

Abstract
Antimicrobial peptides (AMPs) have been proposed as a promising new class of antimicrobials despite warnings that therapeutic use could drive the evolution of pathogens resistant to our own immunity peptides. Using experimental evolution, we demonstrate that Staphylococcus aureus rapidly evolved resistance to pexiganan, a drug-candidate for diabetic leg ulcer infections. Evolved resistance was costly in terms of impaired growth rate, but costs-of-resistance were completely ameliorated by compensatory adaptation. Crucially, we show that, in some populations, experimentally evolved resistance to pexiganan provided S. aureus with cross-resistance to human-neutrophil-defensin-1, a key component of the innate immune response to infection. This unintended consequence of therapeutic use could drastically undermine our innate immune system's ability to control and clear microbial infections. Our results therefore highlight grave potential risks of AMP therapies, with implications for their development.
Keywords: antibiotic resistance, antimicrobial peptide, compensatory adaptation, cost-of-resistance, experimental evolution, innate immunity
1. Introduction
Antimicrobial peptides (AMP) are small cationic molecules produced as part of the innate immune response. AMPs display cidal activity against viral, bacterial and fungal pathogens, but despite exposure to AMPs during infection, levels of resistance in natural microbial populations are typically low [1,2]. This has led to the assumption that resistance to AMPs cannot evolve, or that high costs-of-resistance prevent persistence of resistant strains [2]. Therefore synthetic AMPs, often derived from naturally occurring AMPs, are seen as good alternatives to antibiotics and will be clinically available, by some estimates, within 10 years [1,3]. However, therapeutic use is likely to generate strong directional selection for resistance thereby driving its evolution [4], while compensatory mutations could ameliorate associated costs-of-resistance [2]. Of graver concern than resistance to the therapy itself, though, is the untested potential for evolved cross-resistance to our own immunity peptides. This could undermine our innate immune system's ability to prevent superficial infections progressing into life-threatening systemic disease [2].
Pexiganan is a synthetic AMP derived from frog magainin, and has been proposed as a candidate treatment for diabetic leg ulcer infections [5–7]. While such infections are often polymicrobial, Staphylococcus aureus is the predominant pathogen [7]. Neutrophils are recruited to the site of infection following an inflammatory response [8], where they take up bacterial cells as well as degranulate, releasing a suite of bactericidal proteins and antimicrobial peptides [9]. To determine whether AMP therapy selects for cross-resistance to a host peptide, we experimentally evolved resistance to pexiganan in S. aureus and then tested evolved strains for cross-resistance to human-neutrophil-defensin-1 (HNP-1). It is important to note that HNP-1 and pexiganan belong to structurally distinct classes of antimicrobial peptide with contrasting modes of action [10]. Additionally, we tested whether there were costs associated with pexiganan resistance, and whether costs-of-resistance could be ameliorated by compensatory adaptation in absence of pexiganan.
2. Material and methods
(a). Resistance selection experiment
All experiments were performed in cation-adjusted Mueller–Hinton broth and incubated at 37°C with continuous shaking (200 r.p.m.). A S. aureus nasal carriage isolate was streaked on agar to randomly isolate eight independent colonies that were stored in 15 per cent glycerol at −80°C. Eight 2 ml cultures were founded with approximately 107 isogenic cells of an overnight culture of one of the previously selected colonies and propagated by serial transfer. Six replicate populations were supplemented with increasing concentrations of pexiganan acetate, and two controls were propagated in the absence of AMP. From a starting concentration of 16 μg ml–1 pexiganan acetate, every 48 h we transferred 2 per cent of each culture to two flasks containing fresh medium with either the same concentration of peptide as used previously, or a twofold increased concentration. When growth was observed in the higher concentration this culture was used for subsequent inoculation. This procedure was repeated until populations grew for two consecutive transfers in 1024 μg ml–1 of peptide.
