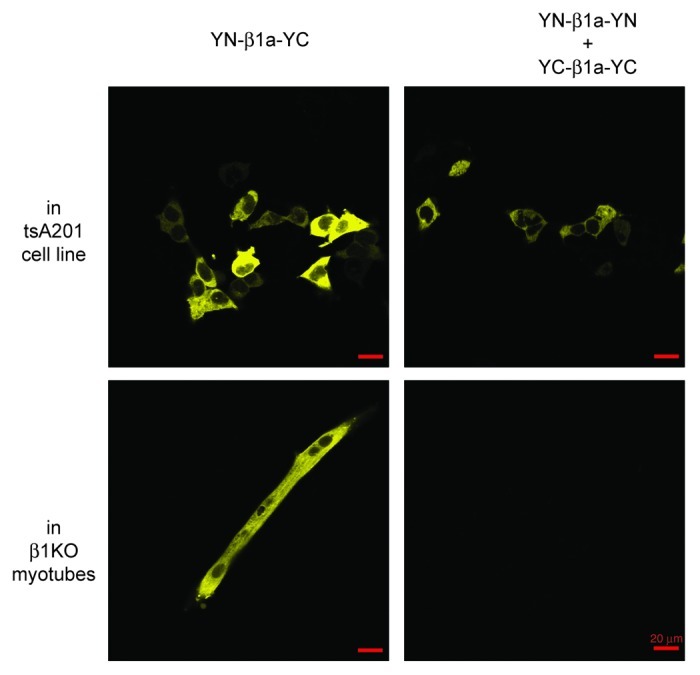
Figure 5. Lack of intermolecular BiFC between β1a subunits associated with CaV1.1 in junctional DHPRs. Top row: tsA201 cells were transfected either with YN-β1a-YC (left) or with YN-β1a-YN plus YC-β1a-YC (right). Yellow fluorescence indicated intramolecular (YN-β1a-YC) or intermolecular (YN-β1a-YN plus YC-β1a-YC) BiFC. Bottom row: after expression in β1KO myotubes, intramolecular BiFC occurred for YN-β1a-YC, but intermolecular BiFC did not occur for YN-β1a-YN plus YC-β1a-YC. All images were obtained at 10X (red scale bar indicates 20 μm) and were typical of the entire dish. The images in horizontal rows were captured with the same confocal settings. Note that at 10X, fluorescent puncta are not resolvable for YN-β1a-YC (compare the 63X image in Figure 3A, top panel).
The results above are consistent with the hypothesis that BiFC does not occur in myotubes co-transfected with YN-β1a-YN plus YC-β1a-YC because the organization of DHPRs in tetrads positions the N- and C-termini of adjacent β1a subunits too far apart for intermolecular fluorescence complementation to occur. If this is correct, then the yellow fluorescent puncta observed in β1KO myotubes transfected with YN-β1a-YC (Fig. 3A) must have arisen intramolecularly between N- and C-termini of individual β1a subunits. We thus tested whether intramolecular BiFC occurred for YN-β1a-YC co-expressed with CaV1.2 in dysgenic (CaV1.1-null) myotubes. Such co-expression resulted in yellow puncta preferentially localized near the surface (Fig. 6), as expected for CaV1.2-containing channels, which target to plasma membrane junctions with the SR.25,35 Importantly, CaV1.2 has substantial homology to CaV1.1, but does not assemble into tetrads.25 Thus, the yellow puncta illustrated in Figure 6 must have arisen from intramolecular BiFC and this is likely also to be the case for the yellow puncta observed when YN-β1a-YC is associated with endogenous CaV1.1 after transfection of β1KO myotubes (Fig. 3). Accordingly, β1a subunits appear to be fully functional when the N- and C-termini are positioned close enough together to permit fluorescence complementation of attached YFP fragments.
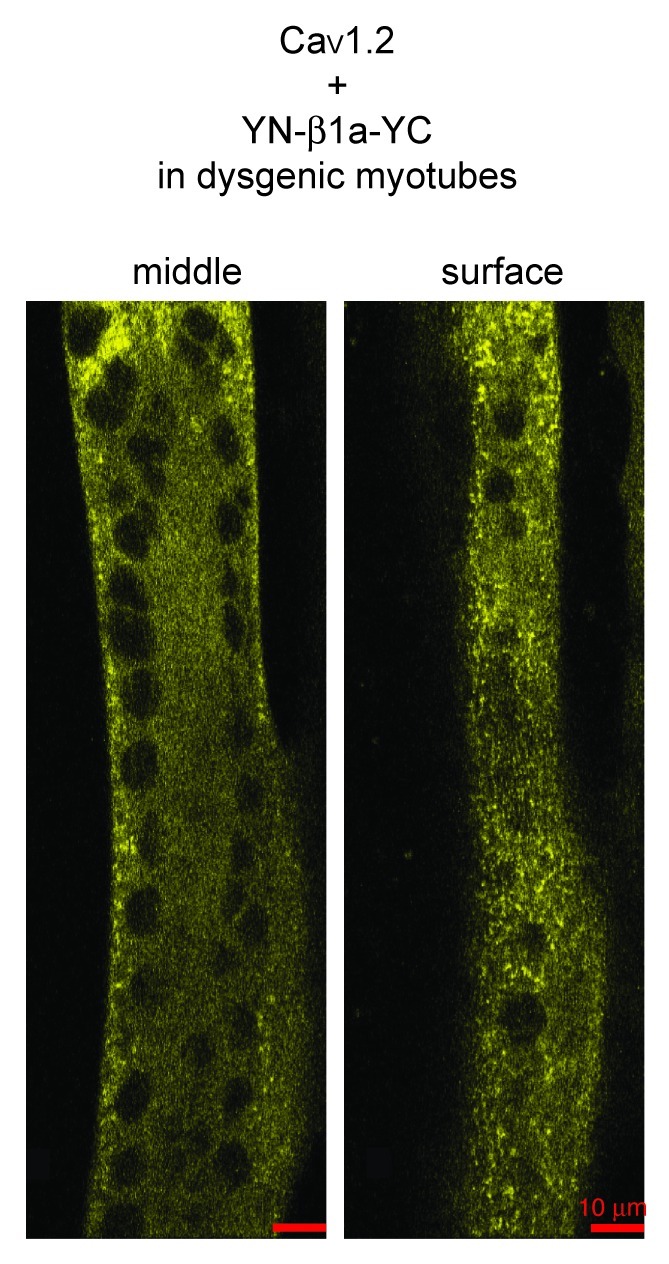
Figure 6. BiFC of YN-β1a-YC co-expressed with CaV1.2 in a dysgenic myotube, as indicated by the presence of yellow fluorescent puncta near the surface. Confocal sections near the middle (left) and surface (right) of the myotube are shown. Since CaV1.2 does not assemble into tetradic arrays,25 the presence of yellow puncta indicates that BiFC arose as a consequence of the close apposition of the N- and C-terminals of individual β1a subunits. Scale bar = 10 μm.
