

Abstract
If one gene regulates another, those two genes are likely to be involved in many of the same biological functions. Conversely, shared biological function may be suggestive of the existence and nature of a regulatory interaction. With this in mind, we develop a measure of functional similarity between genes based on annotations made to the Gene Ontology in which the magnitude of their functional relationship is also indicative of a regulatory relationship. In contrast to other measures that have previously been used to quantify the functional similarity between genes, our measure scales the strength of any shared functional annotation by the frequency of that function's appearance across the entire set of annotations. We apply our method to both Escherichia coli and Saccharomyces cerevisiae gene annotations and find that the strength of our scaled similarity measure is more predictive of known regulatory interactions than previously published measures of functional similarity. In addition, we observe that the strength of the scaled similarity measure is correlated with the structural importance of links in the known regulatory network. By contrast, other measures of functional similarity are not indicative of any structural importance in the regulatory network. We therefore conclude that adequately adjusting for the frequency of shared biological functions is important in the construction of a functional similarity measure aimed at elucidating the existence and nature of regulatory interactions. We also compare the performance of the scaled similarity with a high-throughput method for determining regulatory interactions from gene expression data and observe that the ontology-based approach identifies a different subset of regulatory interactions compared with the gene expression approach. We show that combining predictions from the scaled similarity with those from the reconstruction algorithm leads to a significant improvement in the accuracy of the reconstructed network.
Keywords: Gene Ontology, functional similarity, gene regulatory networks
1. Introduction
1.1. Motivation
The Gene Ontology (GO) [1,2] provides a controlled vocabulary that biologists use to annotate genes with their functional properties. Since its inception, GO has been applied in various ways, including the functional analysis of sets of genes [3], predicting gene function [4,5], and both confirming and predicting regulatory interactions [6–9]. In this paper, we take a complex networks approach to the analysis of annotation data, exploring how different types of network relationships between genes and functions can be combined to give new biological insights. In recent years, complex networks tools have been used alongside traditional bioinformatics techniques to study many different kinds of biological networks [10], including, but not limited to, gene regulatory networks [11,12], protein–protein interaction networks [13,14] and metabolic networks [15,16]. Here, we apply tools from the complex network theory to networks derived from GO annotations.
In the GO, the relationships between terms (representing various biological functions) can be viewed as a directed network in which more specific child terms point to more general parent terms. A second network linking genes to these terms can be built from annotation data that uses the terminology laid out by the ontology. This information can be used to build a third network linking gene–gene pairs based on their functional similarity. However, determining exactly how to calculate this functional similarity is non-trivial. Our goal is to construct a natural weighting scheme in which the functional similarity of two genes is scaled based on the properties of their shared functional annotations and show that this scaled similarity is correlated with that gene-pair's likelihood to have a regulatory relationship (as documented by an experimentally derived network). We aim to develop a measure that will not only accurately represent shared function, but also provide new biological insights by offering a structural interpretation for strong link weights. Figure 1 illustrates our approach.
Figure 1.

An outline of the approach taken in this paper. We use information from the Gene Ontology to construct a functional gene network. We then compare the results to an experimentally derived gene regulatory network both to determine how well our functional measure predicts known interactions and to investigate the implications of high functional similarity for regulatory interactions. DAG, directed acyclic graph.
We focus our study on Escherichia coli, because there exists a high-quality experimental gene regulatory network published by RegulonDB [17] and it has been used extensively in training network reconstruction algorithms. In order to evaluate the predictive power of our approach, we compare our functional gene network with the experimentally determined RegulonDB gene regulatory network. We then further compare the predictive power of our scaled similarity score to several currently established functional similarity measurements as well as to a gene interaction network derived solely from gene expression data. Specifically, we focus our comparison on two contrasting functional similarity methods, one of which relies on the semantic similarity of terms in GO [18] and the other Kappa statistics [19], as well as the well-established, context-likelihood-of-relatedness (CLR) network reconstruction algorithm [20].
In addition to demonstrating that the strength of the links in our annotation-based network is correlated with the existence of known regulatory interactions, we also want to understand what the scaled similarity can tell us about the structural importance of edges in the true regulatory network. Therefore, we expand the comparison of our method to other measures of functional similarity to include not only their relative ability to predict true regulatory interactions but also the extent to which high similarity indicates structural importance in the experimental regulatory network.
1.2. Background
1.2.1. Properties of Gene Ontology annotations
The GO is represented as a directed acyclic graph (DAG) in which nodes of the graph are identified with ‘terms’ representing the different physical and functional roles of genes. Terms are organized hierarchically. For example, a term broadly describing a class of functions may be the ‘parent’ of several more specific ‘child’ terms representing functions belonging to the broad class of the parent term, and these child terms may be the parents of still more specific terms. Links on the GO hierarchy connect child terms to parent terms and are directed from child to parent through the relationships ‘is a’ and ‘part of’. Note that child terms can have more than one parent term. The three most general terms in the GO hierarchy are ‘Biological Process’, ‘Molecular Function’ and ‘Cellular Component’, which are the forebearers of all other terms and may be thought of as the origins of three independent, main branches in the GO hierarchy, formed from the descendants of each of these main terms plus their connections.
Using this structure, GO annotates each gene to a set of ‘terms’ representing that gene’s biological functions. Gene annotations are transitive, meaning a gene annotation to a child term implies annotations to all the parent terms of that child [21]. As a result, all genes contain an annotation to one or more of the three most general terms. The relationship between genes and terms can be represented in the form of a gene-term network, taking the form of a bipartite graph, where each gene is connected to the set of terms to which it is annotated, plus all the parents of those terms.
In order to construct our annotation-based gene network, we used pairs of gene-term annotations downloaded from the GO website (geneontology.org) to first construct a gene-term bipartite graph, represented as an 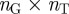 adjacency matrix, where
adjacency matrix, where 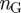 is the total number of genes and
is the total number of genes and 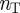 is the total number of terms listed in the annotation file. In this matrix, a value of one indicates a known connection between the corresponding gene and term, and a value of zero indicates that the gene is not associated with that term. We will denote the
is the total number of terms listed in the annotation file. In this matrix, a value of one indicates a known connection between the corresponding gene and term, and a value of zero indicates that the gene is not associated with that term. We will denote the 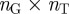 adjacency matrix of this bipartite graph by B and its
adjacency matrix of this bipartite graph by B and its 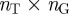 transpose by
transpose by 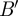 . Thus:
. Thus:
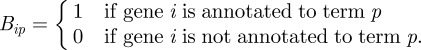 |
1.1 |
Many terms are associated with just a small handful of genes, while some terms are associated with many genes. A histogram of the ‘degree’ of terms (i.e. the number of genes annotated to each term) in E. coli reveals a heavy-tailed relationship (figure 2a). To see whether this distribution follows a power law, we used a Kolmogorov–Smirnov test [22]. Both the overall term degree distribution and the distributions within the individual ontologies approximate a power law very well (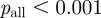 ,
, 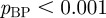 ,
, 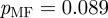 ,
, 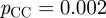 ). Although there are several different reasons for a term to have a large number of genes annotated to it, in the majority of these instances the large number of annotations merely indicates that the functional term is very general and resides at the top levels of the GO hierarchy. We will account for the widely varying degrees of terms in determining the strengths of the functional links between genes in our gene network.
). Although there are several different reasons for a term to have a large number of genes annotated to it, in the majority of these instances the large number of annotations merely indicates that the functional term is very general and resides at the top levels of the GO hierarchy. We will account for the widely varying degrees of terms in determining the strengths of the functional links between genes in our gene network.
Figure 2.

Cumulative degree distribution in E. coli for (a) terms considering all gene-term annotations and just those in each individual ontology, and (b) genes considering all gene-term annotations and just those in each individual ontology. Black denotes all annotations; yellow, Biological Process; magenta, Molecular Function; cyan, Cellular Component.
By contrast, a histogram of the ‘degree’ of genes (i.e. the number of terms to which each gene is annotated) shows that although some genes have many more annotations than others, a large portion of genes in E. coli have approximately the same number of annotations (figure 2b). These properties of annotations are not limited to the GO, but can be found in other functional classification databases as well (electronic supplementary material, figure S1), indicating that these properties should not be ignored when evaluating the functional relationship between genes.
1.2.2. Measures of functional similarity
Many measures have been developed that attempt to accurately quantify the functional similarity between pairs of genes [23]. Measures that involve the concept of semantic similarity focus on the similarity of terms with regard to their information content and relative placement in the GO hierarchy, combining the subset of this information that is also linked to two individual genes in order to derive a gene–gene functional similarity measure. Many standard statistical tools have also been adapted for use on GO annotations. In contrast to the semantic similarity measures, these approaches are often more focused on the annotation properties of the given genes. For simplicity, in this paper, we focus on two of these functional similarity measures, which we believe are a representative sampling of the most widely used and accepted measures. Results for other measures are provided in the electronic supplementary material. All measures were calculated using the csbl.go package in R [24].
In 2003, Lord et al. [18] first applied the information theory concept of semantic similarity to the GO, citing the prior work of Resnik [25], Lin [26] and Jiang & Conrath [27]. A few years later, Schlicker et al. [28] combined the methods of Resnik and Lin in the Relevance measure. Each measure has its own strengths and weaknesses (for previous comparisons and evaluations of semantic similarity measures see [18,28–32]); however, since all rely upon the same basic mathematical concepts, we will choose only one (Resnik's measure) to explore in detail.
All semantic similarity measures begin by defining the probability, p(t), of observing a term, t, as the number of gene annotations (degree) made to that term, divided by the number of gene annotations made to the parent node of the branch to which that term belongs. As a consequence, the parents of the three main branches in GO, ‘Biological Process’, ‘Molecular Function’ and ‘Cellular Component’, will all be given a probability of one. Following Resnik, the semantic similarity between two terms, 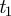 and
and 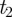 , can then be defined as:
, can then be defined as:
| 1.2 |
where 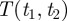 is the set of parent terms shared by the two terms. In order to find the semantic similarity between two genes, G1 and G2, one constructs an
is the set of parent terms shared by the two terms. In order to find the semantic similarity between two genes, G1 and G2, one constructs an 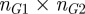 matrix, where
matrix, where 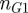 (
( ) are the number of terms annotated to G1 (G2), and populates it with the semantic similarity values between all the pairs of terms. The semantic similarity between the two genes is then determined by taking the average of all values in the matrix.
) are the number of terms annotated to G1 (G2), and populates it with the semantic similarity values between all the pairs of terms. The semantic similarity between the two genes is then determined by taking the average of all values in the matrix.
The use of Kappa statistics as a measure of functional similarity between pairs of genes has recently been popularized because of its inclusion alongside a commonly used gene set enrichment analysis tool [19]. Although slightly different, statistical measures such as a weighted Jaccard [32], cosine similarity [33] and Czekanowski-Dice [34], are similar enough in form that they rank the functional similarity between pairs of genes in approximately the same order as Kappa statistics. Since the following analysis will use rank rather than raw significance, we discuss just the Kappa statistic to illustrate how this type of statistical measure captures functional similarity.
Kappa statistics calculates the agreement between two sets of gene annotations by comparing the actual agreement between the two sets (X) to the average agreement one would expect by chance (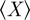 ), given that the two sets are independent:
), given that the two sets are independent:
| 1.3 |
κ will be equal to one for perfect agreement (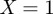 ) and will be close to zero for an actual agreement close to random (
) and will be close to zero for an actual agreement close to random (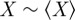 ). X and
). X and 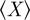 can easily be understood in terms of a Venn diagram (figure 3). The observed agreement (X) is the percentage of annotations the two genes either share or do not share:
can easily be understood in terms of a Venn diagram (figure 3). The observed agreement (X) is the percentage of annotations the two genes either share or do not share:
| 1.4 |
where 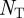 , the total number of terms, is equal to
, the total number of terms, is equal to 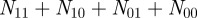 . The agreement expected if the two samples were independent is the sum of the percentage of annotations one would expect the two genes to share or not share, given how many total annotations each gene has:
. The agreement expected if the two samples were independent is the sum of the percentage of annotations one would expect the two genes to share or not share, given how many total annotations each gene has:
| 1.5 |
where 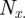 implies a summation over the dotted entry (e.g.
implies a summation over the dotted entry (e.g. 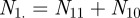 ).
).
Figure 3.
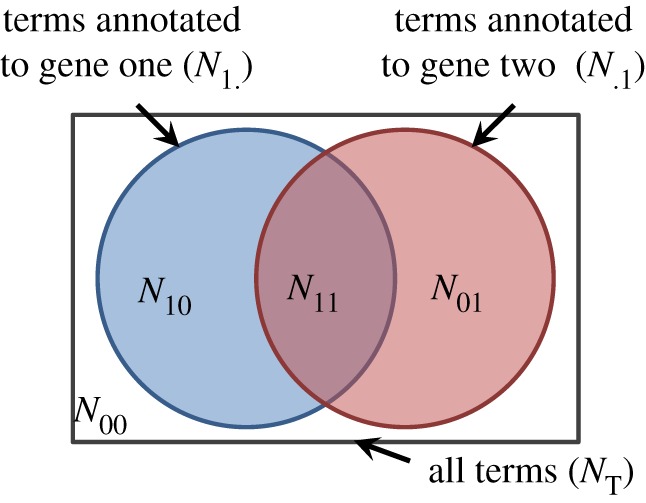
Venn diagram representing the overlap between the annotations of two genes. 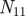 represents the number of annotations both genes share,
represents the number of annotations both genes share, 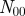 the number annotations neither gene has, and
the number annotations neither gene has, and 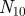 (
(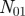 ) the number of annotations that gene one (two) has that gene two (one) does not.
) the number of annotations that gene one (two) has that gene two (one) does not.
1.2.3. Expression-based reconstruction methods
We also compare the predictive value of functional similarity approaches to that of the commonly used high-throughput gene expression approach. In the latter, correlations in gene expression are used to reverse-engineer a regulatory network [35]. Although we note that the values calculated in these approaches do not incorporate functional annotation data and thus are not designed to capture functional similarity, we propose that comparing our measure with the values determined by network reconstruction algorithms will be informative because these algorithms are specifically designed to estimate the probability of a regulatory relationship between two genes, something that we wish to capture in our own functional measure.
Some of the most successful algorithms for generating gene regulatory networks involve the information theory concept of mutual information (MI) [20,36,37]. MI describes the statistical dependence between two variables. However, unlike correlation coefficients, MI captures nonlinear relationships between the tested pair of variables, and has been used to successfully detect regulatory interactions that would have been missed using a linear correlation metric. For comparison with the results of our functional gene network, we focus specifically on the CLR algorithm [20], which is among the set of algorithms that rely on MI calculations. We choose the CLR approach for comparison because of its large number of citations as well as its demonstrated ability to predict edges in RegulonDB [17]. We explore whether the interactions captured by the functional similarity measures are the same as, or different than, those captured by the CLR network.
2. Methods
2.1. Calculating a scaled similarity between genes
We centre our functional similarity measure around the bipartite graph of gene annotations in order to capture information about the types of annotations shared between two genes. As opposed to semantic similarity measures, this distances us from the hierarchical DAG structure of the GO, which should help eliminate issues associated with the DAG's structure, such as its lack of uniformity in depth and the possibility of disjunctive ancestors (i.e. when multiple ancestors of a term can be found based on independent paths). On the other hand, by using the mathematical representation of the bipartite graph, we retain the ability to easily integrate information about the functions (with regard to their GO annotations) into our similarity measure and therefore, as opposed to more gene-centred measures, such as the Kappa statistic, which treat every biological function equally, we are able to better interpret the potential role of individual biological functions.
From the 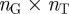 adjacency matrix B of our bipartite graph (see equation (1.1)), we can generate an
adjacency matrix B of our bipartite graph (see equation (1.1)), we can generate an 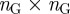 adjacency matrix
adjacency matrix 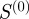 that reflects the relationships between annotated genes:
that reflects the relationships between annotated genes:
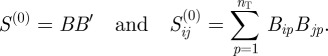 |
2.1 |
In this projection, the value of 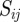 is equal to the total number of functional annotations that are shared between genes i and j (note that this is equal to
is equal to the total number of functional annotations that are shared between genes i and j (note that this is equal to 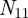 of figure 3). However, as previously discussed, some terms such as ‘Molecular Function’ are quite general and associated with many genes, while more specific terms are often associated with very few genes. It would, therefore, seem inappropriate to weight links between genes i and j simply by the number of their co-associations with terms (as done in the earlier mentioned definition of
of figure 3). However, as previously discussed, some terms such as ‘Molecular Function’ are quite general and associated with many genes, while more specific terms are often associated with very few genes. It would, therefore, seem inappropriate to weight links between genes i and j simply by the number of their co-associations with terms (as done in the earlier mentioned definition of 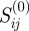 ). For example, one might want to count associations through terms that have many gene annotations less strongly than those associations that occur through more specific terms. Previous works have addressed this issue in several ways, such as weighting the terms by their information content [32], or simply by removing the highest degree terms from the similarity calculation [38]. However, we choose instead to compensate for the variation in the quantity of term annotations by introducing an
). For example, one might want to count associations through terms that have many gene annotations less strongly than those associations that occur through more specific terms. Previous works have addressed this issue in several ways, such as weighting the terms by their information content [32], or simply by removing the highest degree terms from the similarity calculation [38]. However, we choose instead to compensate for the variation in the quantity of term annotations by introducing an 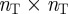 diagonal weighting matrix,
diagonal weighting matrix, 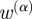 , with elements:
, with elements:
| 2.2. |
where δpq = 1 if p = q and is zero otherwise. Note that the denominator of w(α) is simply the ‘degree’ of term p, or the number of genes k associated with term p, raised to a power, α. Using the matrix w(α), we modify the strength of our gene connections as given in equation (2.1) to obtain a new gene connection matrix S(α) given by:
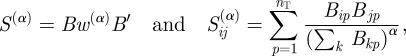 |
2.3 |
where α can be thought of as a weighting parameter such that larger values of α more strongly suppress the weights of terms that have connections to many different genes. Note that for α = 0, the weighting matrix, w(α) reduces to the identity matrix (i.e. uniform weighting of terms). In this case, equations (2.1) and (2.3) are equivalent.
Because the GO has three distinct branches, we will also investigate the effects of considering the individual ontologies represented by each of these branches. Therefore, we will have four versions of S: (i) the reconstruction considering all gene-term annotations (Sall), (ii) considering only gene-term annotations where the term is part of the ‘Biological Process’ ontology (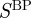 ), (iii) considering only gene-term annotations where the term is part of the ‘Molecular Function’ ontology (
), (iii) considering only gene-term annotations where the term is part of the ‘Molecular Function’ ontology (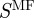 ), and (iv) considering only gene-term annotations where the term is part of the ‘Cellular Component’ ontology (
), and (iv) considering only gene-term annotations where the term is part of the ‘Cellular Component’ ontology (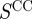 ). Because
). Because 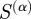 in equation (2.3) is defined as a sum over terms (i.e. the index p in equation (2.3)), for a given α:
in equation (2.3) is defined as a sum over terms (i.e. the index p in equation (2.3)), for a given α:
| 2.4 |
2.2. Suppressing the role of common genetic functions
There are two main limiting cases for α. For 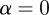 , the weighting matrix reduces to the identity matrix and the calculation is the same as it would have been had we not considered any weighting. In this case, the entries in the matrix
, the weighting matrix reduces to the identity matrix and the calculation is the same as it would have been had we not considered any weighting. In this case, the entries in the matrix 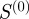 are the number of terms shared between two genes. In the case of large α, the weights of
are the number of terms shared between two genes. In the case of large α, the weights of 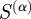 are such that those genes connected through many low degree terms have the highest weight and those connected through only one high degree term have the lowest weight. Low-degree terms (i.e. terms with few gene annotations) are normally lower in the GO hierarchy and in general represent more specific biological functions. On the other hand, the majority of high-degree terms (i.e. terms with many gene annotations) are at the top of the GO hierarchy and are in general more common and less specific, where common often, but does not necessarily mean generic. We note that this association of degree with specificity is an approximation, but believe it can still give a good first-order approximation of whether the biological function represented by the GO term is more specific or more generic. Therefore, by giving the greatest weight to links between genes that share annotations to many low-degree terms, our weights should correspond to a measure of how much specific biological function is shared between the two genes.
are such that those genes connected through many low degree terms have the highest weight and those connected through only one high degree term have the lowest weight. Low-degree terms (i.e. terms with few gene annotations) are normally lower in the GO hierarchy and in general represent more specific biological functions. On the other hand, the majority of high-degree terms (i.e. terms with many gene annotations) are at the top of the GO hierarchy and are in general more common and less specific, where common often, but does not necessarily mean generic. We note that this association of degree with specificity is an approximation, but believe it can still give a good first-order approximation of whether the biological function represented by the GO term is more specific or more generic. Therefore, by giving the greatest weight to links between genes that share annotations to many low-degree terms, our weights should correspond to a measure of how much specific biological function is shared between the two genes.
To determine the consequences of different weighting parameter values, we used GO annotations for E. coli to construct functional gene networks for various values of α and compared these networks to the established regulatory network published by RegulonDB. RegulonDB provides a high-quality TF-gene interaction network, which contains 2341 genes and 6725 regulatory links. Of these RegulonDB-listed genes, 77 per cent (1803) also appear in the GO annotation files, and of the RegulonDB-listed links, 75 per cent (5024) can be assigned a non-zero value in our functional gene network. We believe that this provides sufficient shared information for us to usefully compare our projected gene networks with the experimentally derived RegulonDB gene regulatory network.
We note here that a small percentage of gene-gene pairs are never annotated to a common biological function. In this case, we set the value of their scaled similarity to zero. In addition, when comparing to the experimental regulatory network, we consider only a ‘putative’ set of edges, defined as the set of all edges extending from a transcription factor to a gene, because only these types of edges can exist in a regulatory network.
In order to systematically evaluate the predictive power of each 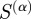 , we calculated the F-score, a statistical measure combining the concepts of precision and recall, using the regulatory interactions provided by RegulonDB as our ‘gold standard’. The F-score is defined as:
, we calculated the F-score, a statistical measure combining the concepts of precision and recall, using the regulatory interactions provided by RegulonDB as our ‘gold standard’. The F-score is defined as:
| 2.5 |
where P is the precision:
| 2.6 |
and R is the recall:
| 2.7 |
In these equations, the numerators (true positives) are those edges above a particular similarity value that are also experimentally verified by RegulonDB, the denominator of P is the total number of edges above that value and the denominator of R is the number of edges that are in RegulonDB. The F-score equals zero for complete lack of precision and recall, and one for simultaneously perfect precision and recall. For each 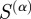 , we calculated the F-score for all possible similarity cut-offs and took the maximum of these values as a measure of the predictive power of that
, we calculated the F-score for all possible similarity cut-offs and took the maximum of these values as a measure of the predictive power of that 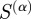 . As a control, we also calculated the maximum F-score for 1000 random orderings of the putative set of edges, taking the average over these randomizations as the expected value of the maximum F-score.
. As a control, we also calculated the maximum F-score for 1000 random orderings of the putative set of edges, taking the average over these randomizations as the expected value of the maximum F-score.
Figure 4 plots the calculated maximum F-score as a function of the weighting parameter, α. The scaled similarity values for high α are the most predictive of true regulatory interactions, in line with our hypothesis that terms with many gene annotations should be suppressed relative to those with only a few gene annotations. In addition, the values for large α approach a steady value. This might be expected since for large α the weights of the edges will take a particular ordering. As a consequence of this analysis, for the remainder of our discussion, we will arbitrarily assign  such that
such that 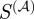 represents the scaled similarity in the case of high α.
represents the scaled similarity in the case of high α.
Figure 4.
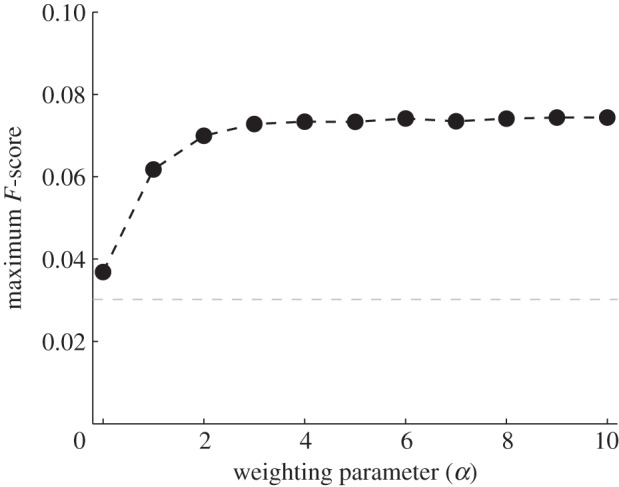
The predictive power of 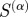 as a function of α calculated by determining maximum F-score for each
as a function of α calculated by determining maximum F-score for each 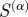 , using the edges reported in RegulonDB as a ‘gold standard’. Higher values of α indicate greater emphasis on biological specificity. The scaled similarity measure is much more predictive of known regulatory interactions when biological functions with high specificity are given a greater weight. For comparison, the expected value of the maximum F-score is shown as a grey dashed line.
, using the edges reported in RegulonDB as a ‘gold standard’. Higher values of α indicate greater emphasis on biological specificity. The scaled similarity measure is much more predictive of known regulatory interactions when biological functions with high specificity are given a greater weight. For comparison, the expected value of the maximum F-score is shown as a grey dashed line.
3. Results
3.1. Comparing measures of functional similarity
Before investigating our scaled similarity measure in a regulatory network context, we wanted to see how it compared with other common measures of functional similarity. Specifically, we compared the results of scaled similarity to two other measures of functional similarity—Resnik's semantic similarity measure and functional similarity as determined by Kappa statistics. To determine whether the different measures were identifying the same gene-pairs as the most functionally similar, we selected the top 5000 most functionally similar pairs of genes according to each measure, and created a Venn diagram of those pairs (figure 5a).
Figure 5.

The scaled similarity predicts known regulatory interactions better than other traditional functional similarity measures. It is also reflective of information about the structural importance of an edge in the regulatory network. (a) Overlap of the top 5000 highest ranking gene-pairs according to the scores given by four different measures of functional similarity. (b) Comparison of the predictive power of 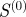 and
and 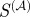 compared with other commonly used measures, calculated by determining the maximum F-score of each measure. The expected value for the maximum F-score is shown as a grey dotted line. (c) The harmonic mean of the new shortest path upon edge removal from the experimental regulatory network published by RegulonDB, plotted as a function of several measures of functional similarity. For each measure edges were sorted, with highest scoring edges given the lowest rank. The harmonic mean of the informational importance (new shortest path upon edge removal) for all edges above each rank was calculated and plotted as a function of the edge rank. Blue denotes scaled similarity (
compared with other commonly used measures, calculated by determining the maximum F-score of each measure. The expected value for the maximum F-score is shown as a grey dotted line. (c) The harmonic mean of the new shortest path upon edge removal from the experimental regulatory network published by RegulonDB, plotted as a function of several measures of functional similarity. For each measure edges were sorted, with highest scoring edges given the lowest rank. The harmonic mean of the informational importance (new shortest path upon edge removal) for all edges above each rank was calculated and plotted as a function of the edge rank. Blue denotes scaled similarity ( ); black, scaled similarity (high α); green, semantic similarity; red, Kappa statistic.
); black, scaled similarity (high α); green, semantic similarity; red, Kappa statistic.
Each measure of functional similarity selects fairly a unique set of gene-pairs as the most functionally related, with the greatest overlap between the scaled similarity in the limit of high α and the semantic similarity measure (1305 common gene-pairs). This is not entirely surprising because both 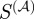 and the semantic similarity take into account some form of the degree of the shared terms. The scaled similarity values
and the semantic similarity take into account some form of the degree of the shared terms. The scaled similarity values 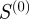 and
and 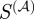 also have a fairly high overlap with 516 common members. By contrast, the gene-pairs selected by Kappa statistics have a relatively poor overlap with the gene-pairs selected by the other three measures. Because the values of
also have a fairly high overlap with 516 common members. By contrast, the gene-pairs selected by Kappa statistics have a relatively poor overlap with the gene-pairs selected by the other three measures. Because the values of 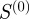 and the Kappa statistic only vary by a normalization factor, much of this difference may be attributable to the gene-focused normalization employed by the Kappa statistics. In fact, the Kappa statistics has a higher overlap with the semantic similarity measure (124 gene-pairs) and
and the Kappa statistic only vary by a normalization factor, much of this difference may be attributable to the gene-focused normalization employed by the Kappa statistics. In fact, the Kappa statistics has a higher overlap with the semantic similarity measure (124 gene-pairs) and 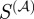 (41 gene-pairs) than
(41 gene-pairs) than 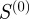 (36 gene-pairs).
(36 gene-pairs).
3.2. Predicting regulatory interactions using measures of functional similarity
We wish to explore how well other measures of functional similarity can predict known regulatory interactions and compare the results with our scaled similarity values, 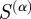 . With this in mind, we determined the predictive ability of the semantic similarity and Kappa statistic by calculating the maximum F-score for these two measures, once again using edges in RegulonDB as a ‘gold standard’, and compared the results with those of the predictive ability of the scaled similarity for the two limiting cases of α. Comparisons with the other measures mentioned in §1.2.2 are shown in the electronic supplementary material, figure S2.
. With this in mind, we determined the predictive ability of the semantic similarity and Kappa statistic by calculating the maximum F-score for these two measures, once again using edges in RegulonDB as a ‘gold standard’, and compared the results with those of the predictive ability of the scaled similarity for the two limiting cases of α. Comparisons with the other measures mentioned in §1.2.2 are shown in the electronic supplementary material, figure S2.
 , which fully accounts for the specificity of the terms in GO, predicts real regulatory relationships better than either the semantic similarity or Kappa statistics (figure 5b). These results are similar if an F-score is calculated at a particular cut-off instead of choosing the maximum (electronic supplementary material, figure S3) and is also true in other functional databases besides the GO (electronic supplementary material, figure S1). Surprisingly, the Kappa statistic predicts true regulatory interactions at a rate comparable to what one would expect by random chance, doing a poorer job of predicting true regulatory interactions than
, which fully accounts for the specificity of the terms in GO, predicts real regulatory relationships better than either the semantic similarity or Kappa statistics (figure 5b). These results are similar if an F-score is calculated at a particular cut-off instead of choosing the maximum (electronic supplementary material, figure S3) and is also true in other functional databases besides the GO (electronic supplementary material, figure S1). Surprisingly, the Kappa statistic predicts true regulatory interactions at a rate comparable to what one would expect by random chance, doing a poorer job of predicting true regulatory interactions than 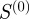 , which represents a count of the number of shared terms between the two genes, without any additional analysis of the statistical significance of this overlap. This suggests that, by failing to take into account the hierarchical nature of the GO, Kappa statistics can miss a large amount of the regulatory information embedded in the functional annotations. This may be owing to the type of normalization employed, which considers the number of terms to which the genes are annotated (the ‘degree’ of the genes) rather than the number of genes annotated to the shared terms (the ‘degree’ of the terms), essentially penalizing the potential existence of an edge between genes involved in many diverse biological functions. A gene with many annotations is either most probably involved in many pathways or is highly studied in the biological community for other reasons. Either way, the large number of annotations is probably indicative of a higher probability that the gene is involved in multiple regulatory interactions, rather than few or none, as suggested by the mathematics of the Kappa statistic.
, which represents a count of the number of shared terms between the two genes, without any additional analysis of the statistical significance of this overlap. This suggests that, by failing to take into account the hierarchical nature of the GO, Kappa statistics can miss a large amount of the regulatory information embedded in the functional annotations. This may be owing to the type of normalization employed, which considers the number of terms to which the genes are annotated (the ‘degree’ of the genes) rather than the number of genes annotated to the shared terms (the ‘degree’ of the terms), essentially penalizing the potential existence of an edge between genes involved in many diverse biological functions. A gene with many annotations is either most probably involved in many pathways or is highly studied in the biological community for other reasons. Either way, the large number of annotations is probably indicative of a higher probability that the gene is involved in multiple regulatory interactions, rather than few or none, as suggested by the mathematics of the Kappa statistic.
It is important to note that neither the semantic similarity measure nor the Kappa statistic was designed with the intent of predicting regulatory interactions. Their relatively poor ability to predict known regulatory interactions compared with the scaled similarity therefore should not necessarily indicate that they are poor functional measures, but rather that the type of shared function they are capturing between two genes is not as important for the existence of a regulatory link between those genes.
3.3. The informational importance of edges with high-functional similarity
Having demonstrated that the weights of edges in our ontology-based network are indicative of real regulatory relationships, we further investigated the significance of these weights for regulatory interactions. To address this question, we considered a metric characterizing how the connectivity of the established RegulonDB network changes when edges are removed. Specifically, we calculated the length of the new shortest path between a pair of genes (A, B) upon the removal of the regulatory link between A and B. We take this value as a measure of the structural importance of the A − B link. Edges whose removal causes little difference in the length of the shortest path between the genes it connects can be thought of as redundant, because the two genes are still closely connected in the regulatory network, and the edge removal may thus be thought to have less effect on the network flow. On the other hand, edges whose removal causes the regulatory path between the two connected genes to increase substantially, or even disappear, are deemed to be informationally important.
The informational importance of an edge is dependent on the direction of regulation of the immediate neighbourhood of the two genes that edge connects. For example, consider a network of only three genes, A, B and C, in which gene A regulates genes B and C, and gene C also regulates gene B. In this network, the regulatory link from gene A to gene B has low informational importance since removing that interaction only increases the shortest path from A to B to two (from A to C to B). On the other hand, removing either the link from A to C or from C to B completely disrupts the information flow. In both cases, the shortest path will increase to infinity, and the edges will be deemed of high informational importance, because there is no longer any way for information to flow from gene A to gene C, or from gene C to gene B in the absence of those regulatory links.
In order to determine whether high annotation weight edges tend to be either redundant or essential to information flow, we ordered the edges in RegulonDB according to their weight in the projected gene network and then calculated the harmonic mean of the new shortest path for edges at or above each indexed value. The results are striking. Those edges with the highest scaled similarity values are of great importance to the information flow in the regulatory network (figure 5c). In contrast to 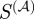 , other measures of functional similarity are not informationally important.
, other measures of functional similarity are not informationally important.
This result is curious because one might initially suspect that genes which share many low-level annotations should be in locally dense regions of the regulatory network and hence exhibit redundancy. It is worthwhile to note that any gene interaction that forms a ‘leaf’ in the regulatory network, i.e. a gene that is only regulated by a single transcription factor, will have high structural importance. Intuitively, we expect the genes in this type of regulatory relationship to have a high functional similarity. A more interesting scenario is when high functional similarity is indicative of multiple regulatory pathways flowing between a pair of genes, for example when both genes are transcription factors. In this case, these pathways may connect communities of genes that are independently involved in only a subset of functions, but which at times must be combined to perform higher order biological tasks.
3.4. Ontology-specific contributions to scaled similarity
In addition to applying our approach to the entire GO hierarchy, we also used it to determine separate functional gene networks for each of the three main branches of the GO hierarchy (see equation (2.4)). Here, we use this information to better understand what type of functional information is contributing to the predictive power and informational importance of our edges. The following analysis is performed only in the case of high α.
We considered the weight contributions from each ontology separately and determined the maximum F-score for the network predicted using annotations unique to that ontology (figure 6a). Edges that have a high weight contribution from the ‘Biological Process’ ontology are more predictive of known regulatory interactions than edges with high weight contributions from either the ‘Molecular Function’ ontology or the ‘Cellular Component’ ontology. A large part of this has to do with the types of functional terms assigned to each ontology, with regulatory terms such as ‘transcription’, ‘gene expression’, and ‘DNA replication’ all belonging to the ‘Biological Process’ ontology. Another more subtle issue is the size of the three ontologies. ‘Biological Process’ and ‘Molecular Function’ contain roughly the same number of annotated terms in E. coli with 1894 and 1784 members, respectively. One reason why the ‘Cellular Component’ ontology does a poorer job in predicting regulatory interactions may not only be owing to the types of functions it describes, but also because E. coli genes are only annotated to 204 terms in this branch of the hierarchy, leading to a sparser and less informational measure of functional similarity.
Figure 6.

The predictive power of each individual ontology and their contribution to the informational importance of the regulatory edges. (a) Comparison of the predictive power of each individual ontology, estimated by calculating the maximum F-score, based on the scaled similarity components originating from each of the three ontologies. (b) A plot of the informational importance of edges in RegulonDB, ordered by the scaled similarity values as calculated using each individual ontology. Black denotes all annotations; yellow, Biological Process; magenta, Molecular Function; cyan, Cellular Component.
We also examined the role of the three ontologies in determining the informational importance of an edge in the true regulatory network (figure 6b). Again, the ‘Biological Process’ carries the bulk, if not all of the information. In the light of the strong role of the ‘Biological Process’ ontology, one might question the need to include the other ontologies in calculating the scaled similarity between genes. However, given that both 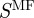 and
and 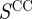 still predict regulatory interactions at a rate greater than random, the authors suggest that including these ontologies in the functional measure has the potential to reveal information about the relationships between genes.
still predict regulatory interactions at a rate greater than random, the authors suggest that including these ontologies in the functional measure has the potential to reveal information about the relationships between genes.
3.5. Regulatory interactions in yeast
To determine whether the scaled similarity measure is predictive of regulatory interactions in organisms other than E. coli, we used GO annotations made to genes in Saccharomyces cerevisiae (bakers yeast) to calculate functional similarity scores. As a ‘gold-standard’, we took interactions predicted by ChIP-chip [39] with a p-value of less than 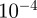 . The results for all the measures mentioned in §1.2.2 are shown in the electronic supplementary material, figure S2.
. The results for all the measures mentioned in §1.2.2 are shown in the electronic supplementary material, figure S2.
The results in yeast are very similar to E. coli. The scaled similarity (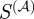 ) predicts real regulatory relationships better than either the semantic similarity or Kappa statistics (figure 7a). The latter once again predicts true regulatory interactions at a rate comparable to chance and slightly worse than
) predicts real regulatory relationships better than either the semantic similarity or Kappa statistics (figure 7a). The latter once again predicts true regulatory interactions at a rate comparable to chance and slightly worse than 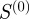 , which represents a count of the number of shared terms between two genes. Similarly, edges with the highest scaled similarity values are of great importance to the information flow in the regulatory network (figure 7b). Semantic similarity is slightly correlated with informational importance. Neither the Kappa statistic nor
, which represents a count of the number of shared terms between two genes. Similarly, edges with the highest scaled similarity values are of great importance to the information flow in the regulatory network (figure 7b). Semantic similarity is slightly correlated with informational importance. Neither the Kappa statistic nor 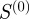 is predictive of the informational importance of an edge.
is predictive of the informational importance of an edge.
Figure 7.

The predictive power of functional similarity measures and their relationship to the informational importance of the edges in the yeast regulatory network. (a) Comparison of the predictive power of the functional similarity measures in yeast, estimated by calculating the maximum F-score. (b) A plot of the informational importance of edges in the yeast regulatory network, ordered by the functional similarity values estimated by each measure. Blue denotes scaled similarity 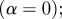 black, scaled similarity (high α); green, semantic similarity; red, Kappa statistic.
black, scaled similarity (high α); green, semantic similarity; red, Kappa statistic.
3.6. Comparison to expression-based network reconstruction methods
We also compared the predictive ability of our scaled similarity measure to that of the widely cited CLR network reconstruction algorithm [20], which returns a Z-score value for each gene-gene pair. In their paper, Faith et al. applied their approach to a compendium of expression profiles in E. coli. We downloaded the results from their paper to use as our comparison against the scaled similarity measure. Only a subset of regulatory edges can be assigned a positive Z-score in the CLR approach. Of the 2341 RegulonDB listed genes, 85 per cent (1979) exist in the CLR reconstructed network and of the 6725 RegulonDB regulatory interactions, 55 per cent (3696) have a non-negative reported Z-score.
According to our analysis, the maximum F-score for the CLR network occurs at the Z-score cut-off of 4.78, slightly lower but similar to the 5.78 cut-off used to define the regulatory network in the original paper [20]. It out-performs our functional measure in its ability to predict true regulatory interactions (figure 8a). However, CLR relies on the collection of many high-throughput experiments, whereas our scaled similarity measure is derived strictly from annotations. With this in mind, we propose that the scaled similarity measure could be used to cheaply reconstruct approximate gene regulatory networks in species that have gene annotations but for which a high-quality collection of experimental expression data does not yet exist.
Figure 8.

Comparison and combination of the scaled similarity with the Z-score calculated by the CLR network reconstruction algorithm. (a) Maximum F-score of the scaled similarity (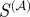 ), the Z-score calculated by CLR, and a combination of these measures. (b) Comparison of edge prediction by
), the Z-score calculated by CLR, and a combination of these measures. (b) Comparison of edge prediction by 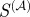 compared with Z-score of the CLR algorithm. We ordered the edges that appear in both networks both by their increasing scaled similarity value and by their Z-score. We coloured each (X, Y) based on the F-score of the set of the edges that have both a scaled similarity value greater than X and a Z-score greater than Y.
compared with Z-score of the CLR algorithm. We ordered the edges that appear in both networks both by their increasing scaled similarity value and by their Z-score. We coloured each (X, Y) based on the F-score of the set of the edges that have both a scaled similarity value greater than X and a Z-score greater than Y.
In order to better estimate the quality of a gene network predicted by our functional score compared with reconstruction approaches using gene expression data, we directly compared the values of the edges as determined by the scaled similarity to the Z-score values reported by the CLR reconstruction algorithm [20]. A little over half (1.2 million) of the edges that can be assigned a non-zero scaled similarity value in our projected gene network also have non-zero Z-score values reported by CLR. We determined the rank order of these edges based on their scaled similarity and also based on their Z-score values. We further identified which of these edges are listed as true regulatory interactions by RegulonDB and used this information to calculate the F-score of edges as a function of both their Z-score and scaled similarity, defining true positives as those edges above both the Z-score and similarity thresholds, which are also experimentally verified by RegulonDB. The results of the F-score calculation are illustrated in figure 8b. Both the scaled similarity 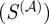 and the Z-score from CLR have similar patterns in their F-score. Curiously, the algorithms are most predictive for independent sets of edges, indicating that if one wished to use functional similarity to improve network reconstruction, it would be most beneficial to take edges predicted by either algorithm, rather than edges predicted by both.
and the Z-score from CLR have similar patterns in their F-score. Curiously, the algorithms are most predictive for independent sets of edges, indicating that if one wished to use functional similarity to improve network reconstruction, it would be most beneficial to take edges predicted by either algorithm, rather than edges predicted by both.
Our method relies upon the assumption that two genes which share many common functions should be involved in a regulatory relationship. Expression-based reconstruction methods, such as CLR, also often rely upon a similar assumption that when two genes are co-expressed one regulates the other. Although these are good first-order approximations, these assumptions can both miss true interactions (false negatives) as well as predict false interactions (false positives). To better understand how these assumptions might effect our predicted regulatory network, we specifically investigated the top edges predicted by our scaled similarity score to see what kinds of regulatory interactions our measure predicts (table 1). Of the top 10 edges by functional weight, half are found in the RegulonDB database. Several edges predicted by the scaled similarity that are not in RegulonDB are annotated with functions related to gene regulation. Interestingly, of the true positives, only three can attribute the majority of their similarity weight to annotations from the Biological Process ontology and these same edges have statistically significant Z-scores 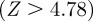 in the CLR algorithm. Edges correctly predicted by the scaled similarity measure that are missed by CLR are predicted based on annotations in the Molecular Function or Cellular Component ontologies.
in the CLR algorithm. Edges correctly predicted by the scaled similarity measure that are missed by CLR are predicted based on annotations in the Molecular Function or Cellular Component ontologies.
Table 1.
Properties of the most functionally similar edges ranked according to the scaled similarity measure. (Equal rank indicates a tie in the similarity score. For edges that appear in RegulonDB the direction of positive versus negative regulation is indicated by an up (↗) or down (↘) arrow, respectively. For all other edges arrows indicate regulation from a transcription factor to a gene. Regulation between two transcription factors is indicated by a bi-directional (↔) arrow. To determine the ontology that contributed most highly to the final score, edges weights were broken into their corresponding ontologies (see equation (2.4)). The percentage annotations from this ontology contributed to the final edge weight is noted in parentheses. BP, Biological Process; CC, Cellular Component; MF, Molecular Function.)
| rank | edge (TF → gene) | in CLR | in RegDB | ontology (%) |
|---|---|---|---|---|
| 1 | FabR → AccC | — | — | BP (100) |
| 2 | AlpA↗IntA | — | ✓ | CC (100) |
| 2 | Lrp ↔ AsnC | — | — | BP (100) |
| 4 | FadR↘FadD | — | ✓ | MF (100 |
| 5 | Ada → Ogt | — | — | MF (100) |
| 6 | Betl↘BetB | ✓ | ✓ | BP (100) |
| 6 | Betl↘BetA | ✓ | ✓ | BP (100) |
| 8 | FlhC↗FliT | ✓ | ✓ | BP (100) |
| 8 | FlhC → YdeH | — | — | BP (100) |
| 10 | FadR → AccC | — | — | BP (97) |
Because the scaled similarity and CLR are identifying a unique subset of true regulatory interactions, we are able to combine them in order to take advantage of the strengths of both approaches. As a simple model, we consider the same fraction of top edges in both CLR and our projected gene network and determined what percentage of this combined set is also in RegulonDB. In other words, we ordered the edges such that in the top N edges, N/2 of the edges are the N/2 edges with the highest scaled similarity value and N/2 of the edges are those with the highest Z-score values in CLR. Using this ranking, we determined the maximum F-score for the combination of the scaled similarity and Z-score values. This combined model outperforms the predictive power of either individual algorithm (figure 8a).
Previous groups have mentioned the power of combining functional and experimental measures in order to improve network reconstruction [7–9]. For example, Marcotte et al. calculated a log-likelihood score to evaluate an experiment's ability to correctly predict shared annotations between gene-pairs and then used this score to more accurately integrate many experiments together into one coherent network. In our case, the GO provides a unique addition to the set of predicted edges because those added are known to be functionally related, and, furthermore, as we demonstrated in §3.3, they are now also known to be links essential to the flow of information within the regulatory network.
4. Discussion
Although using the GO to evaluate the regulatory relationships between genes has many limitations, the method we describe here is inexpensive, computationally simple and takes advantage of a large amount of data that has already been accumulated. Compared with network reconstruction algorithms such as CLR, which focus on one type of biological data, GO annotations include results from many types of experiments. Furthermore, because the annotations are publicly accessible, the wealth of information in GO will continue to grow and be refined as researchers in the community use them for their wide variety of applications.
One advantage of using GO annotations to predict a functional network is that biological meaning can more easily be assigned to a predicted interaction. Although there are many different ways to ascribe functional similarity values to gene-pairs, the fact that the strength of the scaled similarity between two genes is correlated with the likelihood for those genes to be linked by a regulatory relationship and, furthermore, is predictive of that link's importance for information flow through the regulatory network, gives a much wider range of implications for the GO's use not only in constructing, but also in evaluating, regulatory networks.
One potential weakness of using the GO to interpret biological data, especially in the context of a regulatory network, is that there may be subjective biases in its construction. It is possible that a large number of shared specific annotations between two genes is merely a consequence of a large amount of research focused on those genes and/or shared functions. In these cases, the scaled similarity measure results in an overestimate of the ‘true’ functional similarity of the gene-pair. Furthermore, it is likely that many of the annotations in the GO are derived from known regulatory interactions. Therefore, it is virtually impossible to choose an unbiased gold standard with which to evaluate how well various functional measures can predict known regulatory interactions. Fortunately, when functional measures are based in the same database, any bias in their evaluation should effect the measures equally, such that their relative performance can still be used as a guide. We also point out that even if the correlation between known regulatory network interactions and the scaled similarity is a consequence of human bias in annotation (e.g. when it is witnessed that two genes are related in a regulatory fashion, they may be given a common annotation), it does not follow that these regulatory edges must also be the most important for information flow in the established experimental regulatory network, unless the experimental network itself is biased. In addition, biased construction of the experimental network is also possible, as the most informationally important edges may also be the easiest to experimentally verify. However, given the wide acceptance of RegulonDB as a ‘gold standard’, we do not wish to dwell on these possibilities in this analysis. Instead, we look forward to seeing if these striking results continue to manifest themselves as the quality of the network continues to improve, as well as in the experimental regulatory networks of other species as they become available at increasing quality.
Although other measures have previously been proposed to assess the functional similarity between genes, these measures, despite biological intuition, are not as well correlated with the existence of regulatory relationships as the scaled similarity introduced here. We demonstrate that, at least within a regulatory network context, it is critical to consider the specificity of a biological function. Although previously it was unclear how to interpret the meaning of shared annotations between pairs of genes in a regulatory network context, a measure that correlates functional similarity with known regulatory interactions allows us to more accurately assign functional meaning to links in a regulatory context. In particular, we witness that links between genes with high scaled similarity should be more important to information flow in the regulatory network.
The code for calculating the scaled similarity as well as other additional files pertaining to this work can be found at www.networks.umd.edu.
References
- 1.Ashburner M., et al. 2000. Gene ontology: tool for the unification of biology. The Gene Ontology consortium. Nat. Genet. 25, 25–29 10.1038/75556 (doi:10.1038/75556) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Consortium T. G. O. 2010. The Gene Ontology in 2010: extensions and refinements. Nucleic Acids Res. 38(Suppl. 1), D331–D335 10.1093/nar/gkp1018 (doi:10.1093/nar/gkp1018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang D. W., et al. 2007. DAVID Bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 35(Suppl. 2), W169–W175 10.1093/nar/gkm415 (doi:10.1093/nar/gkm415) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mostafavi S., Morris Q. 2010. Fast integration of heterogeneous data sources for predicting gene function with limited annotation. Bioinformatics 26, 1759–1765 10.1093/bioinformatics/btq262 (doi:10.1093/bioinformatics/btq262) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.King O. D., Foulger R. E., Dwight S. S., White J. V., Roth F. P. 2003. Predicting gene function from patterns of annotation. Genome Res. 13, 896–904 10.1101/gr.440803 (doi:10.1101/gr.440803) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Youn A., Reiss D. J., Stuetzle W. 2010. Learning transcriptional networks from the integration of ChIP-chip and expression data in a non-parametric model. Bioinformatics 26, 1879–1886 10.1093/bioinformatics/btq289 (doi:10.1093/bioinformatics/btq289) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee I., Date S. V., Adai A. T., Marcotte E. M. 2004. A probabilistic functional network of yeast genes. Science 306, 1555–1558 10.1126/science.1099511 (doi:10.1126/science.1099511) [DOI] [PubMed] [Google Scholar]
- 8.Franke L., Van Bakel H., Fokkens L., de Jong E. D., Egmont-Petersen M., Wijmenga C. 2006. Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am. J. Hum. Genet. 78, 1011–1025 10.1086/504300 (doi:10.1086/504300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang X., Zhou Y., Jin R., Chan C. 2009. Reconstruct modular phenotype-specific gene networks by knowledge-driven matrix factorization. Bioinformatics 25, 2236–2243 10.1093/bioinformatics/btp376 (doi:10.1093/bioinformatics/btp376) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newman M. E. J. 2003. The structure and function of complex networks. SIAM Rev. 45, 167–256 10.1137/S003614450342480 (doi:10.1137/S003614450342480) [DOI] [Google Scholar]
- 11.Milo R., Shen-Orr S., Itzkovitz S., Kashtan N., Chklovskii D., Alon U. 2002. Network motifs: simple building blocks of complex networks. Science 298, 824–827 10.1126/science.298.5594.824 (doi:10.1126/science.298.5594.824) [DOI] [PubMed] [Google Scholar]
- 12.Solé R. V., Cancho R. F., Montoya J. M., Valverde S. 2002. Selection, tinkering, and emergence in complex networks. Complex 8, 20–33 10.1002/cplx.10055 (doi:10.1002/cplx.10055) [DOI] [Google Scholar]
- 13.Jeong H., Mason S. P., Barabási A. L., Oltvai Z. N. 2001. Lethality and centrality in protein networks. Nature 411, 41–42 10.1038/35075138 (doi:10.1038/35075138) [DOI] [PubMed] [Google Scholar]
- 14.Wagner A. 2001. The yeast protein interaction network evolves rapidly and contains few redundant duplicate genes. Mol. Biol. Evol. 18, 1283–1292 [DOI] [PubMed] [Google Scholar]
- 15.Guimera R., Nunes Amaral L. A. 2005. Functional cartography of complex metabolic networks. Nature 433, 895–900 10.1038/nature03288 (doi:10.1038/nature03288) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao J., Yu H., Luo J., Cao Z., Li Y. 2006. Complex networks theory for analyzing metabolic networks. Chin. Sci. Bull. 51, 1529–1537 [Google Scholar]
- 17.Gama-Castro S., et al. 2008. RegulonDB (version 6.0): gene regulation model of Escherichia coli K-12 beyond transcription, active (experimental) annotated promoters and Textpresso navigation. Nucl. Acids Res. 36(Suppl. 1), D120–D124 10.1093/nar/gkm994 (doi:10.1093/nar/gkm994) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lord P. W., Stevens R. D., Brass A., Goble C. A. 2003. Semantic similarity measures as tools for exploring the Gene Ontology. Pac. Symp. Biocomput. 8, 601–612 [DOI] [PubMed] [Google Scholar]
- 19.Huang D. W., et al. 2007. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 8, R183. 10.1186/gb-2007-8-9-r183 (doi:10.1186/gb-2007-8-9-r183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faith J. J., Hayete B., Thaden J. T., Mogno I., Wierzbowski J., Cottarel G., Kasif S., Collins J. J., Gardner T. S. 2007. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 5, e8. 10.1371/journal.pbio.0050008 (doi:10.1371/journal.pbio.0050008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Gene Ontology Consortium 2001. Creating the Gene Ontology resource: design and implementation. Genome Res. 11, 1425–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clauset A., Shalizi C. R., Newman M. E. J. 2009. Power-law distributions in empirical data. SIAM Rev. 51, 661–703 [Google Scholar]
- 23.Pesquita C., Faria D., Falcão A. O., Lord P., Couto F. M. 2009. Semantic similarity in biomedical ontologies. PLoS Comput. Biol. 5, e1000443. 10.1371/journal.pcbi.1000443 (doi:10.1371/journal.pcbi.1000443) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ovaska K., Laakso M., Hautaniemi S. 2008. Fast gene ontology based clustering for microarray experiments. BioData Mining 1, 11. 10.1186/1756-0381-1-11 (doi:10.1186/1756-0381-1-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Resnik P. 1995. Using information content to evaluate semantic similarity in a taxonomy. In Proc. 14th Int. Joint Conf. on Artificial Intelligence, August 1995, Montreal, Canada, pp. 448–453 [Google Scholar]
- 26.Lin D. 1998. An information-theoretic definition of similarity. In Proc. 15th Int. Conf. on Machine Learning, July 1998, Madison, WI, USA, pp. 296–304 [Google Scholar]
- 27.Jiang J. J., Conrath D. W. 1997. Semantic similarity based on corpus statistics and lexical taxonomy. In Int. Conf. Res. on Computational Linguistics (ROCLING X), 1997, Taiwan, 9008 [Google Scholar]
- 28.Schlicker A., Domingues F., Rahnenfuhrer J., Lengauer T. 2006. A new measure for functional similarity of gene products based on gene ontology. BMC Bioinform. 7, 302. 10.1186/1471-2105-7-302 (doi:10.1186/1471-2105-7-302) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H., Azuaje F., Bodenreider O., Dopazo J. 2004. Gene expression correlation and gene ontology-based similarity: an assessment of quantitative relationships. In CIBCB ‘04. Proc. 2004 IEEE Symp. on Computational Intelligence in Bioinformatics and Computational Biology, La Jolla, San Diego, CA, USA, pp. 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sevilla J. L., Segura V., Podhorski A., Guruceaga E., Mato J. M., Martinez-Cruz L. A., Corrales F. J., Rubio A. 2005. Correlation between gene expression and GO semantic similarity. Comput. Biol. Bioinform. IEEE/ACM Trans. 2, 330–338 [DOI] [PubMed] [Google Scholar]
- 31.Guo X., Liu R., Shriver C. D., Hu H., Liebman M. N. 2006. Assessing semantic similarity measures for the characterization of human regulatory pathways. Bioinformatics 22, 967–973 10.1093/bioinformatics/btl042 (doi:10.1093/bioinformatics/btl042) [DOI] [PubMed] [Google Scholar]
- 32.Pesquita C., Faria D., Bastos H., Ferreira A., Falcao A., Couto F. 2008. Metrics for GO based protein semantic similarity: a systematic evaluation. BMC Bioinform. 9(Suppl. 5), S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chabalier J., Mosser J., Burgun A. 2007. A transversal approach to predict gene product networks from ontology-based similarity. BMC Bioinform. 8, 235. 10.1186/1471-2105-8-235 (doi:10.1186/1471-2105-8-235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin D., Brun C., Remy E., Mouren P., Thieffry D., Jacq B. 2004. GOToolBox: functional analysis of gene datasets based on Gene Ontology. Genome Biol. 5, R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butte A. J., Kohane I. S. 2000. Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements. Pac. Symp. Biocomput. 5, 418–429 [DOI] [PubMed] [Google Scholar]
- 36.Zhu J., et al. 2004. An integrative genomics approach to the reconstruction of gene networks in segregating populations. Cytogenet. Genome Res. 105, 363–374 10.1159/000078209 (doi:10.1159/000078209) [DOI] [PubMed] [Google Scholar]
- 37.Margolin A. A., Nemenman I., Basso K., Wiggins C., Stolovitzky G, Dalla Favera R., Califano A. 2006. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 7(Suppl. 1), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee I., Li Z., Marcotte E. M. 2007. An improved, bias-reduced probabilistic functional gene network of bakers yeast, Saccharomyces cerevisiae. PLoS ONE 2, e988. 10.1371/journal.pone.0000988 (doi:10.1371/journal.pone.0000988) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacIsaac K. D., Wang T., Gordon D. B., Gifford D. K., Stormo G. D., Fraenkel E. 2006. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinform. 7, 113. 10.1186/1471-2105-7-113 (doi:10.1186/1471-2105-7-113) [DOI] [PMC free article] [PubMed] [Google Scholar]