(b). Quantifying pexiganan minimal inhibitory concentration (MIC) and costs-of-resistance
Pexiganan minimal inhibitory concentrations for ancestral and evolved bacteria (stored in 15% glycerol at −80°C) were determined by microtitre broth dilution methods using standard protocols [11] slightly modified to use cation-adjusted Mueller–Hinton broth and 96-well polypropylene microtitre plates. Maximal growth rate (VMAX) of ancestral and evolved bacteria was quantified in the absence of pexiganan acetate in a 96-well plate reader (BioTec ELx808). For each well, optical density (600 nm) was taken every 5 min and VMAX was estimated during exponential growth from log-e transformed optical density values.
(c). Compensatory adaptation selection experiment
To test whether costs-of-resistance could be ameliorated, three independent colonies from the population with the highest cost-of-resistance (SA3; see §3) and its ancestral clone were used to initiate five replicate populations each. Populations were propagated as 10 ml cultures by daily transfer of 0.1 per cent of each population for 10 transfers in the absence of pexiganan acetate. Pexiganan MIC and VMAX were estimated for founding and evolved bacteria.
(d). Quantifying cross-resistance
Evolved bacteria and each founding ancestral clone were examined for susceptibility to HNP-1, which is routinely determined by bactericidal assays [12]. Overnight cultures were diluted 100-fold and allowed to grow for 3 h in order for cells to reach mid log phase. After washing, the cultures were diluted in fresh medium to final cell density approximately 3 × 106 colony forming units ml–1 and added to a 96-well polypropylene microtitre plate. HNP-1 (end concentration: 50 µg ml–1) or purified water (control) was added and cultures were grown for 4 h. At 0 and 4 h, dilutions were plated on brain heart infusion agar plates to estimate cell densities. Growth was calculated by dividing 4 h log-density by 0 h log-density. Resistance was calculated as the ratio of growth in the presence versus absence of HNP-1.
3. Results
Within 14 transfers, five of six populations were capable of growth in 1024 μg ml–1 pexiganan. Evolved bacteria had elevated MICs approximately 10–50x higher than ancestral bacteria (figure 1a; one-sample t-test, t = 3.159, d.f. = 4, p = 0.03). Three replicates displayed impaired maximal growth rates (VMAX) in the absence of pexiganan (figure 1a), and we observed a negative relationship between VMAX and MIC (figure 1a; ρ = −0.877, p = 0.022). This suggests that there were costs associated with resistance to pexiganan, and that more highly resistant bacteria suffered greater costs. However, the cost of pexiganan resistance could be compensated; clones from SA3, the population with the highest cost-of-resistance, attained growth rates comparable to the ancestor while retaining resistance following serial-transfer in the absence of pexiganan (figure 1b).
Figure 1.
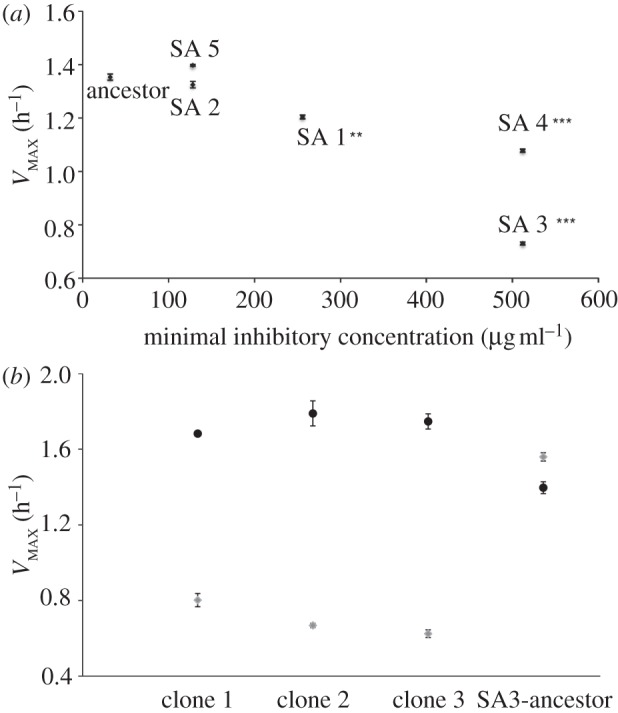
Evolved resistance to pexiganan has associated costs that can be ameliorated by compensatory adaptation. (a) VMAX plotted against pexiganan MIC for ancestral and evolved bacteria. Asterisks indicate significant difference by t-test between ancestral and evolved VMAX (**p ≤ 0.001, *** p ≤ 0.0001). (b) VMAX of ancestral and evolved SA3 bacteria before (light grey) and after (dark grey) selection in the absence of pexiganan. All compensated bacteria retained resistance to pexiganan (data not shown).