Association of β1a with CaV1.1 in junctional DHPRs does not strongly constrain the position of the N- and C-termini
Although the present work indicates that the ability of β1a to function is compatible with its N- and C-termini being in close apposition, previous results indicate that such close apposition is not a requirement for function. In particular, L-type current and EC coupling are supported by β1a subunits with a CFP-YFP tandem attached to either the N- or C-terminus,28 or with CFP attached to one terminus and YFP to the other.27 Additionally, EC coupling appears not be affected by the binding of streptavidin to either the N- or C-termini, respectively, of the constructs YFP-β1a-BAD or BAD-β1a-YFP.29 To obtain additional information about the positional restraints on the N- and C-termini of β1a, we tested whether gold-streptavidin conjugates could bind to N- or C-terminal biotin, using YFP fluorescence to establish the localization of the β1a subunits and Alexa568 or Alexa594 to determine sites at which the conjugates bound. Because of their large size, the conjugates had to be applied to cells that had either been permeabilized with saponin (Fig. 7A) or to cells that had been briefly fixed with paraformaldehyde and then permeabilized with Triton X-100 (Fig. 7B–D). Streptavidin conjugated to 1 nm gold appeared to have good access to the C-terminus (Fig. 7A) and N-terminus (not shown) of β1a in junctional DHPRs. Similarly, 5 nm gold-streptavidin was able to bind to both the N-terminus (Fig. 7B) and C-terminus (Fig. 7C) of β1a in junctional DHPRs. Testing the ability of the 10 nm gold conjugate to bind was problematic because in most instances there was poor penetration and/or diffusion. However, in a single cell (out of seven examined) there appeared to be clear binding to the β1a N-terminus (Fig. 7D). The ability of gold-streptavidin conjugates to bind provides evidence that neither the N-terminus nor C-terminus of β1a is constrained such that is closely opposed by other structures associated with junctional DHPRs.
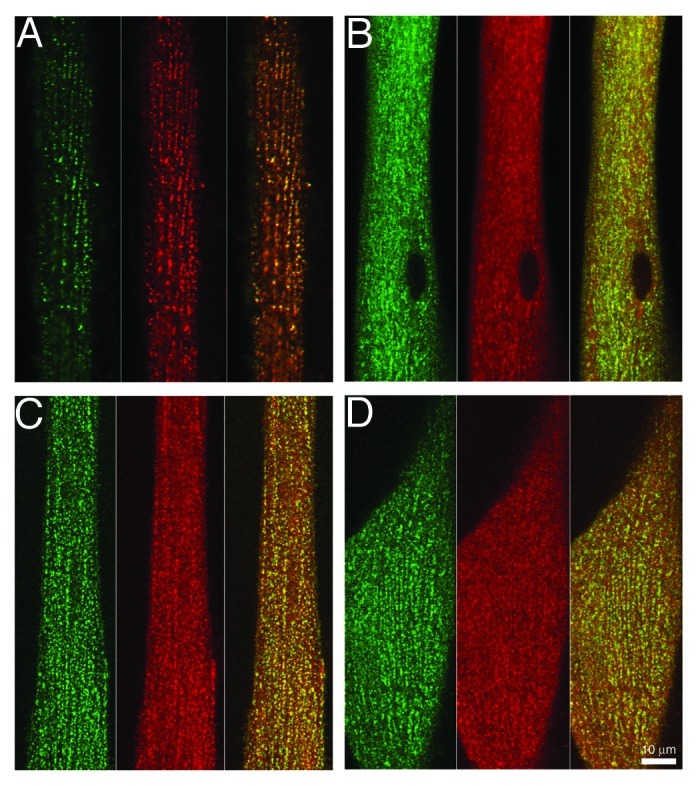
Figure 7. Large probes have access to the N- and C-termini of β1a in junctional DHPRs. β1KO myotubes were transfected with YFP-β1a-BAD (A) and (C) or with BAD-β1a-YFP (B) and (D). The cells were then either permeabilized by a 30 sec exposure to saponin (A) or fixed for 10 min with paraformaldehyde followed by permeabilization with Triton X-100 (C) and (D) and then exposed to 1 nm fluoro-nanogold streptavidin (A), 5 nm gold-streptavidin (B) and (C) or 10 nm gold-streptavidin (D). Except for the 1 nm gold-conjugate which had been pre-labeled with Alexa594, the position of the gold-streptavidin complexes (shown in red, with YFP fluorescence in green) was determined by subsequent labeling with biocytin-Alexa568. The YFP and streptavidin fluorescence images are shown superimposed in the rightmost panels of (A–D). Scale bar = 10 μm.
Discussion
In the current study, we fused non-fluorescent N- and C-terminal fragments of YFP (“YN” and “YC”), or the biotin acceptor domain (“BAD”), to the N- and/or C-termini of the calcium channel β1a subunit to gain insight into its structure and spatial organization within tetrad arrays (1) of CaV1.1-containing calcium channels (DHPRs) in skeletal muscle cells. Addition of the YFP or BAD fragments to β1a did not appear to interfere with β1a function (Figs. 1 and 2; refs. 26 and 27). We found (Fig. 3A) that expression of the construct YN-β1a-YC in β1KO myotubes resulted in the presence of numerous yellow fluorescent puncta, which are presumptive clusters in the plasma membrane of calcium channels that contain β1a and CaV1.1 and which are arranged as tetrads;1,25 these puncta were superposed on a background of diffuse yellow fluorescence, which likely represents β1a subunits not associated with CaV1.1.19,27 The fluorescence complementation was efficient since the fluorescence intensity of myotubes transfected with YN-β1a-YC was only slightly lower than that of myotubes transfected with YFP-β1a (Fig. 3B). To test whether the BiFC observed for YN-β1a-YC depended on the correct folding of β1a, we used the point mutation L134P which lies within the SH3 domain that is highly conserved between CaVβ isoforms.16,17 The L134P mutation strongly impaired the ability of β1a to direct the targeting and function of CaV1.1 (Fig. 4A–D), and abolished BiFC between the N- and C-termini (Fig. 4E), presumably due to a structural alteration of the β1a subunit. BiFC was not observed in β1KO myotubes co-transfected with YN-β1a-YN plus YC-β1a-YC (Fig. 5), which is consistent with the hypothesis that the arrangement of β1a subunits in DHPR tetrads positions the N- and C-termini too far apart to support inter-molecular complementation, and that the punctate fluorescence observed in myotubes transfected with YN-β1a-YC arose intra-molecularly within single β1a subunits. Furthermore, expression in dysgenic myotubes (null for CaV1.1) of YN-β1a-YC plus CaV1.2 resulted in fluorescent puncta (Fig. 6), which presumably were a consequence of intra-molecular BiFC since CaV1.2 produces clusters of channels that are not arranged as tetrads.25 Thus, when β1a is complexed with CaV1.2, and most likely with CaV1.1 as well, the N- and C-termini can closely appose one another. However, such close apposition does not appear to be obligatory because gold-streptavidin probes had access to the N- and C-termini of β1a co-assembled with CaV1.1 into DHPR clusters in myotubes (Fig. 7).
