

Abstract
Recent analyses with high-resolution single-molecule experimental methods have shown highly irregular and variable bursting of mRNA in a wide range of organisms. Noise in gene expression is thought to be beneficial in cell fate specifications, as it can lay a foundation for phenotypic diversification of isogenetic cells in the homogeneous environment. However, because the stability of proteins is, in many cases, higher than that of mRNAs, noise from transcriptional bursting can be considerably buffered at the protein level, limiting the effect of noisy mRNAs at a more global regulation level. This raises a question as to what constructive role noisy mRNAs can play in the system-level dynamics. In this study, we have addressed this question using the computational models that extend the conventional transcriptional bursting model with a post-transcriptional regulation step. Surprisingly, by comparing this stochastic model with the corresponding deterministic model, we find that intrinsic fluctuations can substantially increase the expected mRNA level. Because effects of a higher mRNA level can be transmitted to the protein level even with slow protein degradation rates, this finding suggests that an increase in the protein level is another potential effect of transcriptional bursting. Here, we show that this striking steady state increase is caused by the asynchronous nature of molecular reactions, which allows the transcriptional regulation model to create additional modes of qualitatively distinct dynamics. Our results illustrate non-intuitive effects of reaction asynchronicity on system dynamics that cannot be captured by the traditional deterministic framework. Because molecular reactions are intrinsically stochastic and asynchronous, these findings may have broad implications in modelling and understanding complex biological systems.
Keywords: transcriptional bursting, stochastic chemical kinetics, deviant effects
1. Introduction
The degree of stochasticity displayed in the regulation of biological processes can change based on its network structure as well as other network properties [1–5]. Depending on the characteristics of the underlying reaction network, stochastic effects can play a crucial role in shaping dynamics and functions of a biological system [6–8]. Importantly, such dynamic properties may not be accurately described by continuous-deterministic approximations based on mass-action kinetics or classical chemical kinetics (CCK) [9,10] as it neglects the underlying fluctuations [11]. In order to characterize how dynamical properties are shaped from the network properties given the underlying fluctuations in silico, then, it may be necessary to use a discrete-stochastic modelling approach based on stochastic chemical kinetics (SCK) [12–15].
Gene regulation processes are particularly susceptible to such fluctuations as the molecule counts of participating species (e.g. cis-regulatory units and transcription factors) can be very low. Recent advances in single-molecule experimental methods have made it possible to observe the expression of mRNA in greater detail, enabling discoveries of high variabilities in mRNA levels in a wide range of organisms such as prokaryotes [16–18], primitive eukaryotes [19,20] and higher eukaryotes [21,22]. Furthermore, previous studies have reported that such bursting can be seen in the expression of genes with widely different transcription kinetics [18,22,23]. These suggest that transcriptional bursting is a general biological phenomenon—rather than a property of a specific model system with a specific range of gene expression kinetics. Without relying on any external factors, this phenomenon can be captured by a simple model of mRNA regulation, where transcription is initiated by a two-state promoter that can randomly switch between an active state and an inactive state (figure 1a) [3,24]. Although the underlying mechanism of the stochastic promoter-state transition involved in transcriptional bursting is not decisively known in general, chromatin remodelling [25,26] was experimentally suggested as a cause of stochastic gene expression in some eukaryotes [3,24,27].
Figure 1.

Comparison of the two transcriptional bursting models. Here, species M is mRNA and species P is the protein. The active promoter state is represented by A, and the inactive promoter state is represented by Ac. The active catalyst for mRNA degradation is B, and the inactive catalyst is Bc. The red broken line shows the steady state from the CCK model, whereas the black solid line shows the mean steady state from the SCK model. (a) A model for a gene expression process with a two-state promoter (left). A sample time evolution and the steady state distribution of M (top-right) and P (bottom-right). The mean behaviour of M and P at the stationary regime in the SCK model is accurately captured by the CCK model. The parameter values and the initial condition used for this simulation are as follows: k1 = 2; k2 = 0; k3 = 0.2; k4 = 0.02; k5 = 0; k6 = 0; k7 = 0.06; k8 = 0.00077; k9 = 0.014; A(0) = 1; Ac(0) = 0; B(0) = 0 and Bc(0) = 0. (b) A model for a gene regulation process with two-state promoter and two-state catalyst of mRNA degradation (left). A sample time evolution and the steady state distribution of mRNA (top-right) and the protein (bottom-right). In this model, there is a deviation between the SCK model and the CCK model in that the mean behaviour of M and P at the stationary regime in the SCK model is higher than what is predicted by the CCK model. The parameter values and the initial condition used for this simulation are: k1 = 2; k2 = 0.0137; k3 = 0.2; k4 = 0.02; k5 = 0.01; k6 = 0.001; k7 = 0.06; k8 = 0.00077; k9 = 0.00139; A(0) = 1; Ac(0) = 0; B(0) = 10; and Bc(0) = 0. Here, the units of all of the parameters are min−1. For the justification of the parameter values, see the electronic supplementary material, §1.
While the stochastic two-state promoter model can display irregular, pulsatile gene transcription, the mean time evolution of the mRNA level in this SCK model is accurately captured by the corresponding CCK model as all of the reactions have first-order kinetics [28]. This implies that the average protein count, at the steady state regime, can be accurately captured by the corresponding CCK model. Furthermore, effects of noise in mRNAs can be considerably suppressed at the protein level with a slow protein degradation rate (figure 1a). Thus, this reaction model suggests that transcriptional bursting may not be particularly useful for facilitating phenotypic variations when the stability of a protein is high [21,29].
This observation raises a question as to what effects, if any, the fluctuations in the mRNA regulation can have on protein regulation and more global function given that protein half-life may be very long compared with stochastic fluctuations in mRNA expression level. To address this question, we created a gene expression process model that extends the two-state promoter transcriptional bursting model by including a control of mRNA degradation (figure 1b). This post-transcriptional control can represent, for example, conformational changes of such mRNA decapping enzymes as Dcp2 [30, 31]. Because post-transcriptional controls can play a crucial role in gene regulation [32], we investigated whether or not effects of the fluctuations on the system dynamics can also be altered by considering the control of mRNA stability. The potential importance of post-transcriptional controls in the system-level dynamics has been explored in different contexts in previous theoretical studies [33–35]. For example, a previous study has shown that stochastic effects in a two-gene negative feedback network with a highly cooperative transcriptional repression process can be reduced in both magnitude and frequency, leading to a more coordinated cell-to-cell response by considering the mRNA degradation control step [35].
Here, we explore the role of transcriptional bursting in a more general context by extending the two-state promoter model to consider the expression of a single gene without any feedback loops. Our analysis shows that, unlike the two-state promoter model, our gene expression model can result in steady state behaviours dramatically different from what is predicated by the CCK model. More specifically, we find that the fluctuations in our model can make the expected steady state of mRNA higher, which, in turn, can substantially increase the expected protein level (figure 1b).
While this novel finding suggests that another potential constructive role of underlying fluctuations in mRNA regulation is to increase the expected protein level, how this phenomenon emerges by including a post-transcriptional regulation in the conventional transcriptional bursting model is unclear. To this end, we developed several computational models to specifically analyse how the expected steady state mRNA level can be increased. Our analysis shows that the origin of the steady state gain in the mRNA level in our stochastic gene expression model is the underlying asynchronicity of molecular reactions, which cannot be captured by the traditional deterministic framework. The asynchronous effect in the core part of the mRNA regulatory machinery allows the generation of additional modes of qualitatively distinct dynamics, which in turn can substantially influence the steady state level of mRNA. In particular, we find that, owing to the specific network structure of the gene expression model that we studied, the expected steady state mRNA level can be inevitably increased by this asynchronous effect. By showing that large deviations between stochastic and deterministic steady state predictions are possible with a much simpler mechanism than has previously been considered in gene expression processes, our results imply that similar effects could be a far more widespread phenomenon than is currently appreciated. This observation may have important implications for realistic modelling and better understanding of complex biological reaction systems in general.
2. Results
2.1. Gene expression reaction model
In this study, we extend the two-state promoter gene expression model (figure 1a) by including a catalytic mRNA degradation (figure 1b). The mRNA degradation catalyst has two functionally different states: one is an active state in which the catalyst can degrade mRNA; and the other one is an inactive state in which the catalyst is incapable of degrading mRNA. From this specification, we constructed a reaction model to understand what dynamic behaviours can arise from the given structure of the reaction network. Our reaction model is as follows:
| 2.1 |
| 2.2 |
| 2.3 |
| 2.4 |
| 2.5 |
Reaction (2.1) models the regulation of an mRNA, M, that is constitutively expressed from an active promoter, A, and is down-regulated by the presence of a degradative agent, B. Reaction (2.2) captures the state transition of the promoter between an active state (A) and an inactive state (Ac). The functional changes in the degradation catalyst are described by reaction (2.3), where B is the active state and Bc is the inactive state. Reaction (2.4) models the regulation of protein P, which is synthesized using M as a template and is degraded at a rate proportional to its molecule count, and reaction (2.5) describes the basal degradation of M.
2.2. Steady state gain via fluctuations
To test if the added mRNA degradation control in our reaction model can alter the role the underlying fluctuations play in the steady state behaviour of gene expression, we simulated the corresponding SCK model using the stochastic simulation algorithm (SSA) [36]. The parameter values of our gene expression model are set mainly based on the recent quantitative gene expression results on mouse fibroblasts [22] (for detail see the electronic supplementary material, §1). Our simulation results show that, similar to that of the two-state gene expression model (figure 1a), M(∞), the mRNA count in the stationary regime, has a long-tail distribution with a high variance (figure 1b). However, unlike that of the two-state gene expression model, 〈M(∞)〉 can be higher than what is predicted by the CCK model (figure 1b). This steady state gain in mRNA can lead to a substantially increased steady state level of the protein. Indeed, our simulation results show that, while the high noise level and the long-tail distribution seen in M(∞) is not observed in P(∞), 〈P(∞)〉 for the SCK model can be substantially higher than that for the CCK model (figure 1b). Our simulation results also show that an increase in the steady state of the protein level becomes substantial as the stability of the protein increases (electronic supplementary material, §2 and figure S1).
2.3. Simplified reaction model
In order to gain insights into how the expected mRNA level is increased with the fluctuations, we constructed a simplified reaction model of the gene expression process so that we can extensively analyse what properties of the reaction network give rise to this phenomenon. Thus, in this simplified model, we included a minimal reaction set that we believed could give rise to higher expected mRNA levels with the fluctuations by extending the two-state promoter model. In our analysis of this model, we set the values of the parameters so that the steady state mRNA level is within a biologically relevant range [18,22,23]. Our simplified model has the following reactions:
| 2.6 |
| 2.7 |
| 2.8 |
With reaction (2.6) alone, M(t) follows a simple birth–death process, which is governed by the following master equation:
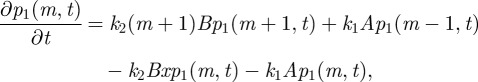 |
2.9 |
where p1(m,t)dt is defined as the probability that M = m in the time interval [t,t + dt). Setting equation (2.9) equal to 0 and solving for p1(m,t) can give us the stationary distribution of this process as follows:
| 2.10 |
which is the Poisson distribution with mean k1A/(k2B). Thus, 〈M(∞)〉 without reactions (2.7) and (2.8) can be obtained by solving for the steady state of the CCK model (electronic supplementary material, §3).
The master equations of reactions (2.7) and (2.8) are identical, and have the following form:
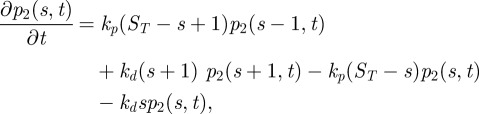 |
2.11 |
where S and Sc are the active form and the inactive form of a molecular species, respectively, p2(s,t)dt is the probability that S = s in the time interval [t,t + dt), and ST is S(0) + Sc(0). By solving this finite birth–death process for the stationary distribution of S, we can obtain:
| 2.12 |
where q = kp/(kp + kd), which is the binomial distribution with n = ST and p = q. Thus, the stationary mean of A and B is equal to STq, which can be accurately predicted by the steady state of the CCK model (electronic supplementary material, §3). In this study, we set A(0) = B(0) = 1 and Ac(0) = Bc(0) = 0 for analysis. Note that these constraints are chosen to make our analysis simpler to give broader conclusions about any potential effects of underlying fluctuations on the steady state behaviour.
2.4. Steady state analysis
The mean evolution of our SCK model is expressed by the following set of differential equations (electronic supplementary material, §4):
| 2.13 |
| 2.14 |
| 2.15 |
Here, we are interested in obtaining M∞, the value of 〈M(∞)〉. By setting equations (2.13)–(2.15) to 0 and using the relation 〈B(t)M(t)〉 = 〈B(t)〉×〈M(t)〉 + Cov(B(t),M(t)), we can derive the following expression for M∞:
| 2.16 |
However, the exact solution of M∞ becomes complicated because of the presence of Cov(B(∞),M(∞)), which essentially comes from the quadratic moment involved in equation (2.13). If we assume that there is no fluctuation in the system (i.e. Cov(B(t),M(t)) = 0), then the expression of M∞ in equation (2.16) becomes simplified. Indeed, this approximation is essentially that made by CCK [28], from which we can write down the expression of ME, the value of 〈M(∞)〉 given by our CCK model, as follows:
| 2.17 |
By assuming there is no fluctuation in the system, the steady state of 〈M(t)〉 thus depends only on two parameters: k1 and k2.
2.5. Deviation from the steady state behaviour of the CCK model
Using the simplified SCK model, we first investigated the cases in which the conformational change reactions of A and B in the simplified model are in the fast time-scale. Our analytical study suggests that ME can accurately predict M∞ when the fast-scale conformational change reaction assumption is valid (electronic supplementary material, §5). This result is in agreement with the basis of the stochastic rapid equilibrium approximation [37, 38], which can substantially lower the computational cost of the SSA.
We next considered cases in which the fast-scale reaction (2.7) assumption is no longer valid, and simulated the SCK model using the SSA (see §4). Our simulation results show that M in the stationary regime can exhibit very infrequent, large bursts and frequent, small bursts, implying a long-tail distribution of M(∞) (figure 2a). Furthermore, the simulation results show that deviant effects of M∞ can become so substantial that the prediction from the CCK model becomes quantitatively and qualitatively misleading when the rapid equilibrium assumption does not hold (figure 2b). This indicates that, unlike ME, which is a function of just k1 and k2, M∞ does in fact depend on the values of kp and kd, and inactivation of A and B can substantially change the steady state behaviour of M.
Figure 2.

Comparison of the behaviour of M(t) between the classical chemical kinetics (CCK) model and the stochastic chemical kinetics (SCK) model of the simplified gene expression reactions. (a) A sample trajectory of M(t) from the SCK model. (b) Comparison between M(t) of the CCK model (red solid line) and 〈M(t)〉 of the SCK model (blue dashed line). (c) Stationary mean of M with respect to various combinations of kp and kd. The circles represent the data points sampled via simulations. (d) Log–log heat map to show the ratio of M∞ to ME for various combinations of kp and kd. The numbers shown in the x- and y-axis are kd and kp in log scale (base 10), respectively. The ratio in the z-axis is given by log10 (M∞/ME). The values of the parameters used in this numerical analysis are k1 = 10, k2 = 1 and M(0) = 100. In the simulation of (a) and (b), the values of kd and kp are set to 0.1 and 0.01, respectively.
In order to gain more insights into how M∞ depends on the two parameters, we performed simulation of the SCK model for various combinations of kp and kd values (see §4). These parameter combinations are in a biologically realistic range in terms of mRNA abundances [18,23,39], which includes parameter regions that have been shown to give transcriptional bursting in some mammalian cells [21,22]. Our results indicate that, while the dependency of M∞ on kp and kd appears to be rather complex, we can find several overall trends (figure 2c). For instance, overall, M∞ is a decreasing function of kp that reaches a plateau when kp is very small. On the other hand, M∞ appears to be a function of kd that has minimum at ME when kd is either very high or very low, and has maximum at kd = kp. Our simulation results also suggest that M∞ never goes below what is predicted by ME (i.e. k1/k2). In other words, underlying fluctuation appears to increase the mean value of M in the stationary regime (figure 2d).
2.6. Role of discreteness in deviant dynamics
One difference between the SCK model and the CCK model is that, while the former is a discrete-state process, the latter is a continuum process. Thus, we analysed whether or not discreteness plays a role in the deviant effect. To do this, we constructed the chemical Langevin equation (CLE) [40] as a continuous-stochastic approximation of our SCK model, and simulated for various combinations of kp and kd values (see §4). Our simulation results show that the continuous approximation still gives rise to deviations, albeit the magnitude of which is not as substantial as with the discrete-stochastic SCK model (figure 3 and the electronic supplementary material, figures S2 and S3). Also, the shape of the steady state distribution of M for each parameter combination is found to resemble what is predicted from the simulation of the SCK model. Our findings suggest that, while it is not the source of the deviant effects in our model, discreteness can substantially amplify the deviation.
Figure 3.
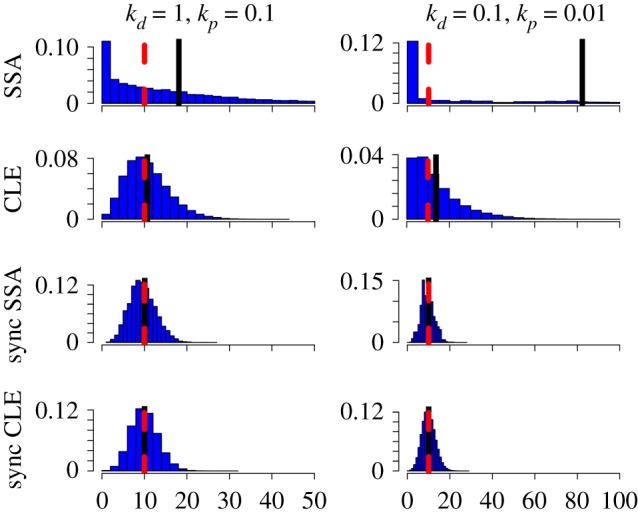
Comparison of steady state distributions of M for the four stochastic models for two sets of kp and kd. The first row (stochastic simulation algorithm, SSA) shows the simulation results of the stochastic chemical kinetics (SCK) model via stochastic simulation algorithm (SSA). The second row (chemical Langevin equation, CLE) shows the results of continuous-stochastic approximation via CLE. The third row (sync SSA) shows the results of the modified SCK model via SSA where the dynamics of A and B is set to be perfectly coordinated. The fourth row (sync CLE) shows the results of the modified CLE where the dynamics of A and B is set to be perfectly coordinated. Here, the red broken line represents ME, whereas the black solid line represents M∞. Here, to show the discrepancies clearly, cutoff lines are used in the histograms. For the histograms on the left column, 50 is used for the cutoff line, whereas for the histograms on the right column, 100 is used for the cutoff line.
2.7. Effects of unsynchronized conformational change reactions on deviation
While A(t) and B(t) have the same time-dependent and steady state probability mass function, the conformational change reaction events of A and B may not be coordinated well, as they are statistically independent. This reaction asynchronicity allows the vector of random variables, V ≡ [A,B], to traverse a Markov chain with four states, namely [1, 1], [1, 0], [0, 1] and [0, 0] (figure 4a). To test if the unsynchronized A and B play a role in deviations of M∞ from what is predicated by ME, we constructed a hypothetical discrete-stochastic model where transitions of A and B are modified so that they are exactly coordinated (i.e. for all t ≥ 0, A(t) = B(t)). In this model, V can be represented by a Markov chain with only two states, namely [1, 1] and [0, 0] (figure 4b). Note that, while this modification changes Cov(A(∞),B(∞)) from 0 to q, it does not have any effect on the time-dependent and steady state probability distributions of A and B. Also note that this modification has no effects on ME, as the CCK model is not capable of capturing reaction asynchronicity of A and B.
Figure 4.

Steady state gain via reaction asynchronicity. (a) The four-state Markov chain for A and B in the SCK model. (b) The two-state Markov chain for synchronized A and B. (c) Stationary means of M conditional on A and B for (i) kd = 0.1 and for (ii) kp = 0.1.
We simulated this modified SCK model using the SSA to estimate the steady state probability distribution and M∞. Our simulation result shows that, like the CCK model, the hypothetical synchronization of the A and B reactions in the SCK model makes M∞ independent of kp and kd (figure 3 and the electronic supplementary material, figure S4). Furthermore, it shows that steady state distribution of M for each parameter combination has a shape that resembles p1(m,∞) (i.e. a Poisson distribution with λ = k1/k2). The steady state behaviour of M in the modified SCK model can also be analytically derived by letting p4(a,b) be the joint probability mass function of A and B in the stationary regime, p5(m) be the probability mass function of M in the stationary regime, and p6(m | a, b) be the conditional steady state probability mass function of M given A = a and B = b in the stationary regime. Then, p5(m) of the modified model can be given by:
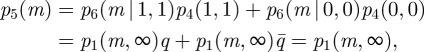 |
2.18 |
where 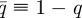 . Thus, while the steady state behaviour of the SCK model may not be captured by the CCK model, that of the modified SCK model is accurately described by the CCK model. This indicates that the discrepancy between M∞ and ME arises solely from the deviations between the SCK model and the modified SCK model, that is, the reaction asynchronicity of A and B.
. Thus, while the steady state behaviour of the SCK model may not be captured by the CCK model, that of the modified SCK model is accurately described by the CCK model. This indicates that the discrepancy between M∞ and ME arises solely from the deviations between the SCK model and the modified SCK model, that is, the reaction asynchronicity of A and B.
In order to understand if this causal relationship can also be applied to the continuous-stochastic model, we modified the CLE to have the synchronized A and B reactions. Our simulation result from the modified CLE indicates that, even with the continuous approximation, the reaction asynchronicity alone can give rise to the deviant effect (figure 3).
2.8. The cause of stochastic steady state gain
We next turn our attention to analysis of the network property that increases the steady state of M via introduction of fluctuations in the system. Because unsynchronized A and B via reaction asynchronicity is the main cause of the deviant effects, the stochastic steady state gain cannot occur without this reaction asynchronicity. However, the conformational switching reactions in our model setting are symmetric and do not seem to have a capacity to bias—one way or another—the directionality of the level of M. Thus, our initial hypothesis is that the steady state gain results from the mRNA synthesis and degradation reactions affected by the asynchronous mechanism.
To this end, we derived an analytical expression for the condition under which M∞ ≥ ME is true as follows (for detail see the electronic supplementary material, §6):
| 2.19 |
where w ≡ 1/(kp + kd) is the average pausing time of V in states [0, 1] and [1, 0] per visit. This condition states that, in our simple gene expression model, the average mRNA synthesis count is always greater than or equal to the average mRNA degradation count in the same time window, given that the mean mRNA count is initially set to ME. Thus, this can give insights into the underlying cause for the emergence of a negative value of Cov(B(∞),M(∞)), which is necessary for M∞ > ME as shown in equation (2.16).
Here, when w is very small, the left-hand side is equal to the right-hand side, in which case there is no discrepancy in the steady states of the stochastic model and the deterministic model (for detail see the electronic supplementary material, §6). That is, when the sum of kp and kd has a large value, M∞ can be accurately captured by ME. Furthermore, this condition shows that in order to have M∞ ≫ ME, the average pausing time for w must be large. However, this does not mean that whenever w is large, we have deviations. Condition (2.19) further explains why M∞ ≈ ME when kp is large (figure 2c,d). When the rate of the activation reaction of A and B is comparable with that of the transcription reaction (i.e. kp ≈ k1), w is very small relative to the transcription rate. In addition, when the activation reaction of A and B is much faster than the inactivation reaction of A and B (i.e. kp ≫ kd), both A and B are almost always active, limiting the effects of asynchronous states.
To gain further insights into the effect of the asynchronous mechanism, we partitioned simulation results into the four 〈M(∞) | a, b〉 values, the stationary mean of M conditional on A = a and B = b, for various combinations of kp and kd (figure 4c). Our analysis shows that there can be a high skewness in the four conditional steady states, where 〈M(∞) | 1, 0〉 is the highest and 〈M(∞) | 0, 1〉 is the lowest for each parameter set. It also indicates that M∞ is bounded by 〈M(∞) | 1, 1〉 and 〈M(∞) | 0, 0 〉, where 〈M(∞) | 1, 1〉 is the lower bound and 〈M(∞) | 0, 0〉 is the upper bound. We observed that 〈M(∞) | 0, 0〉 is the asymptote of M∞ as kp becomes smaller than kd, while 〈M(∞) | 1, 1〉 is the asymptote of M∞ as kd becomes smaller than kp. This result is consistent with our prediction, since when kp ≫ kd the system spends a large portion of time in V = [1, 1], while when kd ≫ kp the system spends a large portion of time in V = [0, 0]. From expression (2.19), a lower bound and an upper bound of 〈M(∞) | 1, 1〉 can be expressed by ME and M∞, respectively. This also means that M∞ is a lower bound of 〈M(∞) | 0, 0〉 because it is an upper bound 〈M(∞) | 1, 1〉. Taken together, these observations indicate that a complex interplay of mRNA synthesis and degradation made possible by the asynchronous mechanism underpins condition 2.19, which, in turn, results in a non-positive value of Cov(B(∞),M(∞)).
3. Discussion
Randomness is an inevitable feature of biological processes. Assuming that fluctuations are subject to selective pressure, biological systems must have evolved to feature molecular reaction interactions that can—to some degree—control and exploit such underlying uncertainties to better meet their objectives [8,41]. For example, previous theoretical studies showed that underlying fluctuations can induce bistable oscillation even when the corresponding deterministic model is monostable [42,43]. Another example of interesting stochastic effects is stochastic focusing where sensitivity of signal-response systems can be amplified with nonlinear, hyperbolic inhibition reactions, whose mean rates become different from the corresponding fluctuation-free, deterministic reaction rates [44]. A qualitative prediction of stochastic focusing was later performed with analysis of the curvature–covariance effect, which examined the deviation of the mean of reaction rate from the corresponding deterministic reaction rate based on the contribution of concentration covariances and the curvature of its kinetic function [45].
A particularly interesting example of stochastic effects is random phenotypic variations of isogenic populations. Indeed, with the recent advances in single-molecule and single-cell experimental techniques, we are able to appreciate, in greater detail, the role the fluctuations play in phenotypic diversifications of isogenic cells in homogeneous environments [46–48]. To diversify cell types, therefore, high transcriptional bursting can be very advantageous when the effect is transmitted to the protein level. However, because a protein's stability is often much higher than that of mRNA, effects of transcriptional bursting are largely buffered at the protein level. Furthermore, widespread observations of transcriptional bursting [18,22,49] suggest that such noisy mRNA molecules may not always be used to facilitate random phenotypic variations.
Here, we have reported another potential role of the fluctuations in the mRNA regulation by considering a post-transcriptional control. Surprisingly, we find that the expected mRNA level can be substantially increased in the presence of the underlying fluctuations. This effect could propagate to the protein level even at a slow protein degradation rate, suggesting that the fluctuations in the mRNA regulation can affect regulations at a more global level. Another implication of this finding is that quantitative measures such as the Fano factor may not accurately estimate the full effects of the intrinsic fluctuations. The strength of intrinsic fluctuations in the regulation of mRNAs is often quantified using the Fano factor [3,24,50]. Assuming mRNA is constitutively synthesized and the turnover rate is proportional to mRNA level, the steady state distribution of this mRNA is found to be Poissonian. Thus, treating the Fano factor of this process as the baseline, the noise strength of a particular mRNA regulation can be measured by comparing its Fano factor with that of the baseline. However, because the expected mRNA level can be substantially increased by the intrinsic fluctuations, such a measure may underestimate the full effects of the fluctuations.
An increase in the mRNA abundance in our stochastic model is a consequence of a negative covariance of the mRNA degradation enzyme and the mRNA. As in the stochastic focusing case, hence, the deviation of the mean of reaction rates from the corresponding fluctuation-free, deterministic reaction rates can lead to an apparent systems-level discrepancy between the stochastic treatment and the hypothetical fluctuation-free treatment of our gene expression system. To unravel how the underlying fluctuations increase the steady state mRNA level, we developed simple computational models to specifically dissect the mechanism of this striking noise effect. Our analysis shows that the steady state gain in mRNA level is caused by underlying asynchronicity of molecular reactions. We find that the asynchronous effect generates additional modes of dynamics in the stationary regime that are qualitatively distinct, and that the longer time the system spends in these asynchronous dynamic modes, the wider the deviation becomes between the stochastic model and the deterministic model. These results illustrate non-intuitive effects of reaction asynchronicity on system dynamics that cannot be captured by the traditional deterministic framework. Because molecular reactions are intrinsically stochastic and asynchronous, such an asynchronous effect could be seen in a wide range of biological systems. Dynamical effects of asynchronous mechanisms on many biological systems can, however, be widely different from what we observed in the gene expression process we have studied. This is because an increase in the expected gene expression level that we observed was directly tied to the specific network structure of the transcription regulation process. In different structures of biochemical networks, reaction asynchronicity may give rise to entirely different dynamical effects. This observation may provide useful guidance for future modelling studies, by showing a new way in which deterministic models of inherently stochastic systems may yield substantially inaccurate results even for outputs that are not themselves apparently stochastic.
4. Methods
4.1. Simulations of the full gene expression model
Each simulation was run for 60 000 min. For steady state distribution plots, data were collected from 10 000 min for every 0.1 min. The simulations of the full SCK model were carried out using the direct method [51] implemented in iBioSim [52].
4.2. Estimation of mean time evolution of the simplified SCK model
The mean time evolution of the SCK model shown in figure 2a is estimated by generating 400 000 SSA trajectories and calculating the sample mean from them.
4.3. Estimation of the steady state distribution and its mean
We generated a sample trajectory of the system, and uniformly sampled at 1 000 000 time points from the trajectory. We discarded the first 5000 samples, and the rest were used to estimate the steady state distribution as well as its mean value.
4.4. Simulation of the CLE
From our simplified SCK model, we derived the following CLE for use in implementing an Euler–Maruyama simulation of the Langevin model:
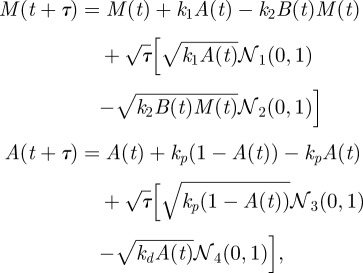 |
and
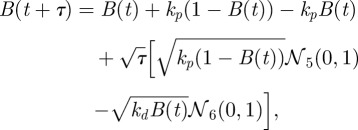 |
where 𝒩j (0, 1) is a unit normal random variable that is statistically independent for each j. In the simulation, we set the time step τ to be 10−4.
4.5. Simulation tools
All of the simulations except for the ones for the full gene expression model are implemented in custom C code in order to achieve high efficiency.
Acknowledgements
H.K. is supported by a Lane Fellowship through the Ray and Stephanie Lane Center for Computational Biology. R.S. is supported by US National Institutes of Health awards 1R01AI076318 and 1R01CA140214.
References
- 1.Pedraza J. M., Van Oudenaarden A. 2005. Noise propagation in gene networks. Science 307, 1965–1969 10.1126/science.1109090 (doi:10.1126/science.1109090) [DOI] [PubMed] [Google Scholar]
- 2.Blake W. J., Balázsi G., Kohanski M. A., Isaacs F. J., Murphy K. F., Kuang Y., Cantor C. R., Walt D. R., Collins J. J. 2006. Phenotypic consequences of promoter-mediated transcriptional noise. Mol. Cell 24, 853–865 10.1016/j.molcel.2006.11.003 (doi:10.1016/j.molcel.2006.11.003) [DOI] [PubMed] [Google Scholar]
- 3.Raj A., Van Oudenaarden A. 2009. Single-molecule approaches to stochastic gene expression. Annu. Rev. Biophys. 38, 255–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kittisopikul M., Süel G. M. 2010. Biological role of noise encoded in a genetic network motif. Proc. Natl Acad. Sci. USA 107, 13 300–13 305 10.1073/pnas.1003975107 (doi:10.1073/pnas.1003975107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy K. F., Adams R. M., Wang X., Balázsi G., Collins J. J. 2010. Tuning and controlling gene expression noise in synthetic gene networks. Nucleic Acids Res. 38, 2712–2726 10.1093/nar/gkq091 (doi:10.1093/nar/gkq091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samoilov M. S., Arkin A. P. 2006. Deviant effects in molecular reaction pathways. Nature Biotechnol. 24, 1235–1240 10.1038/nbt1253 (doi:10.1038/nbt1253) [DOI] [PubMed] [Google Scholar]
- 7.Marquez-Lago T., Stelling J. 2010. Counter-intuitive stochastic behavior of simple gene circuits with negative feedback. Biophys. J. 98, 1742–1750 10.1016/j.bpj.2010.01.018 (doi:10.1016/j.bpj.2010.01.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eldar A., Elowitz M. B. 2010. Functional roles for noise in genetic circuits. Nature 467, 167–173 10.1038/nature09326 (doi:10.1038/nature09326) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keener J., Sneyd J. 1998. Mathematical physiology. New York, NY: Springer [Google Scholar]
- 10.Fall C., Marland E., Wagner J., Tyson J. (eds) 2002. Computational cell biology. New York, NY: Springer [Google Scholar]
- 11.Gillespie D. T. 1992. Markov processes an introduction for physical scientists. Philadelphia, PA: Academic Press [Google Scholar]
- 12.McQuarrie D. A. 1967. Stochastic approach to chemical kinetics. J. Appl. Probab. 4, 413–478 10.2307/3212214 (doi:10.2307/3212214) [DOI] [Google Scholar]
- 13.Van Kampen N. G. 1992. Stochastic processes in physics and chemistry. New York, NY: Academic Press [Google Scholar]
- 14.Gillespie D. T. 2007. Stochastic simulation of chemical kinetics. Annu. Rev. Phys. Chem. 58, 35–55 10.1146/annurev.physchem.58.032806.104637 (doi:10.1146/annurev.physchem.58.032806.104637) [DOI] [PubMed] [Google Scholar]
- 15.Wilkinson D. 2009. Stochastic modelling for quantitative description of heterogeneous biological systems. Nat. Rev. Genet. 10, 122–133 10.1038/nrg2509 (doi:10.1038/nrg2509) [DOI] [PubMed] [Google Scholar]
- 16.Golding I., Paulsson J., Zawilski S., Cox E. 2005. Real-time kinetics of gene activity in individual bacteria. Cell 123, 1025–1036 10.1016/j.cell.2005.09.031 (doi:10.1016/j.cell.2005.09.031) [DOI] [PubMed] [Google Scholar]
- 17.Cai L., Friedman N., Xie X. S. 2006. Stochastic protein expression in individual cells at the single molecule level. Nature 440, 358–362 10.1038/nature04599 (doi:10.1038/nature04599) [DOI] [PubMed] [Google Scholar]
- 18.Taniguchi Y., Choi P. J., Li G. W., Chen H., Babu M., Hearn J., Emili A., Sunney X. 2010. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329, 533–538 10.1126/science.1188308 (doi:10.1126/science.1188308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newman J. R. S., Ghaemmaghami S., Ihmels J., Breslow D. K., Noble M., DeRisi J. L., Weissman J. S. 2006. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 441, 840–846 10.1038/nature04785 (doi:10.1038/nature04785) [DOI] [PubMed] [Google Scholar]
- 20.Zenklusen D., Larson D. R., Singer R. H. 2008. Single-RNA counting reveals alternative modes of gene expression in yeast. Nat. Struct. Mol. Biol. 15, 1263–1271 10.1038/nsmb.1514 (doi:10.1038/nsmb.1514) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raj A., Peskin C., Tranchina D., Vargas D., Tyagi S. 2006. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 4, e309. 10.1371/journal.pbio.0040309 (doi:10.1371/journal.pbio.0040309) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suter D. M., Molina N., Gatfield D., Schneider K., Schibler U., Naef F. 2011. Mammalian genes are transcribed with widely different bursting kinetics. Science 332, 472–474 10.1126/science.1198817 (doi:10.1126/science.1198817) [DOI] [PubMed] [Google Scholar]
- 23.Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. 2011. Global quantification of mammalian gene expression control. Nature 473, 337–342 10.1038/nature10098 (doi:10.1038/nature10098) [DOI] [PubMed] [Google Scholar]
- 24.Kærn M., Elston T., Blake W., Collins J. 2005. Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 6, 451–464 10.1038/nrg1615 (doi:10.1038/nrg1615) [DOI] [PubMed] [Google Scholar]
- 25.Boeger H., Griesenbeck J., Strattan J., Kornberg R. 2003. Nucleosomes unfold completely at a transcriptionally active promoter. Mol. Cell 11, 1587–1598 10.1016/S1097-2765(03)00231-4 (doi:10.1016/S1097-2765(03)00231-4) [DOI] [PubMed] [Google Scholar]
- 26.Steger D. J., Haswell E. S., Miller A. L., Wente S. R., O'Shea E. K. 2003. Regulation of chromatin remodeling by inositol polyphosphates. Science 299, 114–116 10.1126/science.1078062 (doi:10.1126/science.1078062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raser J. M., O'Shea E. K. 2004. Control of stochasticity in eukaryotic gene expression. Science 304, 1811–1814 10.1126/science.1098641 (doi:10.1126/science.1098641) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gillespie D. T. 2005. Handbook of materials modeling, ch. 5.11, pp. 1735–1752 New York, NY: Springer [Google Scholar]
- 29.Shahrezaei V., Swain P. 2008. Analytical distributions for stochastic gene expression. Proc. Natl Acad. Sci. USA 105, 17 256–17 261 10.1073/pnas.0803850105 (doi:10.1073/pnas.0803850105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deshmukh M. et al. 2008. mRNA decapping is promoted by an RNA-binding channel in Dcp2. Mol. Cell 29, 324–336 [DOI] [PubMed] [Google Scholar]
- 31.Floor S., Jones B., Gross J. 2008. Control of mRNA decapping by Dcp2: an open and shut case? RNA Biol. 5, 189–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. 2003. Molecular biology of the cell. Ann. Bot. 91, 401. 10.1093/aob/mcg023 (doi:10.1093/aob/mcg023) [DOI] [Google Scholar]
- 33.Shimoni Y., Friedlander G., Hetzroni G., Niv G., Altuvia S., Biham O., Margalit H. 2007. Regulation of gene expression by small non-coding RNAs: a quantitative view. Mol. Syst. Biol 3, 138. 10.1038/msb4100181 (doi:10.1038/msb4100181) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine E., Zhang Z., Kuhlman T., Hwa T. 2007. Quantitative characteristics of gene regulation by small RNA. PLoS Biol. 5, e229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodríguez Martínez M., Soriano J., Tlusty T., Pilpel Y., Furman I. 2010. Messenger RNA fluctuations and regulatory RNAs shape the dynamics of a negative feedback loop. Phys. Rev. E 81, 031924. 10.1103/PhysRevE.81.031924 (doi:10.1103/PhysRevE.81.031924) [DOI] [PubMed] [Google Scholar]
- 36.Gillespie D. T. 1977. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81, 2340–2361 10.1021/j100540a008 (doi:10.1021/j100540a008) [DOI] [Google Scholar]
- 37.Kuwahara H., Myers C., Samoilov M., Barker N., Arkin A. 2006. Automated abstraction methodology for genetic regulatory networks. In Transactions on computational systems biology VI. Lecture Notes in Computer Science, number 4220 (eds C. Primi & G. Plotkin), pp. 150–175 Berlin, Germany: Springer; 10.1007/11880646 (doi:10.1007/11880646) [DOI] [Google Scholar]
- 38.Kuwahara H., Myers C. J., Samoilov M. S. 2010. Temperature control of fimbriation circuit switch in uropathogenic Escherichia coli: quantitative analysis via automated model abstraction. PLoS Comput. Biol. 6, e1000723. 10.1371/journal.pcbi.1000723 (doi:10.1371/journal.pcbi.1000723) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Belle A., Tanay A., Bitincka L., Shamir R., O'Shea E. K. 2006. Quantification of protein half-lives in the budding yeast proteome. Proc. Natl Acad. Sci. USA 103, 13 004–13 009 10.1073/pnas.0605420103 (doi:10.1073/pnas.0605420103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gillespie D. T. 2000. The chemical Langevin equation. J. Chem. Phys. 113, 297–306 10.1063/1.481811 (doi:10.1063/1.481811) [DOI] [Google Scholar]
- 41.Raser J. M., O'Shea E. K. 2005. Noise in gene expression: origins, consequences, and control. Science 309, 2010–2013 10.1126/science.1105891 (doi:10.1126/science.1105891) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kepler T. B., Elston T. C. 2001. Stochasticity in transcriptional regulation: origins, consequences, and mathematical representations. Biophys. J. 81, 3116–3136 10.1016/S0006-3495(01)75949-8 (doi:10.1016/S0006-3495(01)75949-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samoilov M., Plyasunov S., Arkin A. P. 2005. Stochastic amplification and signaling in enzymatic futile cycles through noise-induced bistability with oscillations. Proc. Natl Acad. Sci. USA 102, 2310–2315 10.1073/pnas.0406841102 (doi:10.1073/pnas.0406841102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paulsson J., Berg O. G., Ehrenberg M. 2000. Stochastic focusing: fluctuation-enhanced sensitivity of intracellular regulation. Proc. Natl Acad. Sci. USA 97, 7148–7153 10.1073/pnas.110057697 (doi:10.1073/pnas.110057697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim K. H., Qian H., Sauro H. M. 2008. Sensitivity regulation based on noise propagation in stochastic reaction networks. Preprint, University of Washington, USA. (http://arxiv.org/abs/arXiv:0805.4455v2) [Google Scholar]
- 46.Samoilov M. S., Price G., Arkin A. P. 2006. From fluctuations to phenotypes: the physiology of noise. Sci. STKE 2006, re17 10.1126/stke.3662006re17 (doi:10.1126/stke.3662006re17) [DOI] [PubMed] [Google Scholar]
- 47.Losick R., Desplan C. 2008. Stochasticity and cell fate. Science 320, 65–68 10.1126/science.1147888 (doi:10.1126/science.1147888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnston R. J., Jr, Desplan C. 2010. Stochastic mechanisms of cell fate specification that yield random or robust outcomes. Annu. Rev. Cell Dev. Biol. 26, 689–719 10.1146/annurev-cellbio-100109-104113 (doi:10.1146/annurev-cellbio-100109-104113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nair G., Raj A. 2011. Time-lapse transcription. Science 332, 431–432 10.1126/science.1205995 (doi:10.1126/science.1205995) [DOI] [PubMed] [Google Scholar]
- 50.Thattai M., Van Oudenaarden A. 2001. Intrinsic noise in gene regulatory networks. Proc. Natl Acad. Sci. USA 98, 8614–8619 10.1073/pnas.151588598 (doi:10.1073/pnas.151588598) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gillespie D. T. 1976. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 22, 403–434 10.1016/0021-9991(76)90041-3 (doi:10.1016/0021-9991(76)90041-3) [DOI] [Google Scholar]
- 52.Myers C., Barker N., Jones K., Kuwahara H., Madsen C., Nguyen N-P. D. 2009. iBioSim: a tool for the analysis and design of genetic circuits. Bioinformatics 25, 2848–2849 10.1093/bioinformatics/btp457 (doi:10.1093/bioinformatics/btp457) [DOI] [PubMed] [Google Scholar]