Two replicate lines selected in the presence of pexiganan displayed increased resistance to HNP-1 compared with ancestral bacteria (figure 2; two-sample t-tests: SA1, t = 5.376, d.f. = 10, p = 0.0003; SA5, t = 6.058, d.f. = 6, p = 0.0009), while control bacteria selected in the absence of pexiganan displayed no change (figure 2). Therefore, evolved resistance to pexiganan could confer cross-resistance to HNP-1, although this was not evident in all our replicate lines (paired-sample t-test of ancestral and evolved means, t = 1.365, d.f. = 4, p = 0.24), and we observed no correlation between the degree of resistance to pexiganan and the degree of cross-resistance to HNP-1 (ρ = 0.049, p = 0.9).
Figure 2.
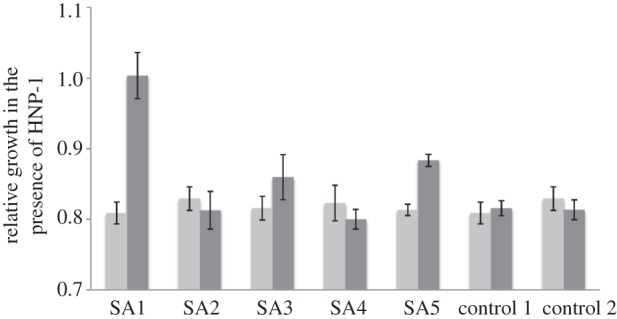
Resistance to pexiganan provides cross-resistance to HNP-1. Bars represent the ratio of growth rates in presence versus absence of HNP-1 for ancestral (light grey) and evolved (dark grey) bacteria; a value of 1 represents no inhibition of growth by HNP-1.
4. Discussion
We demonstrate that S. aureus rapidly evolved resistance to pexiganan and that associated costs-of-resistance could be completely ameliorated by a short period of compensatory adaptation. This confirms that pathogens targeted by AMP therapies are likely to evolve resistance [4] and suggests that associated costs-of-resistance are unlikely to prevent persistence of resistant strains. Crucially, we provide the first evidence that evolved resistance to a therapeutic AMP can provide cross-resistance to a human immunity peptide. Since pexiganan and HNP-1 belong to structurally distinct classes of antimicrobial peptide with contrasting modes of action [10], synclinal selection for resistance to HNP-1 is perhaps even more worrying.
We observed variation between replicate lines in evolved pexiganan MIC, perhaps suggesting that different mechanisms of resistance arose in these independent lineages. Moreover, we observed no correlation between the degree of resistance to pexiganan and the degree of cross-resistance to HNP-1; indeed, cross-resistant bacteria acquired only intermediate levels of pexiganan resistance and at relatively moderate costs. This suggests that resistance mechanisms offering generalized protection against multiple AMPs may actually be less effective at protecting cells against the target therapeutic AMP. Cross-resistance may therefore be selected against under strong directional therapeutic selection, if more specific and effective resistance mechanisms exist within the population. Conversely, cross-resistant genotypes could be favoured if host-peptides also exert appreciable selection at the site of infection.
We chose to use a nasal carriage isolate of S. aureus in our experiments. While this has the advantage of bringing our study closer to clinical reality, it has the disadvantage that no neutrally marked strains were available for use in direct competition experiments. This methodology is the ‘gold-standard’ for quantification of changes in fitness of evolved bacteria relative to their ancestor, and produces more accurate estimates of fitness than the VMAX assays used here. However, as VMAX only estimates one component of fitness (maximal growth rate) it is likely that costs-of-resistance have been underestimated, rather than overestimated in our study.