Although the presence of BiFC is consistent with there being a naturally occurring association between the sites to which the YFP fragments are attached, it was evident that BiFC could occur even when such an association did not seem likely. In particular, we found that yellow fluorescence was present in tsA201 cells co-transfected with YN-β1a-YN plus YC-β1a-YC (Fig. 5, top right). It is possible that this represented intermolecular BiFC dependent on oligomerization of the β1a subunits,33,34 but we also found that yellow fluorescence was present in ~2% of tsA201 cells co-transfected with YN plus YC (not shown). However, even though non-specific fluorescence complementation was an issue, it was nonetheless the case that, compared with tsA201 cells co-transfected with YN-β1a-YN plus YC-β1a-YC (Fig. 5, top right), the yellow fluorescence in cells transfected with YN-β1a-YC (Fig. 5, top left panel) was more frequent and brighter, as would be expected if close apposition can occur between the N- and C-termini of individual β1a subunits which are free in the cytoplasm and not complexed with a CaV subunit. Additionally, the existence of FRET between fluorescent proteins attached to the N- and C-termini of β1a constructs expressed in dysgenic myotubes27 provides further support that the N- and C-termini of non-complexed β1a subunits can be closely apposed.
A variety of evidence suggests that for β1a bound to CaV1.1, just as for free β1a, the N- and C-termini are able to approach one another closely, and that this accounts for the punctate fluorescence observed in β1KO myotubes transfected with YN-β1a-YC. Certainly, the hypothesis that there are no large differences between free and bound β1a in the disposition of the N- and C-termini is consistent with the crystallographic data showing that the conserved, core structure of β2a is very similar whether or not it is bound to the interacting domain of the CaV1.2 I-II loop.13 Indeed, we observed punctate fluorescence in dysgenic myotubes transfected with YN-β1a-YC plus CaV1.2. This punctate fluorescence strongly implies the occurrence of BiFC between N- and C-termini of individual β1a subunits bound to CaV1.2 because the alternative, BiFC between β1a subunits adjacent to one another, seems unlikely given the irregular disposition in the plasma membrane of CaV1.2 expressed in dysgenic myotubes.25
In addition to the “positive” evidence that close apposition can exist between the N- and C-termini of individual β1a subunits complexed with CaV subunits, both our current experiments, and previous work, provide “negative” evidence that close apposition does not occur between the termini of adjacent β1a subunits that are complexed with CaV1.1 in arrays of DHPR tetrads.25 In particular, the earlier work found that measurable FRET was present after expression in β1KO myotubes of YFP-β1a-CFP but not after expression of equimolar mixes of β1a subunits tagged on one terminus with YFP and on the other with CFP.27 Since the presence of FRET indicates that the fluorophores are separated by < 10 nm, the absence of FRET suggests that > 10 nm separates the termini of adjacent β1a subunits in DHPR tetrads. However, because this earlier study relied on modest changes in the intensity of cyan fluorescence after photo-bleaching of YFP, it could have failed to detect weak FRET interactions. Thus, further probing the spatial separation between the termini of adjacent β1a subunits was an important part of our current studies. For this, we co-transfected β1KO myotubes with YN-β1a-YN plus YC-β1a-YC because the use of double tagging, as opposed to single tagging, doubled the number of possible intermolecular complementation events, although the average number of possible complementation events for YN-β1a-YN plus YC-β1a-YC randomly assembled into DHPR tetrads would still be only half that of YN-β1a-YC. Nonetheless, if intermolecular BiFC had occurred, it should have been easily detectable given the bright, yellow fluorescent puncta present in β1KO myotubes after transfection with YN-β1a-YC. However, no fluorescence was detected in a total of eight dishes, containing several hundred myotubes, after transfection with YN-β1a-YN plus YC-β1a-YC. Of course, this test for intermolecular BiFC depended on the successful co-transfection of myotubes with both cDNA constructs. One argument that such co-transfection is likely to have occurred is that yellow fluorescence was present in ~12% of myotubes transfected with YN-β1a-YC (1.5 μg of cDNA per dish). Thus, ~1.5% of the β1KO myotubes co-transfected with YN-β1a-YN and YC-β1a-YC should have expressed both constructs even if the probability of successful transfection of a cell with one of the constructs was independent of transfection with the other construct; such independence seems unlikely given that the two cDNAs were pre-mixed (1.5 μg of each cDNA) prior to addition to the dishes. Additionally, we also examined myotubes that had been co-transfected with free YN plus β1a-YC (1.5 μg of each cDNA per dish). In eight dishes co-transfected with YN plus β1a-YC, yellow fluorescence was detectable in four cells (data not shown), which likely represents a minimal estimate for co-transfection efficiency since the production of yellow fluorescence depended upon both co-transfection of the two constructs and sufficient expression of free YN to produce non-specific fluorescence complementation with YC attached to the C-terminal of β1a. Finally, in experiments not shown, we found successful co-transfection of β1KO myotubes with YFP-β1a and CaV1.1(726-CFP-727). In these experiments, 29% of the yellow fluorescent cells were also fluorescent for cyan, and conversely, 93% of the cyan fluorescent cells also displayed yellow fluorescence, suggesting a high rate of co-transfection. Note that yellow fluorescent myotubes were more abundant than cyan fluorescent myotubes (in two dishes, there were 742 yellow cells and 232 cyan cells). This may be due to the fact that the β1a subunit is ~3–4 fold smaller than CaV1.1 (55 kD vs. 200 kD) and expresses more readily, or the fact that cells weakly expressing the CFP tag are difficult to detect. Whichever the case may be, we conclude that a subset of β1KO myotubes would surely have been co-transfected with YN-β1a-YN and YC-β1a-YC, and that the lack of YFP fluorescence reported is most likely due to the inability of the fragments to complement due to the distances between neighboring β1a subunits in a DHPR tetrad formation.
An important confirmation of the specificity of the BiFC produced by YN-β1a-YC was that this complementation was disrupted by the L134P mutation within the conserved SH3 domain of β1a (Fig. 4E). The absence of yellow fluorescence did not appear to be the result of a lack of expression because the construct YFP-β1aL134P produced substantial fluorescence, although with a much different subcellular distribution than the non-mutated construct, YFP-β1a. In particular, at 2–3 d post-transfection, YFP-β1aL134P was found in juxtanuclear aggregates (Fig. 4A, upper panel), which were never observed after comparable transfection of either YFP-β1a or YN-β1a-YC (Fig. 3A). This distribution of YFP-β1aL134P most likely represented its accumulation into aggresomes, which are cytoplasmic deposits of misfolded and/or overexpressed proteins that have exceeded the capacity of the proteasomal pathway.31 Presumably because the transient expression peaked and then declined, these juxtanuclear aggregates disappeared and YFP-β1aL134P became diffusely distributed by 4–5 d post-transfection (Fig. 4A, lower panel). However, YFP-β1aL134P never produced punctate fluorescence like that resulting from expression of YFP-β1a or YN-β1a-YC in β1KO myotubes. Despite greatly altering trafficking, the L134P mutation did not appear to eliminate membrane targeting entirely because expression of YFP-β1aL134P resulted in the presence of small inward calcium currents in half of all cells 4–5 d post-transfection (Fig. 4B), and caused charge movements to be slightly larger at both ~3 and 4–5 d after transfection than in control β1KO myotubes (Fig. 4C). With β1KO as a reference, a comparison of the magnitude of the charge restored by β1aL134P with that restored by wild-type β1a, indicates that β1aL134P was about 25% as effective as wild-type β1a in supporting membrane trafficking of CaV1.1. This is similar to the ability of β2a bearing the homologous mutation (L93P) to support expression of CaV1.2 in HEK293 cells.17 Interestingly, however, both β1aL134P and β2aL93P appeared to produce normal membrane expression of CaV2.1 in oocytes,16 although endogenously expressed β-subunits that have high sequence similarities to β3 (> 95%) may have contributed to this expression.36
Figure 8 presents a tentative model of the β1a subunit and its arrangement in DHPR tetrads. Because the SH3 and GK domains are highly conserved among all CaVβ subunits,15-17 it is assumed that their structure in β1a is like that determined crystallographically for β2a.13,14 This is indicated in Figure 8A by green α-helices and red β-strands for the SH3 domain, and by blue α-helices and orange β-strands for the GK domain. Because crystallographic information is not available for them, the divergent regions (D1, D3 and D5) are represented with black dots. Although the spatial dimensions and orientations of these divergent regions are presently unknown, we propose on the basis of our current study that the N- and C-termini of a β1a subunit can approach one another closely enough to allow complementation between YN and YC, resulting in the production of a fluorescent YFP molecule. Specifically, given that the fluorescent protein β-barrel is approximately 3 nm × 4 nm,37 and that the YN and YC fragments were each attached via a double glycine linker (~1 nm), it seems likely that the β1a N- and C-termini can be separated by as little as ~5 nm or less.

Figure 8. Models of β1a structure and arrangement in DHPR tetrads. (A) Ribbon structures of the conserved CaVβ core and inferred arrangement of the non-conserved N- and C-termini (D1 and D5, respectively) of β1a. β-sheets and α-helices are shown, respectively, for the SH3 domain (D2) in red and green, and for the GK domain (D4) in orange and blue; continuous gray lines indicate regions of determined structure that are neither α-helical nor β sheet. Regions without crystal structure are indicated by dotted black lines and the L134 mutation site is highlighted in cyan (see text). The results described in this paper indicate that the β1a N- and C-termini are able to allow YN and YC to complement one other and produce a yellow fluorescent protein (yellow ribbon structure). (B) Model for the arrangement of DHPRs within a tetrad (adapted from ref. 27) consistent with the present results on BiFC and gold-streptavidin binding. The RyR1 homotetramer is represented by the black square. Blue ovals represent the entire DHPR complex, including CaV1.1 and the auxiliary subunits β1a, α2-δ1, and γ1. β1a is represented as dark gray ovals with the N and C termini close enough for YN and YC to complement one other and produce a yellow fluorescent protein (YFP, in yellow; shown for three of the four DHPRs). For one DHPR in the tetrad, a position of the N- and C-termini consistent with the binding of gold-streptavidin (gold circle) to YFP-β1a-BAD or to BAD-β1a-YFP is shown. The dotted black circle in the center acts as a reference with a diameter of ~10 nm. (C) DHPR array in which tetrads are opposed to every other RyR1 (dark and light black squares). In myotubes, these arrays may comprise several rows of RyR1.43
Electron microscopic analysis of frozen, hydrated protein purified from skeletal muscle indicates that the DHPR, viewed from the extracellular space, has an oval profile with dimensions of about 13 nm × 15 nm,38 or a little smaller.39 In principle, this information, combined with that on the spatial arrangement of tetradic particles in relationship to the underlying lattice of ryanodine receptors,1 provides an initial framework for assigning the placement of β1a within tetrad arrays. However, the relationship between the individual tetradic particles viewed in freeze-fracture replicas and the DHP structures revealed by single particle analysis remains elusive. In particular, the α2-δ1 subunit should be a major component of the DHPR analyzed by single particle analysis (constituting ~40% of the total mass), but the nearly complete ablation of α2-δ1 has no discernible effect on the size or distribution of tetradic particles.40 Thus, alternative models are possible for the arrangement of DHPRs in tetrads.38,39 Nonetheless, our current results provide a useful constraint for assigning the position of β1a, as illustrated in Figure 8B, which shows an arrangement of DHPRs (denoted by dark blue, oval outlines) relative to RyR1 (black square) similar to that proposed by Wolf et al.38 Within the DHPR, β1a is indicated by the gray oval which has dimensions that would accommodate the CaVβ core structure. For three of the four tetradic DHPRs, β1a is shown in Figure 8B such that BiFC can occur between YN and YC in the construct YN-β1a-YC, but, as shown for the fourth tetradic DHPR, the β1a N- and C-termini have sufficient positional freedom to allow the binding of gold-streptavidin conjugates to BAD-β1a-YFP and to YFP-β1a-BAD in fixed, permeabilized myotubes (Fig. 7). However, the positioning of the β1a subunit appears to be such that this positional freedom is not sufficient to allow BiFC to occur between β1a in DHPRs adjacent to one another in tetrads. Figure 8C illustrates additional constraints: that BiFC not occur between the termini of β1a subunits in neighboring tetrads and that DHPRs in neighboring tetrads not occlude the binding of gold-streptavidin conjugates; these conditions would be violated if the β1a N- and C-termini were shifted ~30° counter-clockwise.
In conclusion, our current results provide constraints that can be used to test models for the positioning of β1a in DHPR tetrads. More generally, the use of BiFC appears to be a useful tool for analyzing the positioning of domains that are sufficiently disordered that they do not produce crystal structures. In the case of CaVβ subunits, the sequence differences in D1, D3 and D5 are likely not only to result in different structures but also to result in the functional differences that have been described for the different β-subunits.41 In future experiments, it will be of interest to determine whether YFP fragments attached to divergent domains of different β-subunits result in BiFC like that we have found for these fragments attached to the N- and C-termini of β1a.
Materials and Methods
cDNA constructs
cDNAs encoding β1a with enhanced yellow fluorescent protein (YFP) and a biotin acceptor domain (BAD) fused to the N- or C-termini (YFP-β1a-BAD, BAD-β1a-YFP) were constructed as described previously.26 YN (YFP amino acids 1–158) and YC (YFP amino acids 159–238) cDNA constructs were originally created by the laboratory of Dr Catherine H. Berlot and described previously.23 Rabbit β1a (GenBankTM number M25514) was previously cloned into pEYFP-C1 (Clontech) and acted as our control.27 To create YN-β1a-YC, we first made YN fused to an N-terminal fragment of β1a (YN-β1afrag) and a C-terminal fragment of β1a fused to YC (β1afrag-YC) with a two-step PCR method. In the first step, a unique XhoI site and double glycine linker were introduced onto the N- and C-termini of YN, respectively, via PCR. The primers used were 1, 5′CTCGAGATGGTGAGCAAGGGC3′ and 2, 5′GACCATTCCGCCCTGCTTGTCGGCCATGATATAGAC3′. The N-terminal β1a fragment was created with the primers 3, 5′AAGCAGGGCGGAATGGTCCAGAAGACCAGC3′ and 4, 5′CTGCAGCAGGCGCAGGCTGTCC3′ and ended at the unique PstI site at nucleotide position 516 of β1a. In the second step, the two fragments were fused together via multiple denaturing and annealing cycles and then amplified with the addition of primers 1 and 4. Likewise, we introduced a double glycine linker to the N-terminus and a stop codon and unique NotI site to the C-terminus of YC with the primers 5, 5′GCCATGGGGGGTAAGAACGGCATCAAGG3′ and 6, 5′GCGGCCGCTCACTTGTACAGCTCGTCCATGC3′. The primers used to create the C-terminal β1a fragment which began at the unique PstI site were 7, 5′CTGCAGGAACAGAAGCTGCGTCAGAGC3′ and 8, 5′GTTCTTACCCCCCATGGCATGTTCCTGC3′. The two fragments were fused together via multiple denaturing and annealing cycles and then amplified with the addition of primers 6 and 7. Both YN-β1afrag and β1afrag-YC fusion fragments were subcloned into the pCR2.1 vector (Invitrogen, Grand Island, NY) and sequenced. Correct fragments were excised with either XhoI and PstI (YN-β1afrag) or PstI and NotI (β1afrag -YC) and ligated into a similarly XhoI and NotI digested unlabeled β1a parent construct27 for the final YN-β1a-YC construct, or a PstI and NotI digested β1a parent construct for the final β1a-YC construct.
The same two-step PCR method was utilized to create both YN-β1a-YN and YC-β1a-YC. A β1afrag -YN fusion fragment was created with primers 7 (above) and 9 5′CACCATACCCCCCATGGCATGTTCCTGC3′ with β1a as a template and primers 10, 5′GCCATGGGGGGTATGGTGAGCAAGGGC3′ and 11, 5′GCGGCCGCTCACTGCTTGTCGGCCATG3′ with YN as a template. These two PCR fragments were fused together via multiple denaturing and annealing cycles and then amplified with the addition of primers 7 and 11. The β1afrag-YN fusion fragment was subcloned into pCR2.1 and sequenced. Correct fragments were excised with PstI and NotI and ligated into a similarly digested YN-β1a-YC parent construct to create the final YN-β1a-YN construct. A YC-β1afrag fusion fragment was created with primers 4 (above) and 12, 5′TACAAGGGCGGAATGGTCCAGAAGACC3′with β1a as a template and primers 13, 5′CTCGAGATGAAGAACGGCATCAAGG3′ and 14, 5′GACCATTCCGCCCTTGTACAGCTCGTCC3′ with YC as a template. These two PCR fragments were fused together via multiple denaturing and annealing cycles and then amplified with the addition of primers 4 and 13. The YC-β1afrag fragment was subcloned into pCR2.1 vector and sequenced. Correct fragments were excised with XhoI and PstI and ligated into a similarly digested YN-β1a-YC parent construct to create the final YC-β1a-YC construct.
YFP-β1aL134P and YN-β1aL134P-YC were created by using the QuikChange® II site-directed mutagenesis kit (Stratagene) using the primers 5′GCCCAAGGACTTCCCGCACATCAAGGAGAAAT3′ and 5′ATTTCTCCTTGATGTGCGGGAAGTCCTTGGGC3′.
To create CaV1.1(726-CFP-727), we initially engineered a unique SpeI site between the triplets encoding for Glu726 and Ser727 via site-directed PCR mutagenesis in a parent YFP-CaV1.1 construct. Next, we created a SpeI-CFP-SpeI fragment using a modified eCFP-C1 (Cerulean) as a template and using the primers 5′ACTAGTGGTGGAGGTGGAAGTGGAGGTGGAGGTACCATGGTGAGCAAGGGCGAGG3′ and 5′ACTAGTACCACCTCCTCCAGATCCTCCACCACCCTTGTACAGCTCGTCCATGCCG3′. This CFP fragment was subcloned into pCR2.1 and sequenced. Correct fragments were excised with SpeI and ligated into the similarly digested YFP-CaV1.1–726 SpeI parent construct to create a YFP-CaV1.1(726-CFP-727) construct. The N-terminal YFP was then excised with AgeI and the enzyme cuts were allowed to self-ligate to produce the final CaV1.1(726-CFP-727) construct. A similar construct, CaV1.1(726-YFP-727), was previously shown to be functional for both EC coupling and L-type Ca2+ current.42
All subcloned fragments and final constructs were sequenced at the DNA Sequencing and Analysis Core of the CU Cancer Center at the University of Colorado Anschutz Medical Campus. All restriction enzymes were purchased from New England Biolabs.
Cell culture and cDNA transfection
Primary cultures of β1KO skeletal myotubes4 were prepared from limbs of E18 fetuses, as described previously43 in accordance with NIH guidelines of the Institutional Animal Care and Use Committee, University of Colorado Anschutz Medical Campus. Briefly, the dissected muscles were digested in 0.125% trypsin (w/v in rodents’ Ringers comprised of (in millimolars): 146 NaCl, 5 KCl, 11 glucose and 10 HEPES, pH 7.4 with NaOH) with gentle shaking at 37°C for 20–25 min. After centrifugation, mononucleated cells were resuspended in plating medium containing Dulbeccos’s modified Eagle’s medium with high glucose (Mediatech) 10% horse serum, and 10% fetal bovine serum, and plated on 35 mm BD Falcon® PrimariaTM plastic culture dishes (Becton Dickinson) or glass coverslip dishes (MatTek) coated with ECL (Upstate Biotechnology) at a density of ~1.5 × 103 cells per dish. Cultures were grown in a humidified incubator at 37°C in 5% CO2. After myoblast fusion had begun (4–6 d), the plating medium was replaced with a differentiation medium (Dulbeccos’s modified Eagle’s medium with high glucose, supplemented with 2% horse serum). Except as noted below, cDNA transfection was performed during the myoblast fusion stage with the polyamine TransIT®-LT1 transfection reagent (Mirus Bio). Cells were exposed to a serum free, OptiMemI (Invitrogen) transfection solution for 2–4 h containing TransIT®-LT1 and the cDNA of interest at a 2–6 μl:1.5 μg ratio per 35 mm dish. For co-transfections with two constructs, each 35 mm dish was transfected with a mix containing 1.5 μg each of both constructs.
All other transfections were performed ~2 d after the change to differentiation medium. Nuclear injection was used for transfection with YN-β1a-YC (15 ng/μl) plus CaV1.2 (50 ng/μl). Transfection with YFP-β1a-BAD or BAD-β1a-YFP was performed either by nuclear injection (5–100 ng/μl) (26) of the cDNAs or by exposure for 2 h to HSV virions (2 × 104 particles/ml; kindly provided by Dr P.D. Allen) containing the cDNAs.44 The cells were then changed into a medium in which the biotin present in the 2% horse serum was supplemented with an additional 1 μM biotin.
tsA201 cells (ECACC) were initially plated on 10 cm Petri dishes (Fisher Scientific) and passaged a minimum of three times prior to experiments. For experiments, the cells were then plated at a density of 3.0 × 105 on 35 mm BD Falcon® plastic culture dishes (Becton Dickinson) in media comprised of Dulbeccos’s modified Eagle’s medium with high glucose and 10% fetal bovine serum. On day 2, cells were transfected with 3 μl of the Lipofectamine 2000 reagent (Invitrogen) and 1.0 μg of the cDNA of interest per 35 mm dish. For co-transfections, each 35 mm dish was transfected with 1.0 μg each of both constructs. On day 3, each dish of transfected tsA201 cells was split and plated on four separate 35 mm glass coverslip dishes (MatTek). On day 4, cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) buffer for 20 min, rinsed, and stored at 4°C for future imaging experiments.
Measurement of ionic currents and intramembrane charge movement
Whole cell voltage clamp analysis of Ca2+ currents and intramembrane charge movements was performed 3–5 d after transfection as described previously.27-29,32 The “external solution” was comprised of sterile filtered (in mM): 145 TEA-Cl, 10 CaCl2, 0.003 tetrodotoxin, and 10 HEPES, pH 7.4 with TEA-OH. The “internal solution” consisted of sterile filtered (in millimolars): 140 CsAsp, 10 Cs2EGTA, 5 MgCl2, and 10 HEPES, pH 7.3 with CsOH. Ionic currents were blocked for intramembrane charge movement studies with addition of 0.5 mM CdCl2 and 0.1 mM LaCl3 (final concentrations) to the external solution. Allmillimolarscharge movements were corrected for linear cell capacitance and leak current using a –P/8 prepulse subtraction protocol (Vhold = -80 mV). Plots of peak calcium current (I) vs. test potential (V) were converted to conductance vs. potential according to
G(V) = I/(V-Vrev) (Eqn. 1)
where the reversal potential (Vrev) was determined visually from the peak I-V plot. The voltage dependence of G and the integral of the ON transient (Qon) for each test potential (V) were fit according to Equation 2:
A = Amax / {1+exp -[(V-V1/2) / k]} (Eqn. 2)
where Amax is the maximal Gmax or Qmax, V1/2 is the potential causing activation of half-maximal conductance or movement of half-maximal charge, and k is a slope parameter. Data analysis and traces for presentation was performed with SigmaPlot 10.0 software (Systat). Figures were arranged for final presentation with Adobe Illustrator CS4.
Application of gold-streptavidin
Gold-conjugated streptavidin was introduced into cells two days after transfection with cDNA for YFP-β1a-BAD or BAD-β1a-YFP. For experiments with 1 nm gold, the cells were permeabilized for 30 sec with saponin (Sigma; 12 μg/ml) in internal solution (composition given above). Following wash in internal solution, cells were incubated in Alexa594-nanogold streptavidin (Invitrogen, 1:500 in internal solution) for 30 min. Excess gold-streptavidin was removed by washing cells with internal solution for 30 min, followed by post-fixation with 4% paraformaldehyde (in PBS). For the 5 or 10 nm gold-streptavidin conjugates (British BioCell Intl.), which likely exceed the size of the pores made by saponin,45 myotubes were first fixed with 4% paraformaldehyde for 10 min, and then permeabilized with Triton X-100 (Sigma; 0.1% in PBS) for 30 min. The cells were then incubated with 5 or 10 nm gold-streptavidin (1:100 in PBS) for 2–12 h followed by incubation with biocytin-Alexa568 (Invitrogen; 1:20,000) for 30 min to fluorescently label the streptavidin-gold conjugate.
Confocal fluorescence microscopy
Cells were imaged with a LSM 510 META laser scanning inverted confocal microscope (Zeiss) in rodents’ Ringers comprised of (in mM): 146 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 11 glucose, and 10 HEPES, pH 7.4 with NaOH. For analysis of yellow or cyan fluorescence, excitation was via a 514 or 458 nm, respectively, line of an argon laser, and directed to the cell via a 458/514 nm dual dichroic mirror. The emitted fluorescence was directed to a photomultiplier equipped with a 530 nm long-pass filter for YFP or a 465–495 band-pass filter for CFP. For analysis of both YFP and Alexa568 or Alexa594, the excitation/emission parameters were: YFP, 488 nm excitation via a 488/543 nm dual dichroic, 505–530 nm band-pass emission filter; Alexa568 or Alexa594: 543 nm excitation via a 488/543 nm dual dichroic, 560 nm long-pass emission filter. Images were acquired as the average of four to eight line scans per pixel and digitized at 8 bits.
All data are reported as the mean ± SEM, unless otherwise noted. Statistical significance was tested with an unpaired Student’s t-test.
KCl membrane depolarization and myotube contractions
The ability of the β1a constructs to support EC coupling after expression in β1KO myotubes was assessed by the presence of evoked contractions in response to microperfusion with rodents’ Ringers in which K+ was increased to 80 mM by equimolar replacement of Na+. The solution was applied via a glass micropipette placed near the cell and controlled by a Picospritzer II (General Valve Co.) set at 5 sec and ~3 psi. Images of the myotubes were acquired at a rate of ~11 Hz and digitized at 8 bits. The contractions were recorded as the movement of a small region of interest on the edge of the myotube.
Protein modeling
Sequences for rabbit β1a and YFP were submitted to the Swiss-Model website at http://swissmodel.expasy.org/workspace. Results were manipulated with the DeepView (Swiss Pdb-View) program.46-49 Final models were arranged in Adobe Photoshop 7.0 and Adobe Illustrator CS4 for figure presentation. The domains of the β1a subunit were oriented to correspond to the published crystal structure of the CaVβ2a subunit.13
Acknowledgments
The authors of the study wish to thank Catherine H. Berlot, MD, PhD (Weis Center for Research at the Geisinger Clinic) for permission to use the YFP-N (YN) and YFP-C (YC) cDNA constructs, Robert Dirksen PhD (University of Rochester Medical Center) for the initial idea to employ the BiFC technique and John Lueck PhD (University of Iowa) for information pertaining to the YN and YC constructs. This study was supported by grants from the Muscular Dystrophy Association (Development Grant MDA4334 to D.C.S. and Research Grant MDA 176448 to K.G.B.) and the National Institutes of Health (AR44750 and AR055104 to K.G.B.). A preliminary version of this work was published as an abstract and presented at a platform session: D.C. Sheridan and K.G. Beam, Abstract 27-Plat, 52nd Annual Meeting of the Biophysical Society, Long Beach, CA, February 2–6, 2008.
Glossary
Abbreviations:
- DHPR
dihydropyridine receptor
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum
- EC
excitation-contraction
- BiFC
bimolecular fluorescence complementation
- FRET
fluorescence resonance energy transfer
- YFP
yellow fluorescent protein
- YN
YFP-N
- YC
YFP-C
- KO
knock-out
- CFP
cyan fluorescent protein
- BAD
biotin acceptor domain
- PBS
phosphate-buffered saline
- Ca2+
calcium
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/18916
References
- 1.Block BA, Imagawa T, Campbell K, Franzini-Armstrong C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol. 1988;107:2587–600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Obermair GJ, Tuluc P, Flucher BE. Auxiliary Ca2+ channel subunits: lessons learned from muscle. Curr Opin Pharmacol. 2008;8:311–8. doi: 10.1016/j.coph.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 3.Arikkath J, Campbell KP. Auxiliary subunits: Essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol. 2003;13:298–307. doi: 10.1016/S0959-4388(03)00066-7. [DOI] [PubMed] [Google Scholar]
- 4.Beam KG, Horowicz P. Excitation-contraction coupling in skeletal muscle. Myology 2004; AG Engel and C Franzini-Armstrong, editors. McGraw Hill, New York. 257-280. [Google Scholar]
- 5.Gregg RG, Messing A, Strube C, Beurg M, Moss R, Behan M, et al. Absence of the beta subunit (cchb1) of the skeletal muscle dihydropyridine receptor alters expression of the alpha1 subunit and eliminates excitation-contraction coupling. Proc Natl Acad Sci USA. 1996;93:13961–6. doi: 10.1073/pnas.93.24.13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strube C, Beurg M, Powers PA, Gregg RG, Coronado R. Reduced Ca2+ current, charge movement, and absence of Ca2+ transients in skeletal muscle deficient in dihydropyridine receptor beta1 subunit. Biophys J. 1996;71:2531–43. doi: 10.1016/S0006-3495(96)79446-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanabe T, Beam KG, Adams BA, Niidome T, Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 1990;346:567–9. doi: 10.1038/346567a0. [DOI] [PubMed] [Google Scholar]
- 8.Pragnell M, DeWaard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- 9.Hofmann F, Biel M, Flockerzi V. Molecular basis for Ca2+ channel diversity. Annu Rev Neurosci. 1994;17:399–418. doi: 10.1146/annurev.ne.17.030194.002151. [DOI] [PubMed] [Google Scholar]
- 10.Perez-Reyes E, Schneider T. Calcium channels: Structure, function, and classification. Drug Dev Res. 1994;33:295–318. doi: 10.1002/ddr.430330311. [DOI] [Google Scholar]
- 11.Beurg M, Sukhareva M, Ahern CA, Conklin MW, Perez-Reyes E, Powers PA, et al. Differential regulation of skeletal muscle L-type Ca2+ current and excitation-contraction coupling by the dihydropyridine receptor beta subunit. Biophys J. 1999;76:1744–56. doi: 10.1016/S0006-3495(99)77336-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Opatowsky Y, Chen CC, Campbell KP, Hirsch JA. Structural analysis of the voltage-dependent calcium channel β subunit functional core and its complex with the α1 interaction domain. Neuron. 2004;42:387–99. doi: 10.1016/S0896-6273(04)00250-8. [DOI] [PubMed] [Google Scholar]
- 13.Van Petegem F, Clark KA, Chatelain FC, Minor DL. Structure of a complex between a voltage-gated calcium channel β subunit and an α subunit domain. Nature. 2004;429:671–5. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen YH, Li MH, Zhang Y, He LI, Yamada Y, Fitzmaurice A, et al. Structural basis of the α1-β subunit interaction of voltage-gated Ca2+ channels. Nature. 2004;429:675–80. doi: 10.1038/nature02641. [DOI] [PubMed] [Google Scholar]
- 15.Hanlon MR, Berrow NS, Dolphin AC, Wallace BA. Modeling of a voltage-dependent Ca2+ channel beta subunit as a basis for understanding its functional properties. FEBS Lett. 1999;445:366–70. doi: 10.1016/S0014-5793(99)00156-8. [DOI] [PubMed] [Google Scholar]
- 16.McGee AW, Nunziato DA, Maltez JM, Prehoda KE, Pitt GS, Bredt DS. Calcium channel function regulated by the SH3-GK module in β subunits. Neuron. 2004;42:89–99. doi: 10.1016/S0896-6273(04)00149-7. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi SX, Miriyala J, Colecraft HM. Membrane-associated guanylate kinase-like properties of β subunits required for modulation of voltage-dependent Ca2+ channels. Proc Natl Acad Sci USA. 2004;101:7193–8. doi: 10.1073/pnas.0306665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chien AJ, Zhao XL, Shirokov RE, Puri TS, Chang CF, Sun K, et al. Roles of a membrane-localized β subunit in the formation and targeting of functional L-type Ca2+ channels. J Biol Chem. 1995;270:30036–44. doi: 10.1074/jbc.270.50.30036. [DOI] [PubMed] [Google Scholar]
- 19.Neuhuber B, Gerster U, Mittendorfer J, Glossmann H, Flucher BE. Differential effects of Ca2+ channel β1a and β2a subunits on complex formation with α1S and on current expression in tsA201 cells. J Biol Chem. 1998;273:9110–8. doi: 10.1074/jbc.273.15.9110. [DOI] [PubMed] [Google Scholar]
- 20.Beurg M, Ahern CA, Vallejo P, Conklin MW, Powers PA, Gregg RG, et al. Involvement of the carboxyl-terminus region of the dihydropyridine receptor β1a subunit in excitation-contraction coupling of skeletal muscle. Biophys J. 1999;77:2953–67. doi: 10.1016/S0006-3495(99)77128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheridan DC, Cheng W, Ahern CA, Mortenson L, Alsammarae D, Vallejo P, et al. Truncation of the carboxyl terminus of the dihydropyridine receptor β1a subunit promotes Ca2+ dependent excitation-contraction coupling in skeletal myotubes. Biophys J. 2003;84:220–37. doi: 10.1016/S0006-3495(03)74844-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheridan DC, Cheng W, Carbonneau L, Ahern CA, Coronado R. Involvement of a heptad repeat in the carboxyl terminus of the dihydropyridine receptor β1a subunit in the mechanism of excitation-contraction coupling in skeletal muscle. Biophys J. 2004;87:929–42. doi: 10.1529/biophysj.104.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hynes TR, Tang L, Mervine SM, Sabo JL, Yost EA, Devreotes PN, et al. Visualization of G protein βγ dimers using bimolecular fluorescence complementation demonstrates roles for both β and γ in subcellular targeting. J Biol Chem. 2004;279:30279–86. doi: 10.1074/jbc.M401432200. [DOI] [PubMed] [Google Scholar]
- 24.Kerppola TK. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat Protoc. 2006;1:1278–86. doi: 10.1038/nprot.2006.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takekura H, Paolini C, Franzini-Armstrong C, Kugler G, Grabner M, Flucher BE. Differential contribution of skeletal and cardiac II-III loop sequences to the assembly of dihydropyridine-receptor arrays in skeletal muscle. Mol Biol Cell. 2004;15:5408–19. doi: 10.1091/mbc.E04-05-0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lorenzon NM, Haarmann CS, Norris EE, Papadopoulos S, Beam KG. Metabolic biotinylation as a probe of supramolecular structure of the triad junction in skeletal muscle. J Biol Chem. 2004;279:44057–64. doi: 10.1074/jbc.M405318200. [DOI] [PubMed] [Google Scholar]
- 27.Leuranguer V, Papadopoulos S, Beam KG. Organization of calcium channel beta1a subunits in triad junctions in skeletal muscle. J Biol Chem. 2006;281:3521–7. doi: 10.1074/jbc.M509566200. [DOI] [PubMed] [Google Scholar]
- 28.Papadopoulos S, Leuranguer V, Bannister RA, Beam KG. Mapping sites of potential proximity between the dihydropyridine receptor and RyR1 in muscle using a cyan fluorescent protein–yellow fluorescent protein tandem as a fluorescence resonance energy transfer probe. J Biol Chem. 2004;279:44046–56. doi: 10.1074/jbc.M405317200. [DOI] [PubMed] [Google Scholar]
- 29.Lorenzon NM, Beam KG. Accessibility of targeted DHPR sites to streptavidin and functional effects of binding on EC coupling. J Gen Physiol. 2007;130:379–88. doi: 10.1085/jgp.200609730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGee AW, Bredt DS. Identification of an intramolecular interaction between the SH3 and guanylate kinase domains of PSD-95. J Biol Chem. 1999;274:17431–6. doi: 10.1074/jbc.274.25.17431. [DOI] [PubMed] [Google Scholar]
- 31.Johnston JA, Ward CL, Kopito RR. Aggresomes: A cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–98. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheridan DC, Takekura H, Franzini-Armstrong C, Beam KG, Allen PD, Perez CF. Bidirectional signaling between calcium channels of skeletal muscle requires multiple direct and indirect interactions. Proc Natl Acad Sci USA. 2006;103:19760–5. doi: 10.1073/pnas.0609473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lao QZ, Kobrinsky E, Liu Z, Soldatov NM. Oligomerization of CaVβ subunits is an essential correlate of Ca2+ channel activity. FASEB J. 2010;24:5013–23. doi: 10.1096/fj.10-165381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miranda-Laferte E, Gonzalez-Gutierrez G, Schmidt S, Zeug A, Ponimaskin EG, Neely A, et al. Homodimerization of the Src homology 3 domain of the calcium channel β-subunit drives dynamin-dependent endocytosis. J Biol Chem. 2011;286:22203–10. doi: 10.1074/jbc.M110.201871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grabner M, Dirksen RT, Beam KG. Tagging with green fluorescent protein reveals a distinct subcellular distribution of L-type and non-L-type Ca2+ channels expressed in dysgenic myotubes. Proc Natl Acad Sci USA. 1998;95:1903–8. doi: 10.1073/pnas.95.4.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tareilus E, Roux M, Qin N, Olcese R, Zhou J, Stefani E, et al. A Xenopus oocyte β subunit: Evidence for a role in the assembly/expression of voltage-gated calcium channels that is separate from its role as a regulatory subunit. Proc Natl Acad Sci USA. 1997;94:1703–8. doi: 10.1073/pnas.94.5.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang F, Moss LG, Phillips GN. The molecular structure of green fluorescent protein. Nat Biotechnol. 1996;14:1246–51. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- 38.Wolf M, Eberhart A, Glossmann H, Striessnig J, Grigorieff N. Visualization of the domain structure of an L-type Ca2+ channel using electron cryo-microscopy. J Mol Biol. 2003;332:171–82. doi: 10.1016/S0022-2836(03)00899-4. [DOI] [PubMed] [Google Scholar]
- 39.Serysheva II, Ludtke SJ, Baker MR, Chiu W, Hamilton SL. Structure of the voltage-gated L-type Ca2+ channel by electron cryomicroscopy. Proc Natl Acad Sci USA. 2002;99:10370–5. doi: 10.1073/pnas.162363499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gach MP, Cherednichenko G, Haarmann C, Lopez JR, Beam KG, Pessah IN, et al. α2δ1 dihydropyridine receptor subunit is a critical element for excitation-coupled calcium entry but not for formation of tetrads in skeletal myotubes. Biophys J. 2008;94:3023–34. doi: 10.1529/biophysj.107.118893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buraei Z, Yang J. The β subunit of voltage-gated Ca2+ channels. Physiol Rev. 2010;90:1461–506. doi: 10.1152/physrev.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bannister RA, Papadopoulus S, Haarmann CA, Beam KG. Effects of inserting fluorescent proteins into the alpha1S II-III loop: Insights into excitation-contraction coupling. J Gen Physiol. 2009;134:35–51. doi: 10.1085/jgp.200910241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beam KG, Franzini-Armstrong C. Functional and structural approaches to the study of excitation-contraction coupling. Methods Cell Biol. 1997;52:283–306. doi: 10.1016/S0091-679X(08)60384-2. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Fraefel C, Protasi F, Moore RA, Fessenden JD, Pessah IN, et al. HSV-1 amplicon vectors are a highly efficient gene delivery system for skeletal muscle myoblasts and myotubes. Am J Physiol Cell Physiol. 2000;278:C619–26. doi: 10.1152/ajpcell.2000.278.3.C619. [DOI] [PubMed] [Google Scholar]
- 45.Bangham AD, Horne RW, Glauert AM, Dingle JT, Lucy JA. Action of saponin on biological cell membranes. Nature. 1962;196:952–5. doi: 10.1038/196952a0. [DOI] [PubMed] [Google Scholar]
- 46.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–5. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 48.Kopp J, Schwede T. The SWISS-MODEL repository of annotated three-dimensional protein structure homology models. Nucleic Acids Res. 2004;32:D230–4. doi: 10.1093/nar/gkh008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modeling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]