

Abstract
Marketed drugs frequently perform worse in clinical practice than in the clinical trials on which their approval is based. Many therapeutic compounds are ineffective for a large subpopulation of patients to whom they are prescribed; worse, a significant fraction of patients experience adverse effects more severe than anticipated. The unacceptable risk–benefit profile for many drugs mandates a paradigm shift towards personalized medicine. However, prior to adoption of patient-specific approaches, it is useful to understand the molecular details underlying variable drug response among diverse patient populations. Over the past decade, progress in structural genomics led to an explosion of available three-dimensional structures of drug target proteins while efforts in pharmacogenetics offered insights into polymorphisms correlated with differential therapeutic outcomes. Together these advances provide the opportunity to examine how altered protein structures arising from genetic differences affect protein–drug interactions and, ultimately, drug response. In this review, we first summarize structural characteristics of protein targets and common mechanisms of drug interactions. Next, we describe the impact of coding mutations on protein structures and drug response. Finally, we highlight tools for analysing protein structures and protein–drug interactions and discuss their application for understanding altered drug responses associated with protein structural variants.
Keywords: bioinformatics, protein structure, drug response, pharmacogenetics
1. Introduction
Population-level statistics irrefutably demonstrate the benefits of pharmaceutical innovation over the past century, which has seen the introduction of antibiotics, statins and cancer therapeutics. Rapid advances in the fields of genomics, proteomics and biotechnology have fuelled the drug discovery process. Yearly from 1982 to 2010, an average of 18 drugs were approved for human use by the US Food and Drug Administration (FDA), with approximately four acting on novel target structures [1].
Yet, in spite of this historical success, the pharmaceutical industry continues to face exceptional challenges. Over the past decade, escalating investments in basic and clinical research have not seen equal returns. Instead, both the developmental rate of new molecular entities and the approval rate of new drugs have dropped by roughly 50 per cent [2,3]. During clinical development, efficacy and safety concerns contribute equally to the attrition of candidate drugs [4]. Even marketed drugs display limited efficacy, with studies showing them to be effective for only 30–60% of the patients to whom they are prescribed [5,6]. Furthermore, for many drugs whose therapeutic windows are narrow and the consequences of adverse events are life-threatening, up to one-third of patients develop unacceptable toxicity [7]. Consequently, a significant number of marketed drugs have poor risk–benefit ratios for diverse patient populations. This occurrence has been termed the ‘efficacy–effectiveness gap’ and is, ultimately, a result of variability in patient–drug responses [8].
The observation that patients are neither equally responsive to beneficial drug effects nor equally susceptible to adverse events motivates the call for a paradigm shift from population-level to patient-specific medicine [6,8]. To address this directive, two cooperative aims have been proposed, (i) determine the detailed molecular mechanisms of drug action and (ii) understand the effect of genetic variants on patient–drug response. The former aim is the focus of the field of pharmacodynamics (reviewed in [9]), and the latter the focus of pharmacogenetics (reviewed in [10,11]). At the intersection lies the challenge of understanding how genetic differences between individuals can translate into structural alterations in protein drug targets and ultimately into variable patient–drug response.
The genetic basis for inter-individual drug response variability has been studied extensively over the last 50 years [12]. While there are numerous behavioural and environmental factors that contribute to patient–drug response, genetic factors also often have a key, if not a dominant, role [7]. Specifically, genetic variants affect gene expression, mRNA processing and stability, and protein structure. Each of these variations can have functionally significant consequences for drug response [12]. Moreover, genetic polymorphisms are observed in all of the principle effectors of therapeutic response, drug transporters, drug-metabolizing enzymes and drug targets. Gaining a detailed understanding of the underlying mechanisms of phenotypic variability in drug response at the protein level is a key factor in the establishment of personalized medicine [13].
Traditionally, the roles of genetic variations in proteins were investigated using sequence analysis tools to predict the tolerability of a given amino acid substitution and its probable effect on protein function [14]. Yet, interpreting the effect of a mutation within the three-dimensional context of the protein structure offers more information [15]. Analysis of three-dimensional structures can provide valuable insight into the mechanisms of drug–target interaction and the relationships between mutations and differential therapeutic responses [16]. Such detailed structural analysis of protein–drug interactions was not always feasible in the past, but structural genomics initiatives have resulted in an explosion of high-resolution structures of known and potential drug target proteins.
In this review, we discuss the relationship between structural protein variations and differences in patient–drug response. The scope is not limited to, but is strongly focused on, the effect of mutations on structures of primary drug targets of human origin. We begin with an overview of protein targets and common mechanisms of drug interactions. Next, we describe the impact of structural mutations on drug response. Finally, tools and databases developed for analysing protein structures and protein–drug interactions are presented and their potential applications for gaining insight into protein structural variants displaying altered drug response are discussed.
2. Small-molecule drugs and their protein targets
2.1. Properties of small-molecule drugs
Structures of therapeutic agents are highly diverse and range from small-molecule compounds, to antibodies, to whole cells [17]. This structural diversity allows them to specifically interact with and modulate the function of their diverse targets. Although therapeutic biologics have seen increased development during the past decade, small-molecule drugs still account for over two-thirds of new molecular entities approved by the US FDA [18]. Thus, this review focuses on small molecular therapeutics (typically 200–550 Da [19]) and the structural mutations in their protein targets that can lead to altered drug response. Yet, it is important to note that such mutations can affect the behaviour of all therapeutic agent classes.
Small-molecule drugs must meet several criteria, including having reasonable solubility and stability levels in aqueous media, appropriate structural and physicochemical features to specifically interact with their targets, and satisfactory pharmacokinetic profiles for clinical use (for recent reviews see [20–22]). Guidelines for evaluating the drug-likeness of a molecule (for example, Lipinski's Rule of Five [23] and its derivatives [24]) have been widely adopted to aid the development process. However, applying these guidelines warrants caution, as the guidelines assume that the target of interest requires a compound whose molecular properties are similar to those of the average drug. In addition, applying drug-like screening criteria to compounds in early stages of development can be disadvantageous because the molecular properties of lead compounds undergo extensive optimization before clinical introduction. On average, the optimization process increases a compound's molecular weight and complexity [25]; thus, an initial compound with drug-like properties would probably lie outside of the desired physicochemical space after development is complete.
Frequently, the universal application of drug-likeness guidelines without regard for the structure of the intended protein target is detrimental. Consideration of the three-dimensional target structure can provide a more accurate understanding of a drug's requisite properties [26,27]. In short, fine-tuning the molecular properties of drugs to the structures of their protein targets promotes binding interactions of high affinity and specificity.
2.2. Three-dimensional structures of protein targets
Ideal therapeutic targets share several features, involvement in a biologically relevant pathway, functional and structural characterization, and druggability [28]. A druggable protein is one possessing structural characteristics that favour interactions with drug-like compounds and whose function can be modulated through such interactions. Inference of druggability historically relied on sequence homology of the protein of interest to known drug targets [29]. However, protein families lacking homology to drug targets have yielded novel targets and not all members of a protein family are equally druggable [30].
Instead, three-dimensional structures can provide information more relevant to protein druggability. In a seminal paper, Cheng et al. [30] applied knowledge derived from biophysical principles and protein structure to accurately predict protein druggability and drug binding affinities. Several subsequent studies then extracted structural and physicochemical descriptors associated with druggability from known protein–ligand complex structures [31–33]. A recent comparison of two protein structure datasets, one comprising drug targets and the other of non-drug targets, revealed drug targets to be more hydrophobic, have lower isoelectric values, be composed of more amino acids and have a higher frequency of beta-sheet secondary structure compared with other proteins [28]. Similarly, some protein tertiary structures are enriched among druggable proteins. Structural classification of drug targets from the Protein Data Bank (PDB [34]) using the Structural Classification of Proteins (SCOP) database [35] showed that the 10 most commonly observed folds are, nuclear receptor ligand-binding domain, ferredoxin-like, C-terminal domain, acid protease, NAD(P)-binding Rossmann-fold domain, TIM beta/alpha-barrel, prealbumin-like, dihydrofolate reductase-like, alpha/beta-hydrolase, and DNA/RNA polymerase.
A nearly ubiquitous structural feature of drug targets is the presence of a solvent accessible cavity or binding pocket. Analysis of 5600 protein–ligand structures from the PDB revealed 95 per cent of binding sites to be within one of the three largest pockets [36]. Yet, the presence of a binding pocket does not, in itself, render a protein druggable. Rather, specific cavity properties strongly affect protein druggability (summarized in table 1).
Table 1.
Properties of druggable protein pockets.
Some proteins of great therapeutic interest (i.e. protein–protein interfaces) lack large binding pockets and/or other structural characteristics associated with druggability [20]. To expand therapeutic protein space to these intractable targets, much effort in the past decade has focused on their structural characterization [37]. The resulting structural insights led to modified drug development approaches, such as expanding the chemical space of drug compound libraries [20,42,43]. The most notable small-molecule success in targeting protein–protein interfaces is the phase II clinical trial drug, ABT-263 [44].
2.3. Classes of protein targets
Considering the structural druggability requirements discussed above, there is surprising diversity among therapeutically targeted proteins. Protein targets of recently approved drugs are found in diverse locations throughout the body; many are secreted (e.g. plasminogen) or transmembrane (e.g. P2Y receptor) proteins, while others are found in specific subcellular locations (e.g. mTORC1). Likewise, their biological functions are varied and include, transmitting signals from the extracellular to the intracellular environment (e.g. thrombopoietin receptor), catalysing biochemical reactions (e.g. dipeptidyl peptidase-4), controlling ion flux across cellular membranes (e.g. KCNQ/Kv7 potassium channel), and directly regulating gene expression (e.g. SERM). In addition to modulating the endogenous functions of wild-type proteins, pharmaceutical efforts have also targeted protein variants (e.g. Bcr-Abl kinase), the altered structures of which confer aberrant biological functions.
Although therapeutic targets are diverse, therapeutic coverage of the human proteome is sparse. There are an estimated 22 000 protein-coding genes in the human genome [45], of which 6000–8000 are probably druggable [46,47]. Currently marketed drugs modulate the functions of only a small number of human proteins, the majority of which are targeted to achieve antihypertensive, antineoplastic or anti-inflammatory effects [1]. The roughly 1400 small-molecule drugs marketed in the US [48] collectively target fewer than 450 unique human proteins [1,46]. Similarly, a recent analysis of 823 179 unique bioactive agents found they correspond to a mere 1654 human protein targets, with a median compound-to-target ratio of 41: 1 [49].
Disparate coverage of potential targets persists even in the genomics era; of the 183 small-molecule drugs approved from 1999 to 2008, only 75 are first-in-class with novel molecular mechanisms of action [18]. Receptors comprise the largest class of drug targets (44% of targets), followed by enzymes (27% of targets) and transporter proteins (15% of targets) [1]. Moreover, half of current small-molecule therapeutics disproportionately target five protein families, rhodopsin-like GPCRs, voltage-gated ion channels, ligand-gated ion channels and kinases [20,50].
3. Mechanisms of drug activity
3.1. Molecular recognition between small molecules and proteins
Binding events, like the formation of protein–drug complexes, are governed by enthalpic and entropic contributions. The former stem from stabilizing interactions with the formation of hydrogen bonds and salt bridges, and the latter involve penalties from the loss of conformational freedom of the protein and drug. The balance between enthalpic and entropic contributions determine the free energy of the interaction and, thereby, the favourability of the binding event at equilibrium. The enthalpy of molecular recognition between a protein and a small molecule depends on two key components, shape complementarity and physicochemical complementarity. Shape complementarity permits the protein and small molecule to achieve sufficient proximity and contact surface area to form stabilizing interactions, while physicochemical complementarity determines the nature of these interactions. Amino acid mutations occurring in target proteins have the ability to disrupt both shape complementarity as well as physicochemical compatibility. Their impact on drug binding, and therefore drug response, depends on the nature of the mutation and its three-dimensional structural context, as discussed in §4.
3.1.1. Models of molecular recognition
Molecular complementarity was first thought of as a lock-and-key fit, where the small molecule (key) possessed perfect shape and physicochemical complementarity to the protein (lock). However, the predominance of structural rearrangements, both minor and major [51], upon ligand binding are better explained by two recently adopted models—induced fit and conformational selection [52,53].
The induced fit model attributes protein structural changes to the binding event of the ligand. The recently solved complex of human neutrophil elastase (HNE) with a dihydropyrimidone inhibitor exemplifies an induced fit binding mechanism, since protein structural rearrangements near the inhibitor differ from the conformations of both ligand-free HNE and other HNE-inhibitor complexes [54].
Conversely, in the conformational selection theory the ligand selects the most complementary conformation from an ensemble of equilibrium structures. This mode of interaction has been implicated in the binding selectivity of imatinib to tyrosine kinases (see §4.3.1.1) [55]. Molecular dynamics (MD) studies reveal that kinases with high imatinib affinity spend more time in conformations compatible with drug binding compared with kinases with low imatinib affinity.
Yet other studies suggest that real interactions reflect a mixture of the two binding models [55–57]. For example, recent in silico work demonstrated that, contrary to previous assumptions, ligand binding to the lysine-, arginine-, ornithine-binding protein proceeds through two stages, initial complex formation by conformational selection followed by ligand-induced transition to the observed bound state [57].
Differences between unbound (apo state) and bound (holo state) protein structures add a transient dimension to molecular complementarity. This has important implications for the use of structural data in detecting and understanding protein–drug interactions. Namely, available protein structures may not display a conformation compatible with small-molecule binding. These non-binding structures can result from protein structural preferences or random chance and crystallization artefacts [58]. Caution must therefore be used when relying on structural data to assess whether a protein binds a drug and the mode of binding. MD simulation (see §5.5) is a useful approach for generating protein structural ensembles and capturing alternative conformations that may be relevant for protein function or small-molecule binding [59,60]. Protein dynamics information enhances structure-based binding site prediction methods and reduces their dependence on experimentally determined target protein structures.
3.1.2. Shape complementarity
Shape complementarity refers to the geometric fit, or steric fitness, of a small molecule and its surrounding protein environment. The importance of shape complementarity in protein–drug interactions is discussed in a recent review by Kortagere et al. [61]. Numerous protein–ligand complexes in the PDB depicting close intermolecular contacts between small molecules and proteins illustrate the importance of shape complementarity for binding. For example, the recently solved complex of the first bromodomain of human Brd4, a transcription factor complex protein and therapeutic target of interest in cancer, with a highly potent and specific inhibitor depicts excellent shape complementarity (figure 1a and b) [62].
Figure 1.

Shape complementarity between small molecules and their protein targets. The structure of Brd4 (grey cartoon) with an inhibitor (sticks) shows excellent shape complementarity when viewed looking (a) into and (b) perpendicular to the binding pocket (grey mesh). In contrast, Cdc34 (grey cartoon) bound to an inhibitor (sticks) has imperfect and incomplete shape complementarity, as depicted looking into (c) and perpendicular to (d) the binding pocket (grey mesh). Water molecules represented as red and white spheres. (Brd4, PDB 3MXF [62]; Cdc34, PDB 3RZ3 [63].) (Online version in colour.)
However, high shape complementarity is not always a requisite for drug binding and is often incomplete or imperfect. This is evident from the crystal structure of the human Cdc34 ubiquitin-conjugating enzyme in complex with a novel allosteric inhibitor showing that the ligand does not fully interact with the protein target [63]. The shape complementarity of this complex is both imperfect, since water-mediated binding interactions introduce unoccupied space between binding partners (figure 1c), and incomplete, as there is a solvent-exposed carboxylic acid group on the ligand (figure 1d).
Shape complementarity dictates whether a ligand is sufficiently close to a protein to form favourable interactions and is therefore a critical determinant of binding. As such, shape complementarity has been applied in virtual screening of drug discovery approaches [64]. Yet, it is important to note that geometric compatibility does not always indicate physicochemical compatibility. Thus, in the following section we describe the role of physicochemical complementarity in molecular recognition.
Numerous shape-matching technologies and protein pocket predictors can be used to gain insights into protein–drug interactions (see §5.3). Ebalunode and Zheng published a recent review on shape complementary methods [65].
3.1.3. Physicochemical complementarity
Physicochemical complementarity refers to non-covalent interactions holding proteins and ligands in a complex. These interactions can involve long-range ionic bonds or weaker short-range interactions including hydrogen bonds, van der Waals forces and hydrophobic packing. Electrostatic complementarity, accounting for both ionic and electrostatic interactions, is one of the most important forces governing protein–ligand complex formation, affecting binding affinity as well as the rate of protein–ligand association (reviewed in [66]). For example, in pancreatic endoplasmic reticulum kinase, electrostatic complementarity to an aspartate in the binding site strongly influences inhibitor affinity, with a lack of complementarity translating to weaker affinity [67]. Likewise, electrostatic steering of a small molecule into the proper orientation for binding enhances complex formation. Protein–drug interactions are further affected by pH owing to the titratable groups (weak acid or weak base) found in many drugs that alter the ionization state of the molecule.
As previously discussed in §2.1, many drugs are hydrophobic in nature and have a partition coefficient greater than one, indicating a preference for solubilizing into octanol versus water [23]. Such compounds are energetically unfavourable in the aqueous compartments of the body, thereby producing a driving force for protein binding. This tendency is responsible for much of the non-specific interactions of hydrophobic drugs. Non-specific interactions are therefore primarily driven by physicochemical compatibility between the compound and protein.
While the effects of both the hydrophobic effect and hydrogen bonds on protein–ligand binding events are well documented, the molecular underpinnings of these phenomena remain the focus of investigation. Of particular importance to understanding protein–ligand interactions, the mechanism of hydrophobicity [68] and the definition of hydrogen bonds [69] have been recently updated.
3.2. Modes of drug binding
Drugs and other chemical compounds interact with proteins in diverse ways. The nature of the interaction may be reversible (e.g. the binding of a competitive antagonist) or irreversible (e.g. the covalent modification of a protein from a suicide inhibitor). The location at which drugs interact with proteins can also vary. Drug binding at the protein's orthosteric site or allosteric site has different implications (figure 2), which we discuss further in the following sections.
Figure 2.
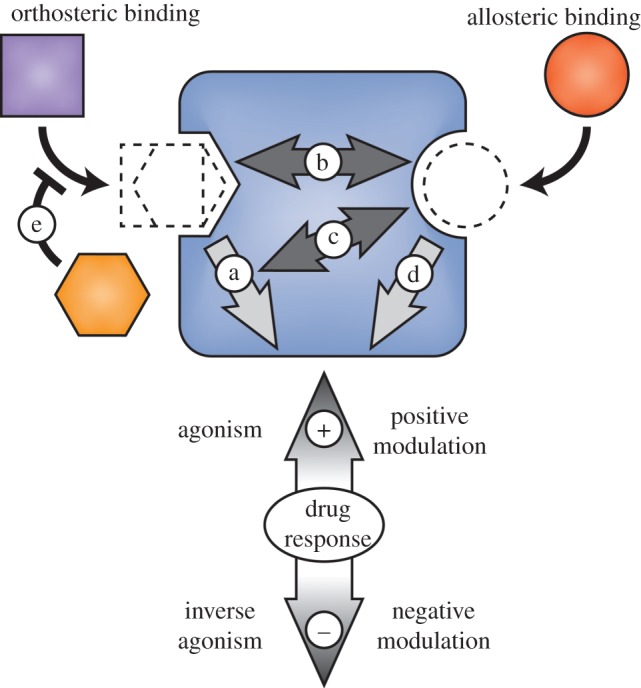
Drug binding modes. Orthosteric and allosteric ligands bind topographically distinct protein sites to positively or negatively affect target protein function. (a) Orthosteric full, partial or inverse agonism; (b) positive or negative affinity modulation; (c) positive or negative efficacy modulation; (d) allosteric full, partial or inverse agonism; (e) competitive drug binding. Target protein (blue rounded rectangle), endogenous ligand (purple square), orthosteric drug (orange hexagon), and allosteric drug (red circle). Adapted with permission from [70]. (Online version in colour.)
3.2.1. Orthosteric binding sites
Classical drug development approaches predominantly focused on targeting the protein orthosteric site (also known as the active site for enzymes). Endogenous ligands bind at the orthosteric site to elicit a biological response. Thus, a popular mechanism of drug action is to occupy the orthosteric site, thereby blocking endogenous ligand binding and modulating protein function (figure 2).
As we noted previously, protein kinases constitute a large protein family of strong pharmaceutical interest. There are two classes of kinase inhibitors. Type 1 inhibitors exert their effects by blocking adenosine triphosphate (ATP) binding to the catalytic kinase domain. Type 2 inhibitors also bind in the active site, but block kinase activity by stabilizing the inactive protein conformation (see §4.3.2). Imatinib, a popular drug used for treating chronic myelogenous leukaemia, is a type 2 inhibitor that binds to the deregulated tyrosine kinase, Bcr-Abl, stabilizing it in an inactive conformation [71].
Because the sequence and structure of active sites are often highly conserved among protein families, cross-reactivity is a significant problem for drugs targeting orthosteric sites. For example, many kinase inhibitors have low selectivity profiles and bind to a variety of family members, as illustrated by the promiscuity of numerous kinase inhibitors for human kinases [72]. Such a broad binding profile may be beneficial for polypharmacology, where inactivation of multiple pathways involved in disease leads to better treatment outcome [73]. However, multi-targeting can incur more side effects owing to drug promiscuity. Thus, there exists a trade-off between drug specificity and efficacy, with the desired balance varying from disease to disease. Tools for selecting compounds with improved selectivity [74] or multi-target binding [75] are both under focused development.
3.2.2. Allosteric binding sites
Allosteric drugs interact with their protein targets at sites spatially distinct from the protein orthosteric site (figure 2). The binding event induces protein conformational rearrangements that lead to altered activity. Allosteric modulators produce a change in affinity or efficacy for the endogenous ligand, while allosteric agonists or antagonists alter the activation state of the protein itself [76].
Targeting protein allosteric sites has received significant attention recently because of the benefits of allosteric modulation versus orthosteric modulation [70,77]. First, in the event of drug overdose, allosteric drugs are likely to pose less heath risk because their effects saturate once full occupancy of targeted sites is reached. Next, allosteric compounds are less likely than orthosteric molecules to desensitize their targets and therefore have decreased tendency for acquired drug tolerance [78,79]. Most importantly, allosteric drugs have enabled highly selective targeting of protein family subtypes. In contrast to orthosteric sites, which are generally highly conserved, allosteric sites have much greater sequence and structural diversity. However, protein allosteric sites are often challenging to locate, characterize and target [80].
The shift towards allosteric therapeutics is evident in the rhodopsin-like GPCR protein family. Numerous drugs, including atenolol (an anti-hypertension drug) and salbutamol (an anti-asthma drug) bind to GPCR orthosteric sites in order to alter the receptor activity. However, like protein kinases, the orthosteric sites of GPCRs are highly conserved [81], leading to problems with off-target activity and thus motivating a move towards development of allosteric compounds to enable targeting of specific GPCR subtypes [70]. GPCR allosteric sites are more diverse, offering more degrees of chemical freedom in developing allosteric drugs compared with orthosteric drugs [82]. Cinacalcet is the first example of an FDA-approved allosteric GPCR modulator, and functions by increasing the sensitivity of its receptor to calcium in the treatment of hyperparathyroidism [70].
4. Protein variants with altered Three-dimensional structures and drug responses
Based on genome sequencing of individuals from different populations, it is estimated that each person's proteome contains roughly 10 000–11 000 mutations compared to the reference proteome [85]. A subset of these mutations (those resulting in premature stop codons, splice-site disruptions, and frame shifts) probably has severe functional consequences, yielding approximately 250–300 loss-of-function protein variants per individual [85]. However, for the great majority of mutations, it is difficult to predict a priori what their effect will be on the resultant protein's structure and function.
4.1. Classes of mutations
Single nucleotide polymorphisms (SNPs) fall either within non-coding (including promoter, operator, enhancer and transcription factor binding regions) or coding regions of DNA. Because of redundancy in the genetic code, some mutations within coding regions (synonymous mutations) do not change the encoded protein sequence. On the other hand, non-synonymous SNPs produce either polypeptide sequences that have an amino acid substitution (missense mutations) or are truncated (nonsense mutations).
Phenotypes resulting from mutations are generally thought of in a protein structural context, where an amino acid substitution or deletion leads to altered protein structure and function. However, some mutations exert their effects via changes to the mRNA that can lead to altered mRNA splicing, folding or stability, and therefore altered protein product [86]. Here we focus our discussion specifically on the structural effects of protein missense mutations, since over half of all known human disease-associated mutations are missense SNPs [87].
Missense mutations can drastically alter protein structure and function, resulting in inter-patient variability in drug response. There are varied structural mechanisms through which missense mutations exert their effects, such as altering the physicochemical or geometric properties of a protein binding pocket, modifying structure dynamics (i.e. conferring or restricting flexibility) or disrupting folding and stability. A missense mutation occurring in the target protein may have a pharmacodynamic effect, while one occurring in a protein involved in drug absorption, distribution, metabolism or excretion may alter drug pharmacokinetics. Protein variants of the former group often impact response to a specific drug class while those of the latter group often affect a diverse array of drug classes.
In the following sections, we describe the mechanisms by which missense mutations alter the three-dimensional protein structure and thereby change drug pharmacodynamics and/or pharmacokinetics. Elucidating the structural underpinning of these effects is important for understanding the mechanisms of drug response and for predicting the clinical implication of novel genetic variants.
4.2. Effects of missense mutations on protein structure
Early approaches for studying the effect of genetic variation on protein function were sequence-based. They relied on the hypothesis that functionally relevant residues would exhibit higher sequence conservation, as mutation would probably be deleterious [88]. Accordingly, several sequence-based approaches harnessing evolutionary conservation information were developed to predict deleterious missense SNPs [89–94].
Although amino acid conservation is a useful metric for identifying functionally important residues, its scope is inherently limited to one-dimensional sequence space. It is much more informative to consider the effect of a mutation within the three-dimensional context of the protein structure. Three-dimensional context of the mutation can reveal the nature of the local environment (solvent-exposed or buried), the proximal interacting residues that are not necessarily contiguous in primary sequence, and the relative position of the mutation to binding or active sites. Furthermore, while SNPs occurring in highly conserved functional sites may directly disrupt protein activity, it does not follow that SNPs in regions under little or no selective pressure are tolerable. Such mutations still have the potential to affect protein folding, dynamics, stability and activity. These effects manifest through changes to the protein structure; thus to accurately predict the effect of a missense mutation, it is necessary to study its structural context.
Recent algorithms for predicting the effects of mutations on protein function [15,95–97] and stability [98–101] are beginning to include protein structure as an input feature. However, high-resolution three-dimensional structures are available only for a subset of known proteins. When an experimentally determined protein structure is unavailable, structure prediction techniques (see §5.2) can provide a model from which to extract structural context information for a mutation.
4.3. Pharmacodynamic effects of missense mutations
Structural variants of target proteins with differential drug response compared with their wild-type counterparts separate into two broad categories, some have mutations affecting the binding site that directly alter drug interaction, while others have mutations distal to the binding region that give rise to long-range structural perturbations or altered protein conformations. Regardless of the mechanism responsible for variable drug outcome, detailed knowledge of the three-dimensional target protein structure is critical for understanding drug–target interactions. Furthermore, once the structures of disease-relevant variants are elucidated, they can be specifically targeted by novel therapeutic strategies for improved outcome. In the following case studies, we briefly discuss the consequences of missense mutations on three-dimensional target protein structures and drug response.
4.3.1. Protein variants with binding site mutations
4.3.1.1. Missense mutations altering drug binding: kinase ‘gatekeeper’ residue substitution
As previously discussed, kinases are important cellular signalling proteins whose aberrant expression and activation is widely implicated in cancer. Kinases are therefore among the most pursued classes of drug targets, with several ATP-competitive inhibitors approved for clinical use. However, the efficacy of these agents is often limited by the subsequent emergence of drug resistance [102]. Such resistance often develops through the acquisition of mutations that abrogate inhibitor binding. The most widely observed of these mutations occur at the ‘gatekeeper’ residue, whose sidechain bulk controls accessibility of the hydrophobic ATP binding pocket [102]. Interestingly, kinase gatekeeper mutations confer drug resistance through two distinct structural mechanisms, (i) sterically blocking binding of the drug to the active site and (ii) decreasing the apparent drug potency by increasing the binding site affinity for ATP.
Patients with the Bcr-Abl oncoprotein frequently acquire mutations in the Abl kinase domain after treatment with ATP-competitive inhibitors, resulting in drug resistance. In particular, the T315I Abl gatekeeper mutation accounts for approximately 20 per cent of clinically observed drug resistance to imatinib, the current gold-standard treatment for Bcr-Abl positive leukaemias [103]. Examination of the crystal structure of the Abl T315I mutant compared with that of the wild-type kinase revealed that gatekeeper residue replacement with isoleucine sterically blocks binding of imatinib in the active site, resulting in drug resistance [104] (figure 3). Structural knowledge of the Abl gatekeeper variant has brought about the development of inhibitors that target alternate Abl kinase druggable pockets or are capable of accommodating the T315I mutation [106–108].
Figure 3.

Mutation of kinase gatekeeper residue confers drug resistance. (a) Overlay of wild-type Abl kinase (light grey cartoon) with bound imatinib (sticks) and T315I variant (dark grey cartoon) shows equivalent global structures. (b) Wild-type Abl binding pocket (yellow mesh) has a threonine gatekeeper residue (grey stick and semitransparent surface) bound to imatinib (yellow sticks). (c) Wild-type Abl binding pocket (yellow mesh) and imatinib (yellow sticks) overlaid on the T315I variant structure shows an inability of the mutant to accommodate the drug owing to protrusion of the isoleucine gatekeeper residue (red stick and semitransparent surface) that sterically prevents drug binding. (Wild-type Abl kinase, PDB 2HYY [105]; Abl kinase T315I, PDB 2Z60 [104].) (Online version in colour.)
Likewise, the epidermal growth factor receptor (EGFR) tyrosine kinase gatekeeper mutation, T790M, is also associated with clinical drug resistance. This mutation typically arises in patients possessing the oncogenic L858R mutation, accounting for approximately half of all clinically observed resistance to gefitinib and erlotinib [109,110]. Prior to attaining the crystal structure of the EGFR T790M/L858R variant, the gatekeeper mutation was proposed to sterically block binding of inhibitors in the active site [111]. More recently, Yun and colleagues solved the crystal structure of the variant and demonstrated that EGFR T790M/L858R is structurally capable of accommodating inhibitors in its kinase active site [112]. In fact, the observed drug resistance of EGFR T790M mutants is owing to increased binding affinity for ATP. Novel inhibitors specifically targeting the EGFR T790M variant are currently in clinical trials [113].
4.3.1.2. Missense mutations altering drug effect: androgen receptor binding pocket expansion
The androgen receptor (AR) is a nuclear hormone receptor essential to normal male development and the maintenance of male-specific organs. Altered AR signalling is implicated in multiple malignancies, including prostate cancer. While the incidence of mutations in the AR ligand-binding domain is low in primary tumours, it increases over the course of treatment with antiandrogens such as bicalutamide and flutamide [114]. Of particular concern are mutations in the AR ligand-binding domain that convert these therapeutic antagonists to partial agonists [115].
One such ligand-binding domain mutation observed in AR-dependent malignancies is T877A. Sack and colleagues first reported the crystal structure of the AR T877A variant and found that the alanine substitution increases the binding pocket volume, allowing bulkier ligands like the antiandrogens to bind to and activate the receptor [116]. Further structural investigations by Bohl et al. [117–119] showed that the AR T877A mutation (figure 4a and b) as well as a similar W741L mutation (figure 4c and d) expand the ligand-binding pocket and alter its physicochemical properties, leading to the conversion of potent antagonist drugs to agonists. Using the crystal structures of AR mutants with expanded binding pockets, drug discovery via structure-based approaches have led to second-generation antiandrogens that are currently undergoing clinical trials [115,122].
Figure 4.

Expansion of AR binding pocket converts antagonist drugs to agonists. (a) Overlay of wild-type AR (light grey cartoon) with bound cyproterone (sticks) and T877A variant (dark grey cartoon) depicts their globally similar structures. (b) Expanded binding pocket of AR T877A variant (red mesh) compared with wild-type (yellow mesh) better accommodates the bulky cyproterone (yellow sticks). (c) Overlay of wild-type AR (light grey cartoon) with bound R-bicalutamide (yellow sticks) and W741L variant (dark grey cartoon) depicts their globally similar structures. (d) Expanded binding pocket of AR W741L variant (red mesh) compared with wild-type (yellow mesh) better accommodates the bulky R-bicalutamide drug (yellow sticks). Substituted sidechains (red sticks and semi-transparent surfaces) compared with wild-type (grey sticks and semi-transparent surfaces) are highlighted. Binding pockets generated using HOLLOW [120]. (Wild-type AR, PDB 2AM9 [121]; AR T877A, PDB 2OZ7 [119]; AR W741L, PDB 1Z95 [117].) (Online version in colour.)
4.3.2. Protein variants with non-binding site mutations
4.3.2.1. Missense mutations altering protein conformation: KIT kinase shifted conformational equilibrium
Approximately 85 per cent of patients with gastrointestinal stromal tumours have activating mutations in KIT receptor tyrosine kinase [123]. Imatinib is an effective first-line treatment, but half of patients on this therapy acquire further KIT mutations conferring drug resistance within two years [124]. A portion of KIT variants with mutations at D816, located in the activation loop of the catalytic domain, are also resistant to second-line treatment with sunitinib [125].
Structural studies by Mol et al. revealed that KIT populates a structural ensemble ranging from an inactive autoinhibited state to an activated conformation [126,127]. Recently, Gajiwala and colleagues found that the D816H/V mutations both shift the conformational equilibrium of KIT variants towards the active form [128]. Yet, both imatinib and sunitinib bind exclusively to the inactive conformation which is less populated in KIT D816 variants, resulting in abrogated clinical efficacy. Similar mechanisms of resistance have been reported with other receptor tyrosine kinases, motivating drug development efforts focused specifically on targeting kinases in the active conformation [129,130].
4.3.2.2. Missense mutations affecting protein stability: p53 decreased thermal stability
Inactivation of the p53 tumour suppressor is an almost universal feature of human cancers [131]. Typically, p53 tumour suppressor functions as a critical barrier to tumour development by binding to DNA and regulating cell cycle progression and apoptosis [132]. Hence, restoration of p53 activity has been the focus of intensive cancer therapeutic efforts [131]. p53 has an extremely limited half-life owing to both its low thermal stability (Tm ≈ 44oC) and targeted ubiquitination by HDM2, its negative regulator [132,133]. Blocking the p53 binding site on HDM2 is sufficient to reactivate the p53 response in cells and induce rapid tumour regression [131,132]. The crystal structure of p53 bound to the mouse homologue of HDM2 (MDM2) showed the presence of a deep druggable pocket at the protein–protein interface and inspired the structure-based development of numerous small-molecule HDM2 inhibitors [43,131,132,134]. Some of these HDM2 inhibitors (e.g. Nutlin-3) have shown promise in preclinical studies and are currently in early clinical trials [135–138].
While HDM2 inhibitors are promising therapeutics for patients with wild-type p53, approximately 50 per cent of human cancers have mutations in the p53 DNA-binding domain that make HDM2-targeted drug treatment ineffective [139,140]. For example, crystallographic studies on p53 Y220C, a common oncogenic variant, revealed that this surface mutation connects two pre-existing clefts to form an extended solvent accessible crevice (figure 5), disrupts packing of the hydrophobic core, and drastically decreases thermodynamic stability [139]. As a result, p53 Y220C is too unstable to function at physiological temperature and is rapidly depleted by denaturation [142]. The extended cleft has been the focus of structure-based drug discovery efforts aimed at rescuing unstable p53 Y220C mutants [133,143].
Figure 5.

Mutation of p53 surface residue decreases protein stability. (a) Overlay of wild-type p53 (light grey) and Y220C variant (dark grey) shows high structural similarity. (b) Mutation of Tyr220 (grey stick and semitransparent surface) to Cys (red stick and semitransparent surface) creates a cleft on the surface of mutant p53 (mesh) that destabilizes the protein structure. (Wild-type p53, PDB 1UOL [141]; p53 Y220C, PDB 2J1X [139].) (Online version in colour.)
4.4. Pharmacokinetic effects of missense mutations
In general, all drugs are slightly promiscuous and the effect they elicit depends on their interaction with numerous proteins throughout the body, not just the target protein. Specifically, the proteins involved in drug pharmacokinetics have an important role in determining clinical outcome. While the focus of this review is primarily on understanding the mechanism by which structural perturbations in the primary drug target can change drug response, the same analysis can be used to gain insight into mutations in proteins involved in drug absorption, distribution, metabolism or excretion.
Cytochrome P450 (CYP) enzymes are responsible for the oxidative metabolism of environmental compounds, pollutants and drugs [144]. Their essential role in drug metabolism makes CYP enzymes of great pharmacokinetic importance. A large number of CYP variants linked to altered drug pharmacokinetics have been reported for several CYP family members, including CYP1A2 [145], CYP2B6 [146], CYP2C9 [147], CYP2C19 [145], CYP2D6 [148], CYP2J2 [149] and CYP3A5 [150]. As a family, CYP enzymes bind a remarkably broad range of ligands. Recent structural studies show that CYP structural flexibility is the major determinant of ligand binding promiscuity [151,152]. Thus, missense mutations that are peripheral to the CYP active site can exert long-range effects that disrupt the enzyme's flexibility or binding pocket structure, resulting in altered drug binding and metabolism [153,154].
The CYP2C9 isoform metabolizes more than 100 drugs in current clinical use [155]. There are large interindividual variations in CYP2C9 activity and, thus, in clinical response to therapeutics metabolized by the enzyme. Specifically, 32 marketed drugs exhibit CYP2C9 variant-dependent metabolism [155]. CYP2C9*3, a common variant carrying an I359L mutation, is correlated with decreased metabolism and clearance for multiple drugs including the anticoagulant warfarin [156]. Williams et al. first reported the crystal structure of apo-CYP2C9 and CYP2C9 in complex with warfarin [144]. Building on this crystallographic data, Sano et al. [157] computationally investigated the structural mechanisms underlying decreased CYP2C9*3 warfarin metabolism. Although the I359L mutation does not directly hinder drug binding, this substitution introduces long-range structural perturbations that result in expansion of the binding pocket volume and increased fluctuations in warfarin-coordinating residues (figure 6). These structural alterations collectively cause warfarin to bind in a region of the binding pocket that is more removed from the active site, leading to decreased enzymatic activity [157]. To account for this altered metabolism, a number of pharmacogenetic algorithms predict warfarin dosing based on patient CYP2C9 genotypes to achieve maximum efficacies with minimum toxicities [158–162].
Figure 6.

CYP2C9 variant displays altered warfarin binding. (a) CYP2C9 (grey) bound to warfarin (yellow sticks) and haem cofactor (blue sticks). (b) Close-up of CYP2C9 warfarin binding pocket and surrounding environment. Structural disturbances in neighbouring residues (cyan sticks and semitransparent surface) resulting from substitution of residue 359 (red stick and semitransparent surface) disrupt the drug binding pocket (yellow mesh). Binding pocket generated using HOLLOW [120]. (Wild-type CYP2C9, PBD 1OG5 [144].) (Online version in colour.)
5. Computational tools
Structure-based computational methods offer molecular insights into drug–protein relationships and the mechanisms by which missense mutations elicit differential drug responses. Below we discuss five major classes of bioinformatics tools, three-dimensional structure visualization, protein structure prediction, binding site detection and comparison, ligand docking and scoring, and MD.
Although none of these tools were specifically developed for studying the structural effects of protein sequence mutations, they can be readily applied for this purpose [163]. For example, protein structure prediction tools have been used to model protein variants given a homologous protein structure [164,165]. Mutations mapped to the protein structure permit analysis of their structural and/or functional consequences using binding site prediction tools [166,167]. Ligand docking studies on protein structures with varied binding site residues have elucidated protein–ligand binding mechanisms [168] and selectivity [169]. In addition, simulation of protein MD and flexibility have provided insight into the effect of mutations on ligand binding [170,171]. Collectively, these bioinformatics tools offer great potential for understanding high-resolution structural details of protein variants that give rise to altered drug response.
It is important to note that biological information can be incorporated into numerous of the tools discussed below. Such prior information can focus research efforts and greatly enhance the accuracy and interpretation of the computational results. Evolutionary conservation information helps define and refine residue contact information, aiding protein structure prediction [172] and both comparative [173] and free-modelling prediction techniques [174]. Such sequence information also contributes to binding pocket and binding residue identification [175–177]. Biophysical data, such as that derived from NMR experiments, can complement standard force fields and bias the sampling of protein conformational space towards regions consistent with experimental observations [178]. In this way, biophysical restraints have been used to assist protein structure predictions [179,180], MD simulations [181,182] and protein docking studies [183,184]. Furthermore, protein mutagenesis studies are routinely applied to both inform [185] and validate [186] docking experiments.
There are an overwhelming number of useful tools for each of the categories described. Highlighted below are examples of programs that are either widely adopted or recently developed. Numerous commercial software packages are also available for each of the categories of tools discussed below; however, we emphasize those methodologies that are freely available to academic researchers. For comprehensive tables of all available programs, refer to the review articles cited within the individual sections.
5.1. Protein structure visualization
There are several widely adopted software programs for visualizing protein structures, such as PyMOL (http://www.pymol.org) [187], UCSF Chimera (http://www.cgl.ucsf.edu/chimera) [188], and VMD (http://www.ks.uiuc.edu/Research/vmd) [189].
5.2. Protein structure prediction
The basic requirement for studying the structural mechanisms underlying variable drug response is a high-resolution protein structure. Three-dimensional structural data are increasingly available from the PDB; however, many drug target structures remain elusive owing to crystallization difficulties and protein size limitations of NMR. Moreover, while the structure of a wild-type target may be available, those of the variant proteins are often unknown. In such scenarios, computational methods can be used to predict three-dimensional protein models [164]. There are two main modelling approaches, comparative (or homology) methods that use structures of homologous proteins as starting templates, and free (or ab initio) methods that use knowledge-based algorithms or first principles [190]. Computational methods for predicting protein structure have been reviewed in detail [190–192], as have automated protein modelling servers [193].
5.2.1. Comparative modelling techniques
Comparative modelling is based on the observation that proteins with similar amino acid sequences have similar structures [194]. Thus, the three-dimensional structure of a protein (model) can be built based on the experimentally determined structure of a homologous protein (template). Comparative modelling methods have been extensively reviewed and evaluated [195–198]. The process of constructing a homology model for a protein of interest consists of the following four stages, template selection, alignment of target and template protein sequences, model generation and model evaluation and refinement [195].
There are four principle approaches for constructing homology models, spatial restraint, segment matching, multiple template and artificial evolution. MODELLER [199] and other spatial restraint techniques extract geometric features (bond lengths, angles, etc.) from the template structure and construct a model by satisfying these restraints. Segment matching tools, including SegMod/ENCAD [200] and the Pfrag extension [201], divide the target protein into fragments, independently align each to a template and assemble the fragment models. In techniques such as SWISS-MODEL [202], multiple template alignments are used to identify conserved structural regions, which are modelled as rigid bodies, while variable regions are built up around them. In artificial evolution programs such as Nest [203], iterative modifications (substitutions, deletions, or insertions) and energy minimizations are applied to the template structure to gradually build up the target protein structure.
Regardless of the modelling approach, model accuracy depends heavily on template selection and sequence alignment quality. Both factors affect the observed sequence similarity between target and template. When sequence similarity is high (more than 50%), homology modelling can reliably generate accurate, high-resolution predictions suitable for drug design, mutational analysis and binding site detection [190,192,204]. Models built using moderate (30–50%) sequence similarity are typically of lower resolution, but offer sufficient detail to assess druggability and generate hypotheses regarding sequence mutations [190,192,204]. For models generated from templates of low (less than 30%) sequence similarity, the resulting structures are generally speculative; however, accuracy can be improved by using multiple template structures, if available [201,204,205].
The ninth edition of the critical assessment of techniques for protein structure prediction (CASP) assessed the state of the art in comparative modelling techniques over a diverse pool of 116 target sequences [206]. Comparison of 61 665 template-based predictions to their experimentally determined structures demonstrated that the best-performing comparative modelling approaches are capable of accurately predicting both overall protein structure and local interactions. Notably, the methods underlying these best-performing technologies are distinct and have different strengths and weaknesses, suggesting that superior models may be attained by integrating different techniques. One commonly observed limitation of comparative modelling techniques is a poor correlation between their estimated and assessed model accuracy.
5.2.2. Free modelling techniques
In the absence of suitable templates for the protein of interest, model creation can be based on experimental data, such as interresidue distance and contact maps, along with secondary structure predictions and advanced force fields [174]. However, experimental contact data are not broadly available, thus requiring that structures be predicted from primary amino acid sequence alone.
Free modelling methods use knowledge-based potentials, physics-based potentials or a hybrid of the two to predict protein structure from first principles. Physics-based approaches, such as QUARK [207,208], perform protein folding using Monte Carlo optimization on physicochemical statistics potentials. These approaches may offer insight into the protein folding pathway, but more importantly require no a priori structural knowledge [209]. In contrast, knowledge-based methods, including ROSETTA [210] and I-TASSER [211], assemble structural fragments with local sequence similarity to the target using Monte Carlo simulation [208,212]. These fragment-assembly tools have been used to predict protein structures with high accuracy [212,213]. Currently, the computational complexity of free modelling methods limits both the model resolution and its applicability to larger protein sequences.
Although free modelling techniques are rapidly advancing, very high accuracy models are rare [214]. Recent CASP results indicate free modelling method performance is highly dependent on target length, with an apparent upper limit of 120 residues [214]. Similarly, accurate prediction of multi-domain structures remains a significant challenge [215]. Much room for improvement remains; evaluation of 16 971 free modelling predictions of 30 target sequences in CASP9 showed that even top-performing technologies produced a number of physically unrealistic models [215]. Despite these challenges, recent years have seen dramatic improvements in the prediction accuracy of free modelling techniques for short target sequences [214].
5.3. Drug binding site analysis
Understanding differential drug outcomes requires high-resolution structural knowledge of the binding site. Computational methods to identify and analyse drug binding sites can supplement experimentally derived structural knowledge. Such tools have multiple applications, including prediction of drug specificity, guidance of drug development and repurposing, and prediction or interpretation of drug response. In addition, binding site prediction and analysis tools offer insights into the effect of mutations on the druggability of the binding pocket [166,167].
5.3.1. Binding site prediction
Binding site detection is challenging because proteins frequently undergo large structural changes upon ligand binding (see §3.1.1). Structure-based computational algorithms for predicting binding sites are described in detail in recent reviews [216–218]. These tools can be divided into four main categories, structural similarity, geometric, energy-based and docking.
Structural similarity approaches, such as 3DLigandSite [219], compare a query structure with a binding site library extracted from protein–ligand complexes, select a subset of similar structures and superimpose the ligands onto the query structure to infer the binding site location. Geometric-based methods, including fpocket [220], are based on shape and assume that the binding site is located within a cavity; potential binding cavities are typically detected by placing or rolling spheres of fixed or variable radii along the protein structure. SiteHound and other energy-based algorithms work on the hypothesis that the energetic properties of binding sites differ from those of the surrounding protein surface [221,222]. Creation of interaction affinity maps between the protein surface and representative chemical probes reveal protein surface patches of high total interaction energy that represent probable ligand binding pockets. Finally, docking methods for predicting binding sites computationally dock libraries of drug-like fragments, as in FTMAP [223], or compounds, as in MolSite [224], to the protein structure. These tools are computational analogues to experimental approaches for studying protein druggability by NMR [40] or X-ray crystallography [225].
Pocket prediction programs vary widely in performance, largely depending on the input structure conformation (apo- or holo-protein) and number of returned sites considered for further analysis [38,226,227]. In addition to predicting their location, many of these tools also provide detailed characterization of binding sites suitable for use in comparative studies, as discussed in the following section.
5.3.2. Binding site structural comparison
Proteins lacking in overall sequence and structural homology can share binding site similarities and therefore bind to common ligands. Algorithms for identifying structurally similar binding sites have broad applications, including evaluating protein druggability and inferring protein–ligand interactions, both on- and off-target, as recently reviewed in [218]. Comparative studies of binding sites typically proceed through three steps. Positional and physicochemical properties of cavity residues are first reduced to simplified geometric patterns. These geometric patterns are then aligned to maximize the overlay of shared features. Finally, scoring metrics assess shared features of the final pattern alignments to quantify binding site similarity.
One aspect in which binding site comparison methods differ is in the identification of the optimal pattern alignment. Comparison of protein active site structures (CPASS) employs a straightforward alignment approach, exhaustively iterating translations and rotations of the query pattern to match a fixed target pattern [228]. More efficient methods group proximal pattern elements into triplets and optimally align these using geometric matching, as in ProSurfer [229], or geometric hashing, as in SiteEngines [230]. CavBase takes advantage of clique detection algorithms to align binding sites by representing patterns as graphs with pattern elements as nodes and element proximities as edges [231]. Other factors impacting the performance of ligand binding site comparison tools include the geometric pattern resolution and incorporation of binding site dynamics [232].
In contrast to geometric-based tools for binding site comparison, PocketFEATURE [233] characterizes protein pockets using the physicochemical microenvironments of the pocket residues. Comparison between two sets of pocket residue microenvironments allows identification of similar pockets that are likely to possess similar binding capabilities. Because the comparison uses only weak geometric restraints, this method is less reliant on the accuracy of crystallographic structures and has improved performance on dynamic binding sites.
A critical factor in binding site comparison methods is the correct identification of the binding sites themselves. Care must be taken to accurately identify entire binding cavities so that relevant comparisons can be gleaned. Furthermore, it is important to note that protein targets may bind a panel of small molecules in the same location and by various binding modes. Such heterogeneity can significantly alter the attributes of the binding site and therefore bias the results of comparison.
5.4. Docking and scoring technologies
Although protein structures are numerous, many protein targets have yet to be co-crystallized with their small-molecule ligands. In such cases, docking technologies (reviewed in [234–237]) allow exploration of the specific structural details of the protein–ligand interaction. The principle contribution of docking approaches towards understanding the molecular details of protein–ligand interactions is to determine the binding pose of a small-molecule ligand. Docking technologies also have important applications in drug discovery, where their aim is to distinguish between true and false positives in lead identification. Docking approaches begin with a docking stage, during which ligand orientations and conformations are sampled within the spatial constraints of the predicted protein binding site. Then, during the scoring stage, the best poses for each ligand are identified and ligands are rank-ordered.
There are numerous docking programs, but none achieve high accuracy across all protein targets. Current methods achieve, at best, 60 per cent accuracy for the ligand-binding conformation [238]. Selection of the best docking algorithm and scoring function is target- and ligand-dependent [238,239].
5.4.1. Docking
Docking a ligand into a protein binding site is a multivariate problem that models several degrees of freedom, including intermolecular translation and rotation and intramolecular conformational changes [239]. There are multiple docking approaches, interaction site matching, incremental construction, genetic algorithms and Monte Carlo searches [234,236]. Interaction site matching approaches, such as FRED [240], represent ligands and protein binding sites as pharamacophores and optimize their overlay to generate a docked ligand pose. FlexX [241] and other incremental construction programs rebuild the ligand in the protein binding site using libraries of preferred ligand fragment conformations. Genetic algorithm docking techniques, including AutoDock [242], mimic natural evolution where ligand conformations are encoded on ‘chromosomes’, diversified by genetic operators (crossovers, mutations and migrations), and subjected to natural selection using a fitness function. Monte Carlo-based docking approaches, like Glide [243], iteratively introduce random perturbations to the ligand pose and accept/reject them using a Monte Carlo criteria. It is important to note that many docking technologies incorporate multiple or blended approaches into their methods.
As discussed earlier in §3.1, protein–drug binding is a structurally dynamic process. While historical docking programs treated the protein and ligand as rigid bodies, recent shifts towards a dynamic representation of the system have improved docking accuracy. However, incorporation of protein flexibility exponentially expands the docking search space [244]. Docking methods address this challenge through varied means, considering a conformational ensemble of target structures (FlexX-Ensemble [245]), softening the binding site through reduced van der Waals penalties (ADAM [246]), and incorporating sidechain or backbone flexibility through rotamer libraries (AutoDock [247]). These methods capture both the induced fit and conformational selection models of protein–ligand binding by accounting for protein flexibility and structural ensembles, respectively.
In addition to protein and ligand flexibility, a number of other considerations also affect docking performance, such as protonation and tautomeric state of the ligand [248] as well as treatment of water molecules and solvent in the binding pocket [249]. These factors continue to limit docking performance today [237].
5.4.2. Scoring
Following docking, the resulting protein–ligand complexes are scored to identify the most biologically probable conformations and estimate their interaction strength. There are three types of scoring functions, force field-based, empirical and knowledge-based [235]. Force field scoring functions assess the physical atomic interactions of the system using van der Waals, electrostatics, and bond stretching/bending/torsional forces that are parameterized from experimental data and quantum mechanical calculations. More simply, empirical scoring functions evaluate ligand–protein complexes using energy terms (van der Waals energy, electrostatics, hydrogen bonding, desolvation, entropy, and hydrophobicity) weighted by fitting to binding affinity data of experimentally determined protein–ligand structures. Knowledge-based scoring functions derive pairwise atomic interaction potentials from experimentally determined protein–ligand complexes, with the assumption that frequently observed interactions are favourable.
The most stringent tests of a scoring function are rank ordering a series of related compounds and predicting their binding affinities [250]. Yet, both goals remain elusive using current scoring algorithms [237,239]. Recent alternative approaches addressing this limitation include use of machine-learning scoring functions [251], machine-learning scoring functions incorporating geometric descriptors [252] and consensus scoring with multiple scoring functions [237]. These approaches occasionally perform well for a particular compound series or target but are not universally applicable. Importantly, as the content of structural databases grows, the performance of machine-learning scoring functions is expected to further improve.
5.5. Molecular dynamic simulation
Protein flexibility and dynamics are fundamental to protein–drug interactions (see §§§3.1.1, 4.3.2.1 and 4.4). Protein dynamics range in scale from rearrangement of binding pocket sidechains to large coordinated movements of entire protein domains [51]. MD simulation is a popular tool for studying the conformational space accessible to proteins and protein–ligand complexes [253–255]. It has been used to refine experimental or modelled protein structures [256], reveal transient binding sites [257], examine the stability and strength of docked protein–ligand conformations [244], aid drug discovery [258,259], and explore altered drug binding profiles of protein variants [169,260,261].
In general, MD simulations depict the physical movements of atoms and molecules as they interact over time. This is accomplished by iteratively calculating the instantaneous forces present in the system (typically protein, ligand, solvent and often a lipid bilayer) and the resultant movements [253]. Forces between atoms and the potential energy of the system are defined by force fields, which contain energy functions with parameters derived from experimental or quantum mechanical studies. Common force fields developed specifically for the simulation of proteins include OPLS-AA [262], CHARMM [263], and AMBER [264]; each contain inherent biases that affect the sampled conformational space [265]. Widely adopted MD software include GROMACS [266], AMBER [267] and NAMD [268]. Thorough reviews on MD simulation methods were recently published [253,254,258].
Chief practical and technical considerations for MD studies are simulation length, system size and system resolution. Ideally, an MD simulation would provide a continuous, atomic-level view of system interactions over a long timescale. Recent methodological progress towards this ideal include the use of graphics processing units [269] and distributed computing [270]. These advances facilitate the simulation of relatively slow processes, such as the large conformational movements of kinases upon drug binding, and of large macromolecular systems, such as solvated GPCR structures embedded in a lipid bilayer [254]. Of exceptional note, Shaw and colleagues recently performed very long (approaching the millisecond and beyond) MD simulations on an all-atom protein system using a special-purpose machine [271]. However, simulation length poses an ongoing challenge, as many important biomolecular motions are slower than even the longest simulation timescales accessible today [272]. Short timescale MD simulations can be misleading, as they may not fully capture the dynamic nature of a protein–ligand system.
Given the limitations discussed above, MD simulation is of principle use when small conformational changes are expected. Normal mode analysis (NMA) is a powerful tool for exploring large conformational changes in protein structures, which are often important for ligand binding events [273–275]. NMA tools, such as The Elastic Network Model (elNémo) webserver [276], use a few low-frequency motions to describe rearrangements of protein domains and other types of large-amplitude MD. In contrast to many MD simulations, NMA offers the potential to extract essential dynamic information for global movements of large protein systems; however, coarse-grained models must often be used in such cases and the resulting studies therefore suffer from lower accuracy and specificity at the local scale [277].
6. Databases
There is currently no single database suitable for a thorough examination of the interplay between coding mutations, protein structure and drug response. Here, we briefly highlight several databases dedicated to individual components of the effect of genetic variation on drug response from the perspective of protein structure (summarized in table 2).
Table 2.
Databases for studying three-dimensional protein structures, genetic variants and drug response.
6.1. Databases of three-dimensional protein structures
The most comprehensive protein structure repository is the PDB, which currently holds approximately 73 000 protein structures (roughly 25% are of human origin) [34]. The PDB is well integrated into the network of bioinformatics tools and contains links to external resources for protein and small-molecule entities, integrated software packages for protein structure comparison and small-molecule similarity searching, and protein structural and functional annotations derived from other databases. Multiple secondary databases then extract and organize the experimentally determined structural data from the PDB using different criteria. sc-PDB collects structural examples of drug binding sites and includes analyses of the binding cavities and ligand chemical structures [299]. SitesBase annotates and compares ligand binding site structural similarities [283]. Druggable Cavity Directory (DCD) is a manually annotated repository of binding sites scored for druggability [41]. FireDB contains structures, ligands and annotated functionally important binding site residues [279]. Catalytic Site Atlas (CSA) annotates enzyme active sites, specifically catalytic residues, as three-dimensional structural templates for structures derived from the PDB [278]. In addition, there are a handful of repositories for protein models, including ModBase [280], Protein Model Portal [281] and SWISS-MODEL Repository [284].
6.2. Databases of protein–ligand interactions
Several databases integrate known protein–ligand interactions with a variety of external data sources. DrugBank is a catalogue of small molecules and integrates target protein structures when available [48]. Other databases, such as Binding MOAD [285] and PDBbind [286], link PDB structures with experimental binding data. Relibase offers tools for comparing ligand binding sites, analysing ligand similarity and searching for binding partners [287]. Similarly, CPASS database contains ligand-defined protein active sites from structures in the PDB [228]. Furthermore, the Structural Biology Knowledgebase (SBKB) [288] and Therapeutic Targets Database (TTD) [289] enhance the study of protein–ligand interactions with information regarding three-dimensional protein structures, ligands, pathways and diseases.
6.3. Databases linking genetic variants and disease
The most comprehensive SNP database is dbSNP, with approximately 20 million validated SNP entries [290]. Online Mendelian Inheritance in Man (OMIM) links genetic disorders with their causative genes [292]. SwissVar is a curated set of annotated missense SNPs linked with protein functional changes and possible disease association [294]. SNP and copy number annotation database (SCAN) annotates SNPs according to physical chromosomal location or effect on gene expression [293]. The catalogue of published genome-wide association studies (GWAS) is a collection of manually curated SNP-trait and SNP-disease associations from nearly 1000 published GWAS [291].
6.4. Databases linking genetic variants and protein structure
The LS-SNP database maps human coding mutations onto protein structures and assesses positional residue conservation patterns within the protein superfamily to predict the mutation's structural and functional impact [295]. Variations and Drugs (VnD) database is a structure-centric database of disease-related protein variants and drugs [296]. Protein family-specific databases also exist. Most notably, SuperCYP database reports manually curated data regarding the effect of SNPs on CYP enzyme structure, activity and drug metabolism [297].
6.5. Databases linking genetic variants and drug response
Parsing the relationship between genetic polymorphisms and drug outcome is complex. A patient's clinical response depends on pharmacodynamic and pharmacokinetic interactions, both of which can be altered by genetic variation and disease state.
PharmGKB is a manually curated knowledgebase of the impact of genetic variation on drug response [298]. It collects information on genes, drugs and diseases and emphasizes the clinical interpretation of the genetic variants, including information on drug dosing and genetic tests. PharmGKB documents approximately 500 genetic variants that significantly affect drug response. Of these variants, 70 per cent alter pharmacodynamic mechanisms, 10 per cent affect pharmacokinetic mechanisms and 10 per cent disrupt both pharmacodynamics and pharmacokinetics. PharmGKB identifies a subpopulation of genetic variants affecting drug response as ‘very important pharmacogenomic (VIP)’ genes. Missense mutations found in these VIP genes are mapped to representative homologous protein structures (where available) and associated drugs in table 3.
Table 3.
PharmGKB missense variants affecting drug response. Column 1 contains the protein encoded by the VIP gene in parentheses. A representative protein structure from the PDB is found in column 2. Missense mutations in the gene are listed by reference SNP ID in column 3 and by amino acid substitution in column 4. Column 5 categorizes the variant's effect as pharmacodynamic (PD) or pharmacokinetic (PK). Associated drugs are listed in column 6.
| protein | PDB ID | rsID | mut | effect | drug |
|---|---|---|---|---|---|
| P-glycoprotein (ABCB1) | 3G60 [300] | rs2032582 | S893T | PD, PK | doxorubicin |
| paclitaxel | |||||
| rs2229107 | S1141T | PK | phenytoin | ||
| β-1 adrenergic Receptor (ADRB1) | 2Y00 [301] | rs1801252 | S39G | PD, PK | atenolol |
| bisoprolol | |||||
| verapamil | |||||
| rs1801253 | G389R | PD | fluorouracil | ||
| metoprolol | |||||
| β-2 adrenergic receptor (ADRB2) | 2R4R [302] | rs1042713 | R16G | PD | salmeterol |
| rs1042714 | Q27E | PD | carvedilol | ||
| catechol O-methyltransferase (COMT) | 3BWM [303] | rs4680 | V158M | PD | antipsychotics |
| nicotine | |||||
| cytochrome P450 2A6 (CYP2A6) | 1Z10 [304] | rs1801272 | L160H | PK | anthracyclines |
| capecitabin | |||||
| cyclosporine | |||||
| cytarabine | |||||
| dexamethasone | |||||
| doxorubicin | |||||
| efavirenz | |||||
| fexofenadine | |||||
| mitoxantrone | |||||
| nicotine | |||||
| paclitaxel | |||||
| platinum | |||||
| taxanes | |||||
| vincristine | |||||
| rs28399468 | R485L | PK | nicotine | ||
| rs5031016 | I471T | PK | nicotine | ||
| cytochrome P450 2B6 (CYP2B6) | 3IBD [305] | rs2279343 | K252R | PK | cyclophosphamide |
| rs28399499 | I328T | PK | efavirenz | ||
| nevirapine | |||||
| rs3211371 | R487C | PD | bupropion | ||
| rs3745274 | Q172H | PK | cyclophosphamide | ||
| efavirenz | |||||
| nevirapine | |||||
| rs8192709 | R22C | PK | cyclophosphamide | ||
| cytochrome P450 2C9 (CYP2C9) | 1OG2 [144] | rs1057910 | I359L | PD, PK | losartan |
| phenytoin | |||||
| sulfonamides and urea deriv. | |||||
| valproic acid | |||||
| warfarin | |||||
| rs1799853 | R144C | PD, PK | phenytoin | ||
| sulfonamides and urea deriv. | |||||
| warfarin | |||||
| rs28371685 | R335W | PD, PK | phenytoin | ||
| warfarin | |||||
| rs28371686 | D360E | PD, PK | phenytoin | ||
| warfarin | |||||
| rs7900194 | R150H | PD | phenytoin | ||
| warfarin | |||||
| cytochrome P450 2D6 (CYP2D6) | 2F9Q [306] | rs1065852 | P34S | PD | tamoxifen |
| rs1135840 | T486S | PD | tamoxifen | ||
| rs16947 | C296R | PD | tamoxifen | ||
| dihydropyrimidine dehydrognase (DPYD) | 1GT8 [307] | rs1801159 | I543V | PD, PK | fluorouracil |
| glutathione S-transferase (GSTP1) | 2A2R [308] | rs1695 | I105V | PD, PK | cisplatin |
| cyclophosphamide | |||||
| doxorubicin | |||||
| fluorouracil | |||||
| mercaptopurine | |||||
| methotrexate | |||||
| oxaliplatin | |||||
| platinum compounds | |||||
| taxanes | |||||
| inward-rectifying potassium channel 11 (B2RC52) | — | rs5219 | K23E | PD | glibenclamide |
| metformin | |||||
| repaglinide | |||||
| sulfonamides & urea deriv. | |||||
| methylenetetrahydrofolate reductase (MTHFR) | — | rs1801131 | E429A | PD | capecitabine |
| cisplatin | |||||
| fluorouracil | |||||
| leucovori | |||||
| mercaptopurine | |||||
| methotrexate | |||||
| nitrous oxide | |||||
| oxaliplatin | |||||
| pemetrexed | |||||
| rs1801133 | A222V | PD, PK | capecitabine | ||
| carboplati | |||||
| cisplatin | |||||
| cyclophosphamide | |||||
| dactinomycin | |||||
| doxorubicin | |||||
| fluorouracil | |||||
| leucovorin | |||||
| methotrexate | |||||
| nitrous oxide | |||||
| oxaliplatin | |||||
| pemetrexed | |||||
| vincristine | |||||
| folate transporter 1 (SLC19A1) | — | rs1051266 | H27R | PD, PK | leucovorin |
| mercaptopurine | |||||
| methotrexate | |||||
| prednisone | |||||
| solute carrier organic anion transporter 1B1 (SLCO1B1) | — | rs11045819 | P155T | PD | fluvastatin |
| rs2306283 | N130D | PK | pravastatin | ||
| rs4149056 | V174A | PD, PK | atorvastatin | ||
| cerivastatin | |||||
| HMG-CoA reductase inhib. | |||||
| methotrexate | |||||
| mycophenolate mofeti | |||||
| repaglinide | |||||
| rifampin | |||||
| simvastatin | |||||
| thiopurine S-methyltransferase (TPMT) | 2BZG [309] | rs1142345 | Y240C | PD, PK | cisplatin |
| mercaptopurine | |||||
| s-adenosylmethionine | |||||
| rs1800460 | A154T | PD, PK | cisplatin | ||
| mercaptopurine | |||||
| s-adenosylmethionine |
7. Outlook
The interplay between genetic variation and protein structure forms the basis of interindividual variability in drug response. As such, this complex relationship is of increasing importance in bioinformatics and drug development. Three-dimensional protein structures are today frequently used in drug development practices, from selection of a therapeutic target, to determination of a molecular mechanism of action, to identification of a target patient population. As a result, great strides have been made in understanding the structural details governing drug–target interactions for recently approved therapeutic agents. Similarly, this information can be harnessed to predict the impact of missense mutations on drug response. In clinical practice protein structural insights along with a patient's genetic profile can be leveraged to improve treatment outcome, as evidenced by the results of gene-based treatment trials [7].
Although the focused study of the interaction of a drug with its target protein can offer insights into treatment outcome, complete understanding of drug response requires a systems pharmacology perspective. For instance, multivariate systems biology approaches that consider multiple proteins, signalling pathways, cell types and tissues can accurately recapitulate and predict therapeutic responses [310–313]. Recently, there has been a call for the inclusion of interpatient variability into these models [312]. This strategy holds tremendous promise for improving drug performance in clinical settings, but will first require extensive structural and mechanistic knowledge. Thus, there is an urgent need for high-throughput methods to systematically analyse drug–target interactions from a structural perspective.
Although the rate of structure deposition to the PDB is ever-increasing [314], deposition of pharmaceutically relevant protein structures remains low, limiting our understanding of variable drug response for many targets. Challenges to attaining crystal structures for protein targets include obtaining requisite large quantities of purified protein and inability to prepare crystals of target proteins in biologically relevant conformations [315]. As a result, three-dimensional structures are unavailable for many therapeutically important proteins. For example, though one-third of clinical drugs target these proteins [1], only 13 human rhodopsin-like GPCR (Pfam PF00001) structures are currently available in the PDB. Furthermore, these structures represent a highly biased sampling of rhodopsin conformations, namely inactive and apoprotein forms, precluding structural understanding of GPCR activation upon agonist binding. Only recently did Standfuss et al. [316] release an agonist-bound rhodopsin structure. Barriers to structural availability are gradually being overcome by structural genomics technology advances such as improved protein production platforms, automated high-throughput crystallization and data collection systems, and advanced software for structure determination [16]. Additionally, recently launched initiatives focused on structural determination of biomedically important proteins, including the protein structure initiative, RIKEN Structural Genomics/Proteomics Initiative and Structural Genomics Consortium, have collectively contributed roughly 10 500 unique protein structures to the PDB [314].
Compounding the problem of incomplete and biased structural data for therapeutic targets, this information is not well linked to experimental and clinical information regarding genetic variants and drug response. Current practice for studying variable drug response in the context of target protein structures with genetic variants requires at least partial manual curation. In the future, we anticipate that a standardized lexicon, improved text-mining algorithms and high-throughput structural analysis or prediction methods will lead to integrated and highly annotated databases. Different information sources must then be integrated into a comprehensive structural database that allows selection of an annotated missense SNP, mapping of the mutation onto a protein structure, evaluation of the interaction between mutated residues and drug-like ligands, and prediction of the effect of novel SNPs on drug activity or metabolism. Such a database will play a critical role in establishing protein structure information as a key tool in pharmacogenetics studies.
Another area in which technological advances are needed is that of high-throughput structural analysis of mutant protein targets and their interactions with therapeutic compounds. Such tools would build upon existing methods that explore the relationship between protein sequence variants, three-dimensional structure and drug response, as discussed previously in this review, and would offer a high-resolution structural understanding of genome-wide pharmacogenetic data. Developments in this area would take advantage of the expansive amount of information anticipated from next-generation sequencing techniques [317] and high-throughput chemical proteomics techniques [318]. Furthermore, the resulting structural technologies could interface with larger systems biology and network pharmacology studies to provide broad insight into variability in drug response among diverse patient populations.
Importantly, structural details of target proteins and their corresponding mutant isoforms can also be applied to inform drug design strategies, as recently reviewed [319–324]. For example, detailed knowledge of a target protein's three-dimensional structure facilitates the development of more selective drugs [169,325,326] and second-generation therapeutics for treating patients with unresponsive disease [327]. In addition, comparative analysis of target protein structures of currently marketed drugs allows for efficient drug repurposing to accelerate drug development for treating rare and orphan diseases [328–330]. Finally, structural knowledge of a drug target is critical for the intelligent combination of therapeutics acting on a single target in order to improve clinical efficacy and reduce the emergence of drug-resistant variants [331,332].
There is immense opportunity in translating information regarding the genetic differences between individuals and the three-dimensional structures of protein targets into drug development and clinical practice. Although the mechanism of many drugs is still not fully understood, we expect that the increasing number of detailed structural studies revealing drug–target interactions will lead to maximized likelihood of patient response, reduced drug resistance, and fewer adverse events. We believe that through the application of our expanding knowledge in this area it will be possible to address the ‘efficacy–effectiveness gap’ of currently marketed drugs and improve clinical outcomes.
Acknowledgements
J.L.L., G.W.T., T.L. and R.B.A. acknowledge support from the SIMBIOS National Center for Physics-based Simulation of Biological Structures (GM-61374) and NIH (LM-05652). G.W.T. also acknowledges support from the Stanford Bio-X Bioengineering Graduate Fellowship. E.C. acknowledges support from the Marie Curie International Outgoing Fellowship program (PIOF-GA-2009-237225) funded by the European Commission's FP7 grant. R.B.A. also acknowledges the NIH/NIGMS Pharmacogenetics Research Network and Database and the PharmGKB resource (GM-61374).
References
- 1.Rask-Andersen M., Almen M. S., Schioth H. B. 2011. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 10, 579–590 10.1038/nrd3478 (doi:10.1038/nrd3478) [DOI] [PubMed] [Google Scholar]
- 2.Pammolli F., Magazzini L., Riccaboni M. 2011. The productivity crisis in pharmaceutical R&D. Nat. Rev. Drug Discov. 10, 428–438 10.1038/nrd3405 (doi:10.1038/nrd3405) [DOI] [PubMed] [Google Scholar]
- 3.Watkins P. B. 2011. Drug safety sciences and the bottleneck in drug development. Clin. Pharmacol. Ther. 89, 788–790 10.1038/clpt.2011.63 (doi:10.1038/clpt.2011.63) [DOI] [PubMed] [Google Scholar]
- 4.Lesko L. J., Woodcock J. 2004. Translation of pharmacogenomics and pharmacogenetics: a regulatory perspective. Nat. Rev. Drug Discov. 3, 763–769 10.1038/nrd1499 (doi:10.1038/nrd1499) [DOI] [PubMed] [Google Scholar]
- 5.Wilkinson G. R. 2005. Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 352, 2211–2221 10.1056/NEJMra032424 (doi:10.1056/NEJMra032424) [DOI] [PubMed] [Google Scholar]
- 6.Sadee W., Dai Z. 2005. Pharmacogenetics/genomics and personalized medicine. Hum. Mol. Genet. 14 Spec No. 2, R207–R214 10.1093/hmg/ddi261 (doi:10.1093/hmg/ddi261) [DOI] [PubMed] [Google Scholar]
- 7.Wilke R. A., Dolan M. E. 2011. Genetics and variable drug response. J. Am. Med. Assoc. 306, 306–307 10.1001/jama.2011.998 (doi:10.1001/jama.2011.998) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eichler H. G., et al. Bridging the efficacy–effectiveness gap: a regulator's perspective on addressing variability of drug response. Nat. Rev. Drug Discov. 10, 495–506 10.1038/nrd3501 (doi:10.1038/nrd3501) [DOI] [PubMed] [Google Scholar]
- 9.Brunton L. L., Chabner B. A., Knollmann B. C. 2011. Pharmacodynamics: molecular mechanisms of drug action. In Goodman and Gilman's the pharmacological basis of therapeutics (ed. Brunton L. L.), 12th edn New York, NY: The McGraw-Hill Companies, Inc [Google Scholar]
- 10.Carlquist J. F., Anderson J. L. 2011. Pharmacogenetic mechanisms underlying unanticipated drug responses. Discov. Med. 11, 469–478 [PubMed] [Google Scholar]
- 11.Roses A. D. 2008. Pharmacogenetics in drug discovery and development: a translational perspective. Nat. Rev. Drug Discov. 7, 807–817 10.1038/nrd2593 (doi:10.1038/nrd2593) [DOI] [PubMed] [Google Scholar]
- 12.Daly A. K. 2010. Pharmacogenetics and human genetic polymorphisms. Biochem. J. 429, 435–449 10.1042/BJ20100522 (doi:10.1042/BJ20100522) [DOI] [PubMed] [Google Scholar]
- 13.Fernald G. H., Capriotti E., Daneshjou R., Karczewski K. J., Altman R. B. 2011. Bioinformatics challenges for personalized medicine. Bioinformatics 27, 1741–1748 10.1093/bioinformatics/btr295 (doi:10.1093/bioinformatics/btr295) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teng S., Michonova-Alexova E., Alexov E. 2008. Approaches and resources for prediction of the effects of non-synonymous single nucleotide polymorphism on protein function and interactions. Curr. Pharm. Biotechnol. 9, 123–133 10.2174/138920108783955164 (doi:10.2174/138920108783955164) [DOI] [PubMed] [Google Scholar]
- 15.Capriotti E., Altman R. B. 2011. Improving the prediction of disease-related variants using protein three-dimensional structure. BMC Bioinformatics 12 (Suppl. 4), S3 10.1186/1471-2105-12-S4-S3 (doi:10.1186/1471-2105-12-S4-S3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weigelt J. 2010. Structural genomics—impact on biomedicine and drug discovery. Exp. Cell Res. 316, 1332–1338 10.1016/j.yexcr.2010.02.041 (doi:10.1016/j.yexcr.2010.02.041) [DOI] [PubMed] [Google Scholar]
- 17.National Drug Code Directory [database on the Internet] 2011. U.S. Food and Drug Administration. [cited August 1, 2011]. Available from: http://www.accessdata.fda.gov/scripts/cder/ndc/default.cfm
- 18.Swinney D. C., Anthony J. 2011. How were new medicines discovered? Nat. Rev. Drug Discov. 10, 507–519 10.1038/nrd3480 (doi:10.1038/nrd3480) [DOI] [PubMed] [Google Scholar]
- 19.Vieth M., Siegel M. G., Higgs R. E., Watson I. A., Robertson D. H., Savin K. A., Durst G. L., Hipskind P. A. 2004. Characteristic physical properties and structural fragments of marketed oral drugs. J. Med. Chem. 47, 224–232 10.1021/jm030267j (doi:10.1021/jm030267j) [DOI] [PubMed] [Google Scholar]
- 20.Bauer R. A., Wurst J. M., Tan D. S. 2010. Expanding the range of ‘druggable’ targets with natural product-based libraries: an academic perspective. Curr. Opin. Chem. Biol. 14, 308–314 10.1016/j.cbpa.2010.02.001 (doi:10.1016/j.cbpa.2010.02.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benjamin B., Barman T. K., Chaira T., Paliwal J. K. 2010. Integration of physicochemical and pharmacokinetic parameters in lead optimization: a physiological pharmacokinetic model based approach. Curr. Drug Discov. Technol. 7, 143–153 [DOI] [PubMed] [Google Scholar]
- 22.Gleeson M. P., Hersey A., Montanari D., Overington J. 2011. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat. Rev. Drug Discov. 10, 197–208 10.1038/nrd3367 (doi:10.1038/nrd3367) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipinski C. A., Lombardo F., Dominy B. W., Feeney P. J. 1997. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23, 3–25 10.1016/S0169-409X(96)00423-1 (doi:10.1016/S0169-409X(96)00423-1) [DOI] [PubMed] [Google Scholar]
- 24.Oprea T. I. 2000. Property distribution of drug-related chemical databases. J. Comput. Aided Mol. Des. 14, 251–264 10.1023/A:1008130001697 (doi:10.1023/A:1008130001697) [DOI] [PubMed] [Google Scholar]
- 25.Hann M. M., Leach A. R., Harper G. 2001. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 41, 856–864 10.1021/ci000403i (doi:10.1021/ci000403i) [DOI] [PubMed] [Google Scholar]
- 26.Mestres J., Gregori-Puigjane E., Valverde S., Sole R. V. 2009. The topology of drug–target interaction networks, implicit dependence on drug properties and target families. Mol. Biosyst. 5, 1051–1057 10.1039/b905821b (doi:10.1039/b905821b) [DOI] [PubMed] [Google Scholar]
- 27.Vieth M., Sutherland J. J. 2006. Dependence of molecular properties on proteomic family for marketed oral drugs. J. Med. Chem. 49, 3451–3453 10.1021/jm0603825 (doi:10.1021/jm0603825) [DOI] [PubMed] [Google Scholar]
- 28.Bakheet T. M., Doig A. J. 2009. Properties and identification of human protein drug targets. Bioinformatics. 25, 451–457 10.1093/bioinformatics/btp002 (doi:10.1093/bioinformatics/btp002) [DOI] [PubMed] [Google Scholar]
- 29.Hopkins A. L., Groom C. R. 2002. The druggable genome. Nat. Rev. Drug Discov. 1, 727–730 10.1038/nrd892 (doi:10.1038/nrd892) [DOI] [PubMed] [Google Scholar]
- 30.Cheng A. C., Coleman R. G., Smyth K. T., Cao Q., Soulard P., Caffrey D. R., Salzberg A. C., Huang E. S. 2007. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 25, 71–75 10.1038/nbt1273 (doi:10.1038/nbt1273) [DOI] [PubMed] [Google Scholar]
- 31.Najmanovich R., Kurbatova N., Thornton J. 2008. Detection of 3D atomic similarities and their use in the discrimination of small molecule protein-binding sites. Bioinformatics 24, i105–i111 10.1093/bioinformatics/btn263 (doi:10.1093/bioinformatics/btn263) [DOI] [PubMed] [Google Scholar]
- 32.Halgren T. A. 2009. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model 49, 377–389 10.1021/ci800324m (doi:10.1021/ci800324m) [DOI] [PubMed] [Google Scholar]
- 33.Nayal M., Honig B. 2006. On the nature of cavities on protein surfaces: application to the identification of drug-binding sites. Proteins 63, 892–906 10.1002/prot.20897 (doi:10.1002/prot.20897) [DOI] [PubMed] [Google Scholar]
- 34.Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. 2000. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 10.1093/nar/28.1.235 (doi:10.1093/nar/28.1.235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murzin A. G., Brenner S. E., Hubbard T., Chothia C. 1995. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247, 536–540 10.1016/S0022-2836(05)80134-2 (doi:10.1016/S0022-2836(05)80134-2) [DOI] [PubMed] [Google Scholar]
- 36.Li B., Turuvekere S., Agrawal M., La D., Ramani K., Kihara D. 2008. Characterization of local geometry of protein surfaces with the visibility criterion. Proteins 71, 670–683 10.1002/prot.21732 (doi:10.1002/prot.21732) [DOI] [PubMed] [Google Scholar]
- 37.Fuller J. C., Burgoyne N. J., Jackson R. M. 2009. Predicting druggable binding sites at the protein–protein interface. Drug Discov. Today 14, 155–161 10.1016/j.drudis.2008.10.009 (doi:10.1016/j.drudis.2008.10.009) [DOI] [PubMed] [Google Scholar]
- 38.Volkamer A., Griewel A., Grombacher T., Rarey M. 2010. Analyzing the topology of active sites, on the prediction of pockets and subpockets. J. Chem. Inf. Model 50, 2041–2052 10.1021/ci100241y (doi:10.1021/ci100241y) [DOI] [PubMed] [Google Scholar]
- 39.Smith R. D., Hu L., Falkner J. A., Benson M. L., Nerothin J. P., Carlson H. A. 2006. Exploring protein–ligand recognition with binding MOAD. J. Mol. Graph Model 24, 414–425 10.1016/j.jmgm.2005.08.002 (doi:10.1016/j.jmgm.2005.08.002) [DOI] [PubMed] [Google Scholar]
- 40.Hajduk P. J., Huth J. R., Fesik S. W. 2005. Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 48, 2518–2525 10.1021/jm049131r (doi:10.1021/jm049131r) [DOI] [PubMed] [Google Scholar]
- 41.Schmidtke P., Barril X. 2010. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J. Med. Chem. 53, 5858–5867 10.1021/jm100574m (doi:10.1021/jm100574m) [DOI] [PubMed] [Google Scholar]
- 42.Wass M. N., David A., Sternberg M. J. 2011. Challenges for the prediction of macromolecular interactions. Curr. Opin. Struct. Biol. 21, 382–390 10.1016/j.sbi.2011.03.013 (doi:10.1016/j.sbi.2011.03.013) [DOI] [PubMed] [Google Scholar]
- 43.Wells J. A., McClendon C. L. 2007. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 450, 1001–1009 10.1038/nature06526 (doi:10.1038/nature06526) [DOI] [PubMed] [Google Scholar]
- 44.Park C. M., et al. 2008. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J. Med. Chem. 51, 6902–6915 10.1021/jm800669s (doi:10.1021/jm800669s) [DOI] [PubMed] [Google Scholar]
- 45.2004. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 10.1038/nature03001 (doi:10.1038/nature03001) [DOI] [PubMed] [Google Scholar]
- 46.Landry Y., Gies J. P. 2008. Drugs and their molecular targets: an updated overview. Fundam. Clin. Pharmacol. 22, 1–18 10.1111/j.1472-8206.2007.00548.x (doi:10.1111/j.1472-8206.2007.00548.x) [DOI] [PubMed] [Google Scholar]
- 47.Plewczynski D., Rychlewski L. 2009. Meta-basic estimates the size of druggable human genome. J. Mol. Model 15, 695–699 10.1007/s00894-008-0353-5 (doi:10.1007/s00894-008-0353-5) [DOI] [PubMed] [Google Scholar]
- 48.Knox C., et al. 2011. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 39, D1035–D1041 10.1093/nar/gkq1126 (doi:10.1093/nar/gkq1126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Southan C., Boppana K., Jagarlapudi S. A., Muresan S. 2011. Analysis of in vitro bioactivity data extracted from drug discovery literature and patents: ranking 1654 human protein targets by assayed compounds and molecular scaffolds. J. Cheminform. 3, 14 10.1186/1758-2946-3-14 (doi:10.1186/1758-2946-3-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Overington J. P., Al-Lazikani B., Hopkins A. L. 2006. How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996 10.1038/nrd2199 (doi:10.1038/nrd2199) [DOI] [PubMed] [Google Scholar]
- 51.Spyrakis F., BidonChanal A., Barril X., Luque F. J. 2011. Protein flexibility and ligand recognition: challenges for molecular modeling. Curr. Top. Med. Chem. 11, 192–210 [DOI] [PubMed] [Google Scholar]
- 52.Okazaki K., Takada S. 2008. Dynamic energy landscape view of coupled binding and protein conformational change: induced-fit versus population-shift mechanisms. Proc. Natl Acad. Sci. USA 105, 11 182–11 187 10.1073/pnas.0802524105 (doi:10.1073/pnas.0802524105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boehr D. D., Nussinov R., Wright P. E. 2009. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 10.1038/nchembio.232 (doi:10.1038/nchembio.232) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hansen G., Gielen-Haertwig H., Reinemer P., Schomburg D., Harrenga A., Niefind K. 2011. Unexpected active-site flexibility in the structure of human neutrophil elastase in complex with a new dihydropyrimidone inhibitor. J. Mol. Biol. 409, 681–691 10.1016/j.jmb.2011.04.047 (doi:10.1016/j.jmb.2011.04.047) [DOI] [PubMed] [Google Scholar]
- 55.Aleksandrov A., Simonson T. 2010. Molecular dynamics simulations show that conformational selection governs the binding preferences of imatinib for several tyrosine kinases. J. Biol. Chem. 285, 13 807–13 815 10.1074/jbc.M110.109660 (doi:10.1074/jbc.M110.109660) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Long D., Bruschweiler R. 2011. In silico elucidation of the recognition dynamics of ubiquitin. PLoS Comput. Biol. 7, e1002035 10.1371/journal.pcbi.1002035 (doi:10.1371/journal.pcbi.1002035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Silva D. A., Bowman G. R., Sosa-Peinado A., Huang X. 2011. A role for both conformational selection and induced fit in ligand binding by the LAO protein. PLoS Comput. Biol. 7, e1002054 10.1371/journal.pcbi.1002054 (doi:10.1371/journal.pcbi.1002054) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chayen N. E., Saridakis E. 2008. Protein crystallization: from purified protein to diffraction-quality crystal. Nat. Methods 5, 147–153 10.1038/nmeth.f.203 (doi:10.1038/nmeth.f.203) [DOI] [PubMed] [Google Scholar]
- 59.Tang G. W., Altman R. B. 2011. Remote thioredoxin recognition using evolutionary conservation and structural dynamics. Structure 19, 461–470 10.1016/j.str.2011.02.007 (doi:10.1016/j.str.2011.02.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Glazer D. S., Radmer R. J., Altman R. B. 2009. Improving structure-based function prediction using molecular dynamics. Structure 17, 919–929 10.1016/j.str.2009.05.010 (doi:10.1016/j.str.2009.05.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kortagere S., Krasowski M. D., Ekins S. 2009. The importance of discerning shape in molecular pharmacology. Trends Pharmacol. Sci. 30, 138–147 10.1016/j.tips.2008.12.001 (doi:10.1016/j.tips.2008.12.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Filippakopoulos P., et al. 2010. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 10.1038/nature09504 (doi:10.1038/nature09504) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ceccarelli D. F., et al. 2011. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell 145, 1075–1087 10.1016/j.cell.2011.05.039 (doi:10.1016/j.cell.2011.05.039) [DOI] [PubMed] [Google Scholar]
- 64.Ballester P. J., Westwood I., Laurieri N., Sim E., Richards W. G. 2010. Prospective virtual screening with Ultrafast Shape Recognition: the identification of novel inhibitors of arylamine N-acetyltransferases. J. R. Soc. Interface 7, 335–342 10.1098/rsif.2009.0170 (doi:10.1098/rsif.2009.0170) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ebalunode J. O., Zheng W. 2010. Molecular shape technologies in drug discovery: methods and applications. Curr. Top. Med. Chem. 10, 669–679 10.2174/156802610791111489 (doi:10.2174/156802610791111489) [DOI] [PubMed] [Google Scholar]
- 66.Kukic P., Nielsen J. E. 2010. Electrostatics in proteins and protein–ligand complexes. Future Med. Chem. 2, 647–666 10.4155/fmc.10.6 (doi:10.4155/fmc.10.6) [DOI] [PubMed] [Google Scholar]
- 67.Wang H., Blais J., Ron D., Cardozo T. 2010. Structural determinants of PERK inhibitor potency and selectivity. Chem. Biol. Drug Des. 76, 480–95 10.1111/j.1747-0285.2010.01048.x (doi:10.1111/j.1747-0285.2010.01048.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Snyder P. W., et al. 2011. Mechanism of the hydrophobic effect in the biomolecular recognition of arylsulfonamides by carbonic anhydrase. Proc. Natl Acad. Sci. USA 108, 17 889–17 894 10.1073/pnas.1114107108 (doi:10.1073/pnas.1114107108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arunan E., et al. 2011. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 83, 1637–1641 10.1351/PAC-REC-10-01-02 (doi:10.1351/PAC-REC-10-01-02) [DOI] [Google Scholar]
- 70.Urwyler S. 2011. Allosteric modulation of family C G-protein-coupled receptors: from molecular insights to therapeutic perspectives. Pharmacol. Rev. 63, 59–126 10.1124/pr.109.002501 (doi:10.1124/pr.109.002501) [DOI] [PubMed] [Google Scholar]
- 71.Schindler T., Bornmann W., Pellicena P., Miller W. T., Clarkson B., Kuriyan J. 2000. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 289, 1938–1942 10.1126/science.289.5486.1938 (doi:10.1126/science.289.5486.1938) [DOI] [PubMed] [Google Scholar]
- 72.Karaman M. W., et al. 2008. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132 10.1038/nbt1358 (doi:10.1038/nbt1358) [DOI] [PubMed] [Google Scholar]
- 73.Branca M. A. 2005. Multi-kinase inhibitors create buzz at ASCO. Nat. Biotechnol. 23, 639 10.1038/nbt0605-639 (doi:10.1038/nbt0605-639) [DOI] [PubMed] [Google Scholar]
- 74.Sessel S., Fernandez A. 2011. Selectivity filters to edit out deleterious side effects in kinase inhibitors. Curr. Top. Med. Chem. 11, 788–799 10.2174/156802611795165089 (doi:10.2174/156802611795165089) [DOI] [PubMed] [Google Scholar]
- 75.Ma X. H., Shi Z., Tan C., Jiang Y., Go M. L., Low B. C., Chen Y. Z. 2010. In-silico approaches to multi-target drug discovery: computer aided multi-target drug design, multi-target virtual screening. Pharm. Res. 27, 739–749 10.1007/s11095-010-0065-2 (doi:10.1007/s11095-010-0065-2) [DOI] [PubMed] [Google Scholar]
- 76.Schwartz T. W., Holst B. 2007. Allosteric enhancers, allosteric agonists and ago-allosteric modulators: where do they bind and how do they act? Trends Pharmacol. Sci. 28, 366–373 10.1016/j.tips.2007.06.008 (doi:10.1016/j.tips.2007.06.008) [DOI] [PubMed] [Google Scholar]
- 77.Eglen R., Reisine T. 2011. Drug discovery and the human kinome: recent trends. Pharmacol. Ther. 130, 144–156 10.1016/j.pharmthera.2011.01.007 (doi:10.1016/j.pharmthera.2011.01.007) [DOI] [PubMed] [Google Scholar]
- 78.Gjoni T., Urwyler S. 2008. Receptor activation involving positive allosteric modulation, unlike full agonism, does not result in GABAB receptor desensitization. Neuropharmacology 55, 1293–1299 10.1016/j.neuropharm.2008.08.008 (doi:10.1016/j.neuropharm.2008.08.008) [DOI] [PubMed] [Google Scholar]
- 79.Bowery N. G. 2006. Allosteric receptor modulation in drug targeting. New York, NY: Taylor and Francis Group [Google Scholar]
- 80.Sadowsky J. D., Burlingame M. A., Wolan D. W., McClendon C. L., Jacobson M. P., Wells J. A. 2011. Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc. Natl Acad. Sci. USA 108, 6056–6061 10.1073/pnas.1102376108 (doi:10.1073/pnas.1102376108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Warne T., Serrano-Vega M. J., Baker J. G., Moukhametzianov R., Edwards P. C., Henderson R., Leslie A. G., Tate C. G., Schertler G. F. 2008. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454, 486–491 10.1038/nature07101 (doi:10.1038/nature07101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lewis J. A., Lebois E. P., Lindsley C. W. 2008. Allosteric modulation of kinases and GPCRs: design principles and structural diversity. Curr. Opin. Chem. Biol. 12, 269–280 10.1016/j.cbpa.2008.02.014 (doi:10.1016/j.cbpa.2008.02.014) [DOI] [PubMed] [Google Scholar]
- 83.Ashley E. A., et al. 2010. Clinical assessment incorporating a personal genome. Lancet 375, 1525–1535 10.1016/S0140-6736(10)60452-7 (doi:10.1016/S0140-6736(10)60452-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dewey F. E., et al. 2011. Phased whole-genome genetic risk in a family quartet using a major allele reference sequence. PLoS Genet. 7, e1002280 10.1371/journal.pgen.1002280 (doi:10.1371/journal.pgen.1002280) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.2010. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 10.1038/nature09534 (doi:10.1038/nature09534) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mendell J. T., Dietz H. C. 2001. When the message goes awry: disease-producing mutations that influence mRNA content and performance. Cell 107, 411–414 10.1016/S0092-8674(01)00583-9 (doi:10.1016/S0092-8674(01)00583-9) [DOI] [PubMed] [Google Scholar]
- 87.Stenson P. D., Mort M., Ball E. V., Howells K., Phillips A. D., Thomas N. S., Cooper D. N. 2009. The Human Gene Mutation Database: 2008 update. Genome Med. 1, 13 10.1186/gm13 (doi:10.1186/gm13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Capriotti E., Nehrt N. L., Kann M. G., Bromberg Y. 2012. Bioinformatics for personal genome interpretation. Brief Bioinform . 10.1093/bib/bbr070 (doi:10.1093/bib/bbr070) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Calabrese R., Capriotti E., Fariselli P., Martelli P. L., Casadio R. 2009. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 30, 1237–44 10.1002/humu.21047 (doi:10.1002/humu.21047) [DOI] [PubMed] [Google Scholar]
- 90.Capriotti E., Altman R. B. 2011. A new disease-specific machine learning approach for the prediction of cancer-causing missense variants. Genomics 98, 310–317 10.1016/j.ygeno.2011.06.010 (doi:10.1016/j.ygeno.2011.06.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Capriotti E., Arbiza L., Casadio R., Dopazo J., Dopazo H., Marti-Renom M. A. 2008. Use of estimated evolutionary strength at the codon level improves the prediction of disease-related protein mutations in humans. Hum. Mutat. 29, 198–204 10.1002/humu.20628 (doi:10.1002/humu.20628) [DOI] [PubMed] [Google Scholar]
- 92.Capriotti E., Calabrese R., Casadio R. 2006. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 22, 2729–2734 10.1093/bioinformatics/btl423 (doi:10.1093/bioinformatics/btl423) [DOI] [PubMed] [Google Scholar]
- 93.Li B., Krishnan V. G., Mort M. E., Xin F., Kamati K. K., Cooper D. N., Mooney S. D., Radivojac P. 2009. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25, 2744–2750 10.1093/bioinformatics/btp528 (doi:10.1093/bioinformatics/btp528) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ng P. C., Henikoff S. 2003. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 10.1093/nar/gkg509 (doi:10.1093/nar/gkg509) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Karchin R., Diekhans M., Kelly L., Thomas D. J., Pieper U., Eswar N., Haussler D., Sali A. 2005. LS-SNP: large-scale annotation of coding non-synonymous SNPs based on multiple information sources. Bioinformatics 21, 2814–2820 10.1093/bioinformatics/bti442 (doi:10.1093/bioinformatics/bti442) [DOI] [PubMed] [Google Scholar]
- 96.Ramensky V., Bork P., Sunyaev S. 2002. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 30, 3894–900 10.1093/nar/gkf493 (doi:10.1093/nar/gkf493) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yue P., Moult J. 2006. Identification and analysis of deleterious human SNPs. J. Mol. Biol. 356, 1263–1274 10.1016/j.jmb.2005.12.025 (doi:10.1016/j.jmb.2005.12.025) [DOI] [PubMed] [Google Scholar]
- 98.Dehouck Y., Grosfils A., Folch B., Gilis D., Bogaerts P., Rooman M. 2009. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 25, 2537–2543 10.1093/bioinformatics/btp445 (doi:10.1093/bioinformatics/btp445) [DOI] [PubMed] [Google Scholar]
- 99.Capriotti E., Fariselli P., Rossi I., Casadio R. 2008. A three-state prediction of single point mutations on protein stability changes. BMC Bioinformatics 9 (Suppl. 2), S6 10.1186/1471-2105-9-S2-S6 (doi:10.1186/1471-2105-9-S2-S6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L. 2005. The FoldX web server: an online force field. Nucleic Acids Res. 1; 33. (Web Server issue; W382–W388) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Capriotti E., Fariselli P., Casadio R. 2005. I Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 1, 33. (Web Server issue;W306–W310) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang J., Yang P. L., Gray N. S. 2009. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39 10.1038/nrc2559 (doi:10.1038/nrc2559) [DOI] [PubMed] [Google Scholar]
- 103.Fei F., Stoddart S., Groffen J., Heisterkamp N. 2010. Activity of the Aurora kinase inhibitor VX-680 against Bcr/Abl-positive acute lymphoblastic leukemias. Mol. Cancer Ther. 9, 1318–1327 10.1158/1535-7163.MCT-10-0069 (doi:10.1158/1535-7163.MCT-10-0069) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou T., et al. 2007. Crystal structure of the T315I mutant of AbI kinase. Chem. Biol. Drug Des. 70, 171–181 10.1111/j.1747-0285.2007.00556.x (doi:10.1111/j.1747-0285.2007.00556.x) [DOI] [PubMed] [Google Scholar]
- 105.Cowan-Jacob S. W., et al. 2007. Structural biology contributions to the discovery of drugs to treat chronic myelogenous leukaemia. Acta Crystallogr. D Biol. Crystallogr. 63, 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chan W. W., et al. 2011. Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036. Cancer Cell 19, 556–568 10.1016/j.ccr.2011.03.003 (doi:10.1016/j.ccr.2011.03.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhou T., et al. 2011. Structural mechanism of the Pan-BCR-ABL inhibitor ponatinib (AP24534): lessons for overcoming kinase inhibitor resistance. Chem. Biol. Drug Des. 77, 1–11 10.1111/j.1747-0285.2010.01054.x (doi:10.1111/j.1747-0285.2010.01054.x) [DOI] [PubMed] [Google Scholar]
- 108.Cheetham G. M., Charlton P. A., Golec J. M., Pollard J. R. 2007. Structural basis for potent inhibition of the Aurora kinases and a T315I multi-drug resistant mutant form of Abl kinase by VX-680. Cancer Lett. 251, 323–329 10.1016/j.canlet.2006.12.004 (doi:10.1016/j.canlet.2006.12.004) [DOI] [PubMed] [Google Scholar]
- 109.Kosaka T., Yatabe Y., Endoh H., Yoshida K., Hida T., Tsuboi M., Tada H., Kuwano H., Mitsudomi T. 2006. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin. Cancer Res. 12, 5764–5769 10.1158/1078-0432.CCR-06-0714 (doi:10.1158/1078-0432.CCR-06-0714) [DOI] [PubMed] [Google Scholar]
- 110.Balak M. N., et al. 2006. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin. Cancer Res. 12, 6494–6501 10.1158/1078-0432.CCR-06-1570 (doi:10.1158/1078-0432.CCR-06-1570) [DOI] [PubMed] [Google Scholar]
- 111.Kobayashi S., et al. 2005. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792 10.1056/NEJMoa044238 (doi:10.1056/NEJMoa044238) [DOI] [PubMed] [Google Scholar]
- 112.Yun C. H., Mengwasser K. E., Toms A. V., Woo M. S., Greulich H., Wong K. K., Meyerson M., Eck M. J. 2008. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl Acad. Sci. USA 105, 2070–2075 10.1073/pnas.0709662105 (doi:10.1073/pnas.0709662105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhou W., et al. 2009. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462, 1070–1074 10.1038/nature08622 (doi:10.1038/nature08622) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.van de Wijngaart D. J., Molier M., Lusher S. J., Hersmus R., Jenster G., Trapman J., Dubbink M. J. 2010. Systematic structure-function analysis of androgen receptor Leu701 mutants explains the properties of the prostate cancer mutant L701H. J. Biol. Chem. 285, 5097–5105 10.1074/jbc.M109.039958 (doi:10.1074/jbc.M109.039958) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tran C., et al. 2009. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787–790 10.1126/science.1168175 (doi:10.1126/science.1168175) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sack J. S., et al. 2001. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc. Natl Acad. Sci. USA 98, 4904–4909 10.1073/pnas.081565498 (doi:10.1073/pnas.081565498) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bohl C. E., Gao W., Miller D. D., Bell C. E., Dalton J. T. 2005. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl Acad. Sci. USA 102, 6201–6206 10.1073/pnas.0500381102 (doi:10.1073/pnas.0500381102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bohl C. E., Miller D. D., Chen J., Bell C. E., Dalton J. T. 2005. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J. Biol. Chem. 280, 37 747–37 754 10.1074/jbc.M507464200 (doi:10.1074/jbc.M507464200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bohl C. E., Wu Z., Miller D. D., Bell C. E., Dalton J. T. 2007. Crystal structure of the T877A human androgen receptor ligand-binding domain complexed to cyproterone acetate provides insight for ligand-induced conformational changes and structure-based drug design. J. Biol. Chem. 282, 13 648–13 655 10.1074/jbc.M611711200 (doi:10.1074/jbc.M611711200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ho B. K., Gruswitz F. 2008. HOLLOW, generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct. Biol. 8, 49 10.1186/1472-6807-8-49 (doi:10.1186/1472-6807-8-49) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pereira de Jesus-Tran K., Cote P. L., Cantin L., Blanchet J., Labrie F., Breton R. 2006. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci. 15, 987–999 10.1110/ps.051905906 (doi:10.1110/ps.051905906) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Scher H. I., et al. 2010. Antitumour activity of MDV3100 in castration-resistant prostate cancer, a phase 1–2 study. Lancet 375, 1437–1446 10.1016/S0140-6736(10)60172-9 (doi:10.1016/S0140-6736(10)60172-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Corless C. L., Fletcher J. A., Heinrich M. C. 2004. Biology of gastrointestinal stromal tumors. J. Clin. Oncol. 22, 3813–3825 10.1200/JCO.2004.05.140 (doi:10.1200/JCO.2004.05.140) [DOI] [PubMed] [Google Scholar]
- 124.Verweij J., et al. 2004. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 364, 1127–1134 10.1016/S0140-6736(04)17098-0 (doi:10.1016/S0140-6736(04)17098-0) [DOI] [PubMed] [Google Scholar]
- 125.Heinrich M. C., et al. 2008. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J. Clin. Oncol. 26, 5352–5359 10.1200/JCO.2007.15.7461 (doi:10.1200/JCO.2007.15.7461) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mol C. D., et al. 2003. Structure of a c-Kit product complex reveals the basis for kinase transactivation. J. Biol. Chem. 278, 31 461–31 464 10.1074/jbc.C300186200 (doi:10.1074/jbc.C300186200) [DOI] [PubMed] [Google Scholar]
- 127.Mol C. D., et al. 2004. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J. Biol. Chem. 279, 31 655–31 663 10.1074/jbc.M403319200 (doi:10.1074/jbc.M403319200) [DOI] [PubMed] [Google Scholar]
- 128.Gajiwala K. S., et al. 2009. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc. Natl Acad. Sci. USA 106, 1542–1547 10.1073/pnas.0812413106 (doi:10.1073/pnas.0812413106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pan B. S., et al. 2010. MK-2461, a novel multitargeted kinase inhibitor, preferentially inhibits the activated c-Met receptor. Cancer Res. 70, 1524–1533 10.1158/0008-5472.CAN-09-2541 (doi:10.1158/0008-5472.CAN-09-2541) [DOI] [PubMed] [Google Scholar]
- 130.Tsai J., et al. 2008. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl Acad. Sci. USA 105, 3041–3046 10.1073/pnas.0711741105 (doi:10.1073/pnas.0711741105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lane D. P., Cheok C. F., Lain S. 2010. p53-based cancer therapy. Cold Spring Harb. Perspect. Biol. 2, a001222 10.1101/cshperspect.a001222 (doi:10.1101/cshperspect.a001222) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wiman K. G. 2010. Pharmacological reactivation of mutant p53: from protein structure to the cancer patient. Oncogene 29, 4245–4252 10.1038/onc.2010.188 (doi:10.1038/onc.2010.188) [DOI] [PubMed] [Google Scholar]
- 133.Boeckler F. M., Joerger A. C., Jaggi G., Rutherford T. J., Veprintsev D. B., Fersht A. R. 2008. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl Acad. Sci. USA 105, 10 360–10 365 10.1073/pnas.0805326105 (doi:10.1073/pnas.0805326105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kussie P. H., Gorina S., Marechal V., Elenbaas B., Moreau J., Levine A. J., Pavletich N. P. 1996. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948–953 10.1126/science.274.5289.948 (doi:10.1126/science.274.5289.948) [DOI] [PubMed] [Google Scholar]
- 135.Secchiero P., Bosco R., Celeghini C., Zauli G. 2011. Recent advances in the therapeutic perspectives of Nutlin-3. Curr. Pharm. Des. 17, 569–577 10.2174/138161211795222586 (doi:10.2174/138161211795222586) [DOI] [PubMed] [Google Scholar]
- 136.Azmi A. S., Philip P. A., Almhanna K., Beck F. W., Sarkar F. H., Mohammad R. M. 2010. MDM2 inhibitors for pancreatic cancer therapy. Mini Rev. Med. Chem. 10, 518–526 10.2174/138955710791384054 (doi:10.2174/138955710791384054) [DOI] [PubMed] [Google Scholar]
- 137.Pishas K. I., Al-Ejeh F., Zinonos I., Kumar R., Evdokiou A., Brown M. P., Callan D. F., Neilsen P. M. 2011. Nutlin-3a is a potential therapeutic for ewing sarcoma. Clin. Cancer Res. 17, 494–504 10.1158/1078-0432.CCR-10-1587 (doi:10.1158/1078-0432.CCR-10-1587) [DOI] [PubMed] [Google Scholar]
- 138.Azmi A. S., et al. 2010. Reactivation of p53 by novel MDM2 inhibitors: implications for pancreatic cancer therapy. Curr. Cancer Drug Targets 10, 319–331 10.2174/156800910791190229 (doi:10.2174/156800910791190229) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Joerger A. C., Ang H. C., Fersht A. R. 2006. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl Acad. Sci. USA 103, 15 056–15 061 10.1073/pnas.0607286103 (doi:10.1073/pnas.0607286103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Saddler C., et al. 2008. Comprehensive biomarker and genomic analysis identifies p53 status as the major determinant of response to MDM2 inhibitors in chronic lymphocytic leukemia. Blood 111, 1584–1593 10.1182/blood-2007-09-112698 (doi:10.1182/blood-2007-09-112698) [DOI] [PubMed] [Google Scholar]
- 141.Joerger A. C., Allen M. D., Fersht A. R. 2004. Crystal structure of a superstable mutant of human p53 core domain. Insights into the mechanism of rescuing oncogenic mutations. J. Biol. Chem. 279, 1291–1296 10.1074/jbc.M309732200 (doi:10.1074/jbc.M309732200) [DOI] [PubMed] [Google Scholar]
- 142.Bullock A. N., Henckel J., Fersht A. R. 2000. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 19, 1245–1256 10.1038/sj.onc.1203434 (doi:10.1038/sj.onc.1203434) [DOI] [PubMed] [Google Scholar]
- 143.Basse N., Kaar J. L., Settanni G., Joerger A. C., Rutherford T. J., Fersht A. R. 2010. Toward the rational design of p53-stabilizing drugs: probing the surface of the oncogenic Y220C mutant. Chem. Biol. 17, 46–56 10.1016/j.chembiol.2009.12.011 (doi:10.1016/j.chembiol.2009.12.011) [DOI] [PubMed] [Google Scholar]
- 144.Williams P. A., Cosme J., Ward A., Angove H. C., Matak Vinkovic D., Jhoti H. 2003. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 424, 464–468 10.1038/nature01862 (doi:10.1038/nature01862) [DOI] [PubMed] [Google Scholar]
- 145.Thorn C. F., Aklillu E., Klein T. E., Altman R. B. 2011. PharmGKB summary: very important pharmacogene information for CYP1A2. Pharmacogenet. Genom. 22, 73–77 10.1097/FPC.0b013e32834c6efd (doi:10.1097/FPC.0b013e32834c6efd) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Thorn C. F., Lamba J. K., Lamba V., Klein T. E., Altman R. B. 2010. PharmGKB summary: very important pharmacogene information for CYP2B6. Pharmacogenet. Genom. 20, 520–523 10.1097/FPC.0b013e32833947c2 (doi:10.1097/FPC.0b013e32833947c2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Van Booven D., Marsh S., McLeod H., Carrillo M. W., Sangkuhl K., Klein T. E., Altman R. B. 2010. Cytochrome P450 2C9-CYP2C9. Pharmacogenet. Genom. 20, 277–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Owen R. P., Sangkuhl K., Klein T. E., Altman R. B. 2009. Cytochrome P450 2D6. Pharmacogenet. Genom. 19, 559–562 10.1097/FPC.0b013e32832e0e97 (doi:10.1097/FPC.0b013e32832e0e97) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Berlin D. S., Sangkuhl K., Klein T. E., Altman R. B. 2011. PharmGKB summary: cytochrome P450, family 2, subfamily, J., polypeptide 2: CYP2J2. Pharmacogenet. Genom. 21, 308–311 10.1097/FPC.0b013e32833d1011 (doi:10.1097/FPC.0b013e32833d1011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schuetz E. G., et al. 2004. PharmGKB update. II. CYP3A5, cytochrome P450, family 3, subfamily, A., polypeptide 5. Pharmacol. Rev. 56, 159 10.1124/pr.56.2.1 (doi:10.1124/pr.56.2.1) [DOI] [PubMed] [Google Scholar]
- 151.Hendrychova T., Anzenbacherova E., Hudecek J., Skopalik J., Lange R., Hildebrandt P., Otyepka M., Anzenbacher P. 2011. Flexibility of human cytochrome P450 enzymes: molecular dynamics and spectroscopy reveal important function-related variations. Biochim. Biophys. Acta 1814, 58–68 [DOI] [PubMed] [Google Scholar]
- 152.Skopalik J., Anzenbacher P., Otyepka M. 2008. Flexibility of human cytochromes P450: molecular dynamics reveals differences between CYPs 3A4, 2C9, and 2A6, which correlate with their substrate preferences. J. Phys. Chem. B 112, 8165–8173 10.1021/jp800311c (doi:10.1021/jp800311c) [DOI] [PubMed] [Google Scholar]
- 153.Wilderman P. R., Gay S. C., Jang H. H., Zhang Q., Stout C. D., Halpert J. R. 2012. Investigation by site-directed mutagenesis of the role of cytochrome P450 2B4 non-active site residues in protein–ligand interactions based on crystal structures of the ligand-bound enzyme. FEBS J. 279, 1607–1620 10.1111/j.1742-4658.2011.08411.x (doi:10.1111/j.1742-4658.2011.08411.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhang T., Liu L. A., Lewis D. F., Wei D. Q. 2011. Long-range effects of a peripheral mutation on the enzymatic activity of cytochrome P450 1A2. J. Chem. Inf. Model 51, 1336–1346 10.1021/ci200112b (doi:10.1021/ci200112b) [DOI] [PubMed] [Google Scholar]
- 155.He S. M., Zhou Z. W., Li X. T., Zhou S. F. 2011. Clinical drugs undergoing polymorphic metabolism by human cytochrome P450 2C9 and the implication in drug development. Curr. Med. Chem. 18, 667–713 10.2174/092986711794480131 (doi:10.2174/092986711794480131) [DOI] [PubMed] [Google Scholar]
- 156.Takanashi K., Tainaka H., Kobayashi K., Yasumori T., Hosakawa M., Chiba K. 2000. CYP2C9 Ile359 and Leu359 variants: enzyme kinetic study with seven substrates. Pharmacogenetics 10, 95–104 10.1097/00008571-200003000-00001 (doi:10.1097/00008571-200003000-00001) [DOI] [PubMed] [Google Scholar]
- 157.Sano E., Li W., Yuki H., Liu X., Furihata T., Kobayashi K., Chiba K., Neya S., Hoshino T. 2010. Mechanism of the decrease in catalytic activity of human cytochrome P450 2C9 polymorphic variants investigated by computational analysis. J. Comput. Chem. 31, 2746–2758 10.1002/jcc.21568 (doi:10.1002/jcc.21568) [DOI] [PubMed] [Google Scholar]
- 158.Pavani A., Naushad S. M., Rupasree Y., Kumar T. R., Malempati A. R., Pinjala R. K., Mishra R. C., Kutala V. K. 2011. Optimization of warfarin dose by population-specific pharmacogenomic algorithm. Pharmacogenom. J. 10.1038/tpj.2011.4 (doi:10.1038/tpj.2011.4) [DOI] [PubMed] [Google Scholar]
- 159.Sagreiya H., Berube C., Wen A., Ramakrishnan R., Mir A., Hamilton A., Altman R. B. 2010. Extending and evaluating a warfarin dosing algorithm that includes CYP4F2 and pooled rare variants of CYP2C9. Pharmacogenet. Genom. 20, 407–413 10.1097/FPC.0b013e328338bac2 (doi:10.1097/FPC.0b013e328338bac2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wells P. S., Majeed H., Kassem S., Langlois N., Gin B., Clermont J., Taljaard M. 2010. A regression model to predict warfarin dose from clinical variables and polymorphisms in CYP2C9, CYP4F2, and VKORC1: derivation in a sample with predominantly a history of venous thromboembolism. Thromb. Res. 125, e259–e264 10.1016/j.thromres.2009.11.020 (doi:10.1016/j.thromres.2009.11.020) [DOI] [PubMed] [Google Scholar]
- 161.Tan G. M., Wu E., Lam Y. Y., Yan B. P. 2010. Role of warfarin pharmacogenetic testing in clinical practice. Pharmacogenomics 11, 439–448 10.2217/pgs.10.8 (doi:10.2217/pgs.10.8) [DOI] [PubMed] [Google Scholar]
- 162.Roper N., Storer B., Bona R., Fang M. 2010. Validation and comparison of pharmacogenetics-based warfarin dosing algorithms for application of pharmacogenetic testing. J. Mol. Diagn. 12, 283–291 10.2353/jmoldx.2010.090110 (doi:10.2353/jmoldx.2010.090110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Schwede T., et al. 2009. Outcome of a workshop on applications of protein models in biomedical research. Structure 17, 151–159 10.1016/j.str.2008.12.014 (doi:10.1016/j.str.2008.12.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tarn C., et al. 2005. Analysis of KIT mutations in sporadic and familial gastrointestinal stromal tumors: therapeutic implications through protein modeling. Clin. Cancer Res. 11, 3668–3677 10.1158/1078-0432.CCR-04-2515 (doi:10.1158/1078-0432.CCR-04-2515) [DOI] [PubMed] [Google Scholar]
- 165.Tang Y., Poustovoitov M. V., Zhao K., Garfinkel M., Canutescu A., Dunbrack R., Adams P. D., Marmorstein R. 2006. Structure of a human ASF1a-HIRA complex and insights into specificity of histone chaperone complex assembly. Nat. Struct. Mol. Biol. 13, 921–929 10.1038/nsmb1147 (doi:10.1038/nsmb1147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Witkowski W. A., Hardy J. A. 2009. L2’ loop is critical for caspase-7 active site formation. Protein Sci. 18, 1459–1468 10.1002/pro.151 (doi:10.1002/pro.151) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhang T., Chen M., Lu Y., Xing Q., Chen W. 2011. A novel mutation of the PTCH1 gene activates the Shh/Gli signaling pathway in a Chinese family with nevoid basal cell carcinoma syndrome. Biochem. Biophys. Res. Commun. 409, 166–170 10.1016/j.bbrc.2011.04.047 (doi:10.1016/j.bbrc.2011.04.047) [DOI] [PubMed] [Google Scholar]
- 168.Parthiban M., Shanmughavel P., Sowdhamini R. 2010. In silico point mutation and evolutionary trace analysis applied to nicotinic acetylcholine receptors in deciphering ligand-binding surfaces. J. Mol. Model 16, 1651–1670 10.1007/s00894-010-0670-3 (doi:10.1007/s00894-010-0670-3) [DOI] [PubMed] [Google Scholar]
- 169.Sabbah D. A., Vennerstrom J. L., Zhong H. 2010. Docking studies on isoform-specific inhibition of phosphoinositide-3-kinases. J. Chem. Inf. Model 50, 1887–1898 10.1021/ci1002679 (doi:10.1021/ci1002679) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang N., Zhong R. 2010. Structural basis for decreased affinity of Emodin binding to Val66-mutated human CK2 alpha as determined by molecular dynamics. J. Mol. Model 16, 771–780 10.1007/s00894-009-0582-2 (doi:10.1007/s00894-009-0582-2) [DOI] [PubMed] [Google Scholar]
- 171.Dirauf P., Meiselbach H., Sticht H. 2010. Effects of the V82A and I54V mutations on the dynamics and ligand binding properties of HIV-1 protease. J. Mol. Model 16, 1577–1583 10.1007/s00894-010-0677-9 (doi:10.1007/s00894-010-0677-9) [DOI] [PubMed] [Google Scholar]
- 172.Gobel U., Sander C., Schneider R., Valencia A. 1994. Correlated mutations and residue contacts in proteins. Proteins 18, 309–317 10.1002/prot.340180402 (doi:10.1002/prot.340180402) [DOI] [PubMed] [Google Scholar]
- 173.Michino M., Brooks C. L., 3rd 2009. Predicting structurally conserved contacts for homologous proteins using sequence conservation filters. Proteins 77, 448–453 10.1002/prot.22456 (doi:10.1002/prot.22456) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wu S., Szilagyi A., Zhang Y. 2011. Improving protein structure prediction using multiple sequence-based contact predictions. Structure 19, 1182–1191 10.1016/j.str.2011.05.004 (doi:10.1016/j.str.2011.05.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lichtarge O., Bourne H. R., Cohen F. E. 1996. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 257, 342–358 10.1006/jmbi.1996.0167 (doi:10.1006/jmbi.1996.0167) [DOI] [PubMed] [Google Scholar]
- 176.Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. 2010. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 10.1093/nar/gkq399 (doi:10.1093/nar/gkq399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Capra J. A., Laskowski R. A., Thornton J. M., Singh M., Funkhouser T. A. 2009. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput. Biol. 5, e1000585 10.1371/journal.pcbi.1000585 (doi:10.1371/journal.pcbi.1000585) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Vendruscolo M., Dobson C. M. 2005. Towards complete descriptions of the free-energy landscapes of proteins. Phil. Trans. R. Soc. A. 363, 433–450; discussion 50–52 10.1098/rsta.2004.1501 (doi:10.1098/rsta.2004.1501) [DOI] [PubMed] [Google Scholar]
- 179.Tress M. L., Valencia A. 2010. Predicted residue-residue contacts can help the scoring of 3D models. Proteins 78, 1980–1991 10.1002/prot.22714 (doi:10.1002/prot.22714) [DOI] [PubMed] [Google Scholar]
- 180.Zhang Y., Kolinski A., Skolnick J. 2003. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys, J. 85, 1145–1164 10.1016/S0006-3495(03)74551-2 (doi:10.1016/S0006-3495(03)74551-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lindorff-Larsen K., Best R. B., Depristo M. A., Dobson C. M., Vendruscolo M. 2005. Simultaneous determination of protein structure and dynamics. Nature 433, 128–132 10.1038/nature03199 (doi:10.1038/nature03199) [DOI] [PubMed] [Google Scholar]
- 182.Lange O. F., et al. 2008. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 320, 1471–1475 10.1126/science.1157092 (doi:10.1126/science.1157092) [DOI] [PubMed] [Google Scholar]
- 183.de Vries S. J., van Dijk M., Bonvin A. M. 2010. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 10.1038/nprot.2010.32 (doi:10.1038/nprot.2010.32) [DOI] [PubMed] [Google Scholar]
- 184.Bertini I., Fragai M., Giachetti A., Luchinat C., Maletta M., Parigi G., Yeo K. J. 2005. Combining in silico tools and NMR data to validate protein–ligand structural models: application to matrix metalloproteinases. J. Med. Chem. 48, 7544–7559 10.1021/jm050574k (doi:10.1021/jm050574k) [DOI] [PubMed] [Google Scholar]
- 185.Vajda S., Kozakov D. 2009. Convergence and combination of methods in protein–protein docking. Curr. Opin. Struct. Biol. 19, 164–170 10.1016/j.sbi.2009.02.008 (doi:10.1016/j.sbi.2009.02.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kaufmann K. W., Dawson E. S., Henry L. K., Field J. R., Blakely R. D., Meiler J. 2009. Structural determinants of species-selective substrate recognition in human and Drosophila serotonin transporters revealed through computational docking studies. Proteins 74, 630–642 10.1002/prot.22178 (doi:10.1002/prot.22178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.DeLano W. L. 2002. The PyMOL Molecular Graphics System, version 1.2r3pre. Schrodinger, LLC [Google Scholar]
- 188.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T.E. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 (doi:10.1002/jcc.20084) [DOI] [PubMed] [Google Scholar]
- 189.Humphrey W., Dalke A., Schulten K. 1996. VMD: visual molecular dynamics. J. Mol. Graph 14, 27–28, 33–38 [DOI] [PubMed] [Google Scholar]
- 190.Kryshtafovych A., Fidelis K. 2009. Protein structure prediction and model quality assessment. Drug Discov. Today 14, 386–393 10.1016/j.drudis.2008.11.010 (doi:10.1016/j.drudis.2008.11.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Pierri C. L., Parisi G., Porcelli V. 2010. Computational approaches for protein function prediction: a combined strategy from multiple sequence alignment to molecular docking-based virtual screening. Biochim. Biophys. Acta 1804, 1695–1712 [DOI] [PubMed] [Google Scholar]
- 192.Zhang Y. 2009. Protein structure prediction: when is it useful? Curr. Opin. Struct. Biol. 19, 145–155 10.1016/j.sbi.2009.02.005 (doi:10.1016/j.sbi.2009.02.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kopp J., Schwede T. 2004. Automated protein structure homology modeling: a progress report. Pharmacogenomics 5, 405–416 10.1517/14622416.5.4.405 (doi:10.1517/14622416.5.4.405) [DOI] [PubMed] [Google Scholar]
- 194.Chothia C., Lesk A. M. 1986. The relation between the divergence of sequence and structure in proteins. Embo J. 5, 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Daga P. R., Patel R. Y., Doerksen R. J. 2010. Template-based protein modeling: recent methodological advances. Curr. Top. Med. Chem. 10, 84–94 10.2174/156802610790232314 (doi:10.2174/156802610790232314) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Qu X., Swanson R., Day R., Tsai J. 2009. A guide to template based structure prediction. Curr. Protein Pept. Sci. 10, 270–285 10.2174/138920309788452182 (doi:10.2174/138920309788452182) [DOI] [PubMed] [Google Scholar]
- 197.Wallner B., Elofsson A. 2005. All are not equal: a benchmark of different homology modeling programs. Protein Sci. 14, 1315–1327 10.1110/ps.041253405 (doi:10.1110/ps.041253405) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Liu T., Tang G. W., Capriotti E. 2011. Comparative modeling: the state of the art and protein drug target structure prediction. Comb. Chem. High Throughput Screen 14, 532–547 10.2174/138620711795767811 (doi:10.2174/138620711795767811) [DOI] [PubMed] [Google Scholar]
- 199.Sali A., Blundell T. L. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 10.1006/jmbi.1993.1626 (doi:10.1006/jmbi.1993.1626) [DOI] [PubMed] [Google Scholar]
- 200.Levitt M. 1992. Accurate modeling of protein conformation by automatic segment matching. J. Mol. Biol. 226, 507–533 10.1016/0022-2836(92)90964-L (doi:10.1016/0022-2836(92)90964-L) [DOI] [PubMed] [Google Scholar]
- 201.Larsson P., Wallner B., Lindahl E., Elofsson A. 2008. Using multiple templates to improve quality of homology models in automated homology modeling. Protein Sci. 17, 990–1002 10.1110/ps.073344908 (doi:10.1110/ps.073344908) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Schwede T., Kopp J., Guex N., Peitsch M. C. 2003. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 10.1093/nar/gkg520 (doi:10.1093/nar/gkg520) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Petrey D., et al. 2003. Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins 53 (Suppl. 6), 430–435 10.1002/prot.10550 (doi:10.1002/prot.10550) [DOI] [PubMed] [Google Scholar]
- 204.Cavasotto C. N., Phatak S. S. 2009. Homology modeling in drug discovery: current trends and applications. Drug Discov. Today 14, 676–683 10.1016/j.drudis.2009.04.006 (doi:10.1016/j.drudis.2009.04.006) [DOI] [PubMed] [Google Scholar]
- 205.Capriotti E., Marti-Renom M. A. 2008. Assessment of protein structure predictions. In Computational structural biology: methods and applications (eds Schwede T., Peitsch M. C.), pp. 89–109 Singapore, Republic of Singapore: World Scientific Publishing Company [Google Scholar]
- 206.Mariani V., Kiefer F., Schmidt T., Haas J., Schwede T. 2011. Assessment of template based protein structure predictions in CASP9. Proteins 79 (Suppl. 10), 37–58 10.1002/prot.23177 (doi:10.1002/prot.23177) [DOI] [PubMed] [Google Scholar]
- 207.Xu D., Zhang Y. 2012. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins. 10.1002/prot.24065 (doi:10.1002/prot.24065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Xu D., Zhang J., Roy A., Zhang Y. 2011. Automated protein structure modeling in CASP9 by I-TASSER pipeline combined with QUARK-based ab initio folding and FG-MD-based structure refinement. Proteins 79, 147–160 10.1002/prot.23111 (doi:10.1002/prot.23111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kihara D., Chen H., Yang Y. D. 2009. Quality assessment of protein structure models. Curr. Protein Pept. Sci. 10, 216–228 10.2174/138920309788452173 (doi:10.2174/138920309788452173) [DOI] [PubMed] [Google Scholar]
- 210.Simons K. T., Kooperberg C., Huang E., Baker D. 1997. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 268, 209–225 10.1006/jmbi.1997.0959 (doi:10.1006/jmbi.1997.0959) [DOI] [PubMed] [Google Scholar]
- 211.Zhang Y., Skolnick J. 2004. Automated structure prediction of weakly homologous proteins on a genomic scale. Proc. Natl Acad. Sci. USA 101, 7594–7599 10.1073/pnas.0305695101 (doi:10.1073/pnas.0305695101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhang Y. 2008. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 18, 342–348 10.1016/j.sbi.2008.02.004 (doi:10.1016/j.sbi.2008.02.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bradley P., Misura K. M., Baker D. 2005. Toward high-resolution de novo structure prediction for small proteins. Science 309, 1868–1871 10.1126/science.1113801 (doi:10.1126/science.1113801) [DOI] [PubMed] [Google Scholar]
- 214.Kryshtafovych A., Fidelis K., Moult J. 2011. CASP9 results compared to those of previous CASP experiments. Proteins 79 (Suppl. 10), 196–207 10.1002/prot.23182 (doi:10.1002/prot.23182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kinch L., Yong Shi S., Cong Q., Cheng H., Liao Y., Grishin N. V. 2011. CASP9 assessment of free modeling target predictions. Proteins 79 (Suppl. 10), 59–73 10.1002/prot.23181 (doi:10.1002/prot.23181) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ghersi D., Sanchez R. 2011. Beyond structural genomics: computational approaches for the identification of ligand binding sites in protein structures. J. Struct. Funct. Genom. 12, 109–117 10.1007/s10969-011-9110-6 (doi:10.1007/s10969-011-9110-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Henrich S., Salo-Ahen O. M., Huang B., Rippmann F. F., Cruciani G., Wade R. C. 2010. Computational approaches to identifying and characterizing protein binding sites for ligand design. J. Mol. Recognit. 23, 209–219 [DOI] [PubMed] [Google Scholar]
- 218.Perot S., Sperandio O., Miteva M. A., Camproux A. C., Villoutreix B. O. 2010. Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery. Drug Discov. Today. 15, 656–667 10.1016/j.drudis.2010.05.015 (doi:10.1016/j.drudis.2010.05.015) [DOI] [PubMed] [Google Scholar]
- 219.Wass M. N., Kelley L. A., Sternberg M. J. 2010. 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res. 38, W469–W473 10.1093/nar/gkq406 (doi:10.1093/nar/gkq406) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Le Guilloux V., Schmidtke P., Tuffery P. 2009. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics 10, 168 10.1186/1471-2105-10-168 (doi:10.1186/1471-2105-10-168) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ghersi D., Sanchez R. 2009. EasyMIFS and SiteHound: a toolkit for the identification of ligand-binding sites in protein structures. Bioinformatics 25, 3185–3186 10.1093/bioinformatics/btp562 (doi:10.1093/bioinformatics/btp562) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hernandez M., Ghersi D., Sanchez R. 2009. SITEHOUND-web: a server for ligand binding site identification in protein structures. Nucleic Acids Res. 37, W413–W416 10.1093/nar/gkp281 (doi:10.1093/nar/gkp281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Brenke R., Kozakov D., Chuang G. Y., Beglov D., Hall D., Landon M. R., Mattos C., Vajda S. 2009. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics 25, 621–627 10.1093/bioinformatics/btp036 (doi:10.1093/bioinformatics/btp036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Fukunishi Y., Nakamura H. 2011. Prediction of ligand-binding sites of proteins by molecular docking calculation for a random ligand library. Protein Sci. 20, 95–106 10.1002/pro.540 (doi:10.1002/pro.540) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Mattos C., Ringe D. 1996. Locating and characterizing binding sites on proteins. Nat. Biotechnol. 14, 595–599 10.1038/nbt0596-595 (doi:10.1038/nbt0596-595) [DOI] [PubMed] [Google Scholar]
- 226.Fauman E. B., Rai B. K., Huang E. S. 2011. Structure-based druggability assessment-identifying suitable targets for small molecule therapeutics. Curr. Opin. Chem. Biol. 15, 463–468 10.1016/j.cbpa.2011.05.020 (doi:10.1016/j.cbpa.2011.05.020) [DOI] [PubMed] [Google Scholar]
- 227.Schmidtke P., Souaille C., Estienne F., Baurin N., Kroemer R. T. 2010. Large-scale comparison of four binding site detection algorithms. J. Chem. Inf. Model 50, 2191–2200 10.1021/ci1000289 (doi:10.1021/ci1000289) [DOI] [PubMed] [Google Scholar]
- 228.Powers R., Copeland J. C., Stark J. L., Caprez A., Guru A., Swanson D. 2011. Searching the protein structure database for ligand-binding site similarities using CPASS v.2. BMC Res. Notes 4, 17 10.1186/1756-0500-4-17 (doi:10.1186/1756-0500-4-17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Minai R., Matsuo Y., Onuki H., Hirota H. 2008. Method for comparing the structures of protein ligand-binding sites and application for predicting protein–drug interactions. Proteins. 72, 367–381 10.1002/prot.21933 (doi:10.1002/prot.21933) [DOI] [PubMed] [Google Scholar]
- 230.Shulman-Peleg A., Nussinov R., Wolfson H. J. 2005. SiteEngines: recognition and comparison of binding sites and protein–protein interfaces. Nucleic Acids Res. 33, W337–W341 10.1093/nar/gki482 (doi:10.1093/nar/gki482) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Schmitt S., Kuhn D., Klebe G. 2002. A new method to detect related function among proteins independent of sequence and fold homology. J. Mol. Biol. 323, 387–406 10.1016/S0022-2836(02)00811-2 (doi:10.1016/S0022-2836(02)00811-2) [DOI] [PubMed] [Google Scholar]
- 232.Kellenberger E., Schalon C., Rognan D. 2008. How to measure the similarity between protein ligand-binding sites? Curr. Comput. Aided Drug Des. 4 10.2174/157340908785747401 (doi:10.2174/157340908785747401) [DOI] [Google Scholar]
- 233.Liu T., Altman R. B. 2011. Using multiple microenvironments to find similar ligand-binding sites: application to kinase inhibitor binding. PLoS Comput. Biol. [Research Support, N.I.H., Extramural] 7, e1002326 10.1371/journal.pcbi.1002326 (doi:10.1371/journal.pcbi.1002326) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Corbeil C. R., Therrien E., Moitessier N. 2009. Modeling reality for optimal docking of small molecules to biological targets. Curr. Comput. Aided Drug Des. 5, 241–263 10.2174/157340909789577856 (doi:10.2174/157340909789577856) [DOI] [Google Scholar]
- 235.Huang S. Y., Grinter S. Z., Zou X. 2010. Scoring functions and their evaluation methods for protein–ligand docking: recent advances and future directions. Phys. Chem. Chem. Phys. 12, 12 899–12 908 10.1039/c0cp00151a (doi:10.1039/c0cp00151a) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Moitessier N., Englebienne P., Lee D., Lawandi J., Corbeil C. R. 2008. Towards the development of universal, fast and highly accurate docking/scoring methods: a long way to go. Br. J. Pharmacol. 153 (Suppl. 1), S7–S26 10.1038/sj.bjp.0707515 (doi:10.1038/sj.bjp.0707515) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Yuriev E., Agostino M., Ramsland P. A. 2011. Challenges and advances in computational docking: 2009 in review. J. Mol. Recognit. 24, 149–164 10.1002/jmr.1077 (doi:10.1002/jmr.1077) [DOI] [PubMed] [Google Scholar]
- 238.Plewczynski D., Lazniewski M., Augustyniak R., Ginalski K. 2011. Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database. J. Comput. Chem. 32, 742–755 10.1002/jcc.21643 (doi:10.1002/jcc.21643) [DOI] [PubMed] [Google Scholar]
- 239.Leach A. R., Shoichet B. K., Peishoff C. E. 2006. Prediction of protein–ligand interactions. Docking and scoring: successes and gaps. J. Med. Chem. 49, 5851–5855 10.1021/jm060999m (doi:10.1021/jm060999m) [DOI] [PubMed] [Google Scholar]
- 240.McGann M. R., Almond H. R., Nicholls A., Grant J. A., Brown F. K. 2003. Gaussian docking functions. Biopolymers 68, 76–90 10.1002/bip.10207 (doi:10.1002/bip.10207) [DOI] [PubMed] [Google Scholar]
- 241.Rarey M., Kramer B., Lengauer T., Klebe G. 1996. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 261, 470–489 10.1006/jmbi.1996.0477 (doi:10.1006/jmbi.1996.0477) [DOI] [PubMed] [Google Scholar]
- 242.Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J. 1998. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662 (doi:10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B) [DOI] [Google Scholar]
- 243.Friesner R. A., et al. 2004. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 10.1021/jm0306430 (doi:10.1021/jm0306430) [DOI] [PubMed] [Google Scholar]
- 244.B-Rao C., Subramanian J., Sharma S. D. 2009. Managing protein flexibility in docking and its applications. Drug Discov. Today 14, 394–400 10.1016/j.drudis.2009.01.003 (doi:10.1016/j.drudis.2009.01.003) [DOI] [PubMed] [Google Scholar]
- 245.Claussen H., Buning C., Rarey M., Lengauer T. 2001. FlexE: efficient molecular docking considering protein structure variations. J. Mol. Biol. 308, 377–395 10.1006/jmbi.2001.4551 (doi:10.1006/jmbi.2001.4551) [DOI] [PubMed] [Google Scholar]
- 246.Mizutani M. Y., Takamatsu Y., Ichinose T., Nakamura K., Itai A. 2006. Effective handling of induced-fit motion in flexible docking. Proteins 63, 878–891 10.1002/prot.20931 (doi:10.1002/prot.20931) [DOI] [PubMed] [Google Scholar]
- 247.Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. 2009. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 10.1002/jcc.21256 (doi:10.1002/jcc.21256) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Feher M., Williams C. I. 2009. Effect of input differences on the results of docking calculations. J. Chem. Inf. Model 49, 1704–1714 10.1021/ci9000629 (doi:10.1021/ci9000629) [DOI] [PubMed] [Google Scholar]
- 249.Corbeil C. R., Moitessier N. 2009. Docking ligands into flexible and solvated macromolecules. 3. Impact of input ligand conformation, protein flexibility, and water molecules on the accuracy of docking programs. J. Chem. Inf. Model 49, 997–1009 10.1021/ci8004176 (doi:10.1021/ci8004176) [DOI] [PubMed] [Google Scholar]
- 250.Kolb P., Ferreira R. S., Irwin J. J., Shoichet B. K. 2009. Docking and chemoinformatic screens for new ligands and targets. Curr. Opin. Biotechnol. 20, 429–436 10.1016/j.copbio.2009.08.003 (doi:10.1016/j.copbio.2009.08.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Ballester P. J., Mitchell J. B. 2010. A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking. Bioinformatics 26, 1169–1175 10.1093/bioinformatics/btq112 (doi:10.1093/bioinformatics/btq112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Deng W., Breneman C., Embrechts M. J. 2004. Predicting protein–ligand binding affinities using novel geometrical descriptors and machine-learning methods. J. Chem. Inf. Comput. Sci. 44, 699–703 10.1021/ci034246+ (doi:10.1021/ci034246+) [DOI] [PubMed] [Google Scholar]
- 253.Adcock S. A., McCammon J. A. 2006. Molecular dynamics: survey of methods for simulating the activity of proteins. Chem. Rev. 106, 1589–1615 10.1021/cr040426m (doi:10.1021/cr040426m) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lee E. H., Hsin J., Sotomayor M., Comellas G., Schulten K. 2009. Discovery through the computational microscope. Structure 17, 1295–1306 10.1016/j.str.2009.09.001 (doi:10.1016/j.str.2009.09.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Schlick T., Collepardo-Guevara R., Halvorsen L. A., Jung S., Xiao X. 2011. Biomolecularmodeling and simulation: a field coming of age. Q. Rev. Biophys. 44, 191–228 10.1017/S0033583510000284 (doi:10.1017/S0033583510000284) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Fan H., Irwin J. J., Webb B. M., Klebe G., Shoichet B. K., Sali A. 2009. Molecular docking screens using comparative models of proteins. J. Chem. Inf. Model 49, 2512–2527 10.1021/ci9003706 (doi:10.1021/ci9003706) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Ivetac A., McCammon J. A. 2010. Mapping the druggable allosteric space of G-protein coupled receptors: a fragment-based molecular dynamics approach. Chem. Biol. Drug Des. 76, 201–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Salsbury F. R., Jr. 2010. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr. Opin. Pharmacol. 10, 738–744 10.1016/j.coph.2010.09.016 (doi:10.1016/j.coph.2010.09.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Shan Y., Kim E. T., Eastwood M. P., Dror R. O., Seeliger M. A., Shaw D. E. 2011. How does a drug molecule find its target binding site? J. Am. Chem. Soc. 133, 9181–9183 10.1021/ja202726y (doi:10.1021/ja202726y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Rajasekaran R., Sethumadhavan R. 2010. Exploring the cause of drug resistance by the detrimental missense mutations in KIT receptor: computational approach. Amino Acids 39, 651–660 10.1007/s00726-010-0486-6 (doi:10.1007/s00726-010-0486-6) [DOI] [PubMed] [Google Scholar]
- 261.Shan Y., Seeliger M. A., Eastwood M. P., Frank F., Xu H., Jensen M. O., Dror R. O., Kuriyan J., Shaw D. E. 2009. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl Acad. Sci. USA 106, 139–144 10.1073/pnas.0811223106 (doi:10.1073/pnas.0811223106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Jorgensen W. L., Maxwell D. S., Tirado-Rives J. 1996. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11 225–11 236 10.1021/ja9621760 (doi:10.1021/ja9621760) [DOI] [Google Scholar]
- 263.MacKerell A. D. J., et al. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 10.1021/jp973084f (doi:10.1021/jp973084f) [DOI] [PubMed] [Google Scholar]
- 264.Cornell W. D., et al. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117, 5179–5197 10.1021/ja00124a002 (doi:10.1021/ja00124a002) [DOI] [Google Scholar]
- 265.Freddolino P. L., Park S., Roux B., Schulten K. 2009. Force field bias in protein folding simulations. Biophys. J. 96, 3772–3780 10.1016/j.bpj.2009.02.033 (doi:10.1016/j.bpj.2009.02.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J. 2005. GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 10.1002/jcc.20291 (doi:10.1002/jcc.20291) [DOI] [PubMed] [Google Scholar]
- 267.Pearlman D. A., Case D. A., Caldwell J. W., Ross W. S., Cheatham T. E., III, DeBolt S., Ferguson D., Seibel G., Kollman P. 1995. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91, 1–41 10.1016/0010-4655(95)00041-D (doi:10.1016/0010-4655(95)00041-D) [DOI] [Google Scholar]
- 268.Kalé L., et al. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151, 283–312 10.1006/jcph.1999.6201 (doi:10.1006/jcph.1999.6201) [DOI] [Google Scholar]
- 269.Friedrichs M. S., Eastman P., Vaidyanathan V., Houston M., Legrand S., Beberg A. L., Ensign D. L., Bruns C. M., Pande V. S. 2009. Accelerating molecular dynamic simulation on graphics processing units. J. Comput. Chem. 30, 864–872 10.1002/jcc.21209 (doi:10.1002/jcc.21209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Pande V. S., et al. 2003. Atomistic protein folding simulations on the submillisecond timescale using worldwide distributed computing. Biopolymers 68, 91–109 10.1002/bip.10219 (doi:10.1002/bip.10219) [DOI] [PubMed] [Google Scholar]
- 271.Shaw D. E., et al. 2010. Atomic-level characterization of the structural dynamics of proteins. Science 330, 341–346 10.1126/science.1187409 (doi:10.1126/science.1187409) [DOI] [PubMed] [Google Scholar]
- 272.Mobley D. L. 2012. Let's get honest about sampling. J. Comput. Aided Mol. Des. 26, 93–95 10.1007/s10822-011-9497-y (doi:10.1007/s10822-011-9497-y) [DOI] [PubMed] [Google Scholar]
- 273.Bray J. K., Weiss D. R., Levitt M. 2011. Optimized torsion-angle normal modes reproduce conformational changes more accurately than cartesian modes. Biophys. J. 101, 2966–2969 10.1016/j.bpj.2011.10.054 (doi:10.1016/j.bpj.2011.10.054) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Yang L., Song G., Jernigan R. L. 2007. How well can we understand large-scale protein motions using normal modes of elastic network models? Biophys. J. 93, 920–929 10.1529/biophysj.106.095927 (doi:10.1529/biophysj.106.095927) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Hinsen K. 1998. Analysis of domain motions by approximate normal mode calculations. Proteins 33, 417–429 (doi:10.1002/(SICI)1097-0134(19981115)33:3<417::AID-PROT10>3.0.CO;2-8) [DOI] [PubMed] [Google Scholar]
- 276.Suhre K., Sanejouand Y. H. 2004. ElNemo, a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 32, W610–W614 10.1093/nar/gkh368 (doi:10.1093/nar/gkh368) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Bahar I., Lezon T. R., Bakan A., Shrivastava I. H. 2010. Normal mode analysis of biomolecular structures, functional mechanisms of membrane proteins. Chem. Rev. 110, 1463–1497 10.1021/cr900095e (doi:10.1021/cr900095e) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Torrance J. W., Bartlett G. J., Porter C. T., Thornton J. M. 2005. Using a library of structural templates to recognise catalytic sites and explore their evolution in homologous families. J. Mol. Biol. 347, 565–581 10.1016/j.jmb.2005.01.044 (doi:10.1016/j.jmb.2005.01.044) [DOI] [PubMed] [Google Scholar]
- 279.Lopez G., Valencia A., Tress M. 2007. FireDB—a database of functionally important residues from proteins of known structure. Nucleic Acids Res. 35, D219–D223 10.1093/nar/gkl897 (doi:10.1093/nar/gkl897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Pieper U., et al. 2011. ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 39, D465–D474 10.1093/nar/gkq1091 (doi:10.1093/nar/gkq1091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Arnold K., Kiefer F., Kopp J., Battey J. N., Podvinec M., Westbrook J. D., Nerman H. M., Bordoli L., Schwede T. 2009. The protein model portal. J. Struct. Funct. Genom. 10, 1–8 10.1007/s10969-008-9048-5 (doi:10.1007/s10969-008-9048-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Kellenberger E., Muller P., Schalon C., Bret G., Foata N., Rognan D. 2006. sc-PDB: an annotated database of druggable binding sites from the Protein Data Bank. J. Chem. Inf. Model 46, 717–727 10.1021/ci050372x (doi:10.1021/ci050372x) [DOI] [PubMed] [Google Scholar]
- 283.Gold N. D., Jackson R. M. 2006. SitesBase: a database for structure-based protein–ligand binding site comparisons. Nucleic Acids Res. 34, D231–D234 10.1093/nar/gkj062 (doi:10.1093/nar/gkj062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Kiefer F., Arnold K., Kunzli M., Bordoli L., Schwede T. 2009. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 37, D387–D392 10.1093/nar/gkn750 (doi:10.1093/nar/gkn750) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Hu L., Benson M. L., Smith R. D., Lerner M. G., Carlson H. A. 2005. Binding MOAD (Mother Of All Databases). Proteins