Our findings raise serious concerns about the long-term risks associated with the development and the use of AMP therapies. Unlike resistance to traditional antibiotics, which may select solely for cross-resistance to other antibiotics, resistance to therapeutic AMPs may commonly compromise our natural immune defences. This unintended consequence of therapeutic use may fundamentally alter the interaction with our commensal microbiome, and potentiate a rise in opportunistic infection. Therefore, while AMP therapies may prove to be efficacious in the short term, their use could have catastrophic longer term consequences for our ability to control microbial infections.
Acknowledgements
We acknowledge funding from The Leverhulme Trust (F/00025/AD), the kind gift of pexiganan from M. Zasloff, technical assistance from D. Rozen, and helpful comments on the manuscript by G. Hurst, M. Begon, C. Winstanley and A. Cossins.
References
- 1.Hancock R. E. W., Sahl H. G. 2006. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557 10.1038/nbt1267 (doi:10.1038/nbt1267) [DOI] [PubMed] [Google Scholar]
- 2.Bell G., Gouyon P. H. 2003. Arming the enemy: the evolution of resistance to self-proteins. Microbiology 149, 1367–1375 10.1099/mic.0.26265-0 (doi:10.1099/mic.0.26265-0) [DOI] [PubMed] [Google Scholar]
- 3.Eckert R. 2011. Road to clinical efficacy: challenges and novel strategies for antimicrobial peptide development. Future Microbiol. 6, 635–651 10.2217/FMB.11.27 (doi:10.2217/FMB.11.27) [DOI] [PubMed] [Google Scholar]
- 4.Bell G., Perron G. G., Zasloff M. 2006. Experimental evolution of resistance to an antimicrobial peptide. Proc. R. Soc. B 273, 251–256 10.1098/rspb.2005.3301 (doi:10.1098/rspb.2005.3301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ge Y. G., MacDonald D., Henry M. M., Halt H. I., Nelson K. A., Lipsky B. A., Zasloff M. A., Holroyd K. J. 1999. In vitro susceptibility to pexiganan of bacteria isolated from infected diabetic foot ulcers. Diagn. Microbiol. Infect. Dis. 35, 45–53 10.1016/S0732-8893(99)00056-5 (doi:10.1016/S0732-8893(99)00056-5) [DOI] [PubMed] [Google Scholar]
- 6.Ge Y. G., MacDonald D. L., Holroyd K. J., Thornsberry C., Wexler H., Zasloff M. 1999. In vitro antibacterial properties of pexiganan, an analog of magainin. Antimicrob. Agents Chemother. 43, 782–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ge Y., MacDonald D., Hait H., Lipsky B., Zasloff M., Holroyd K. 2002. Microbiological profile of infected diabetic foot ulcers. Diabetic Med. 19, 1032–1034 10.1046/j.1464-5491.2002.00696_1.x (doi:10.1046/j.1464-5491.2002.00696_1.x) [DOI] [PubMed] [Google Scholar]
- 8.Lee J. C., Rich J. 2005. The pathogenesis of Staphylococcus aureus infection in the diabetic NOD mouse. Diabetes 54, 2904–2910 10.2337/diabetes.54.10.2904 (doi:10.2337/diabetes.54.10.2904) [DOI] [PubMed] [Google Scholar]
- 9.Nathan C. 2006. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 6, 173–182 10.1038/Nri1785 (doi:10.1038/Nri1785) [DOI] [PubMed] [Google Scholar]
- 10.Brogden K. A. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria. Nat. Rev. Microbiol. 3, 238–250 10.1038/nrmicro1098 (doi:10.1038/nrmicro1098). [DOI] [PubMed] [Google Scholar]
- 11.Amsterdam D. 1996. Susceptibility testing of antimicrobials in liquid media. In Antibiotics in laboratory medicine (ed. Lorain V.), pp. 52–111 Philadelphia, PA: Williams and Wilkins [Google Scholar]
- 12.Midorikawa K., et al. 2003. Staphylococcus aureus susceptibility to innate antimicrobial peptides, beta-defensins and CAP18, expressed by human keratinocytes. Infect. Immun. 71, 3730–3739 10.1128/IAI.71.7.3730-3739.2003 (doi:10.1128/IAI.71.7.3730-3739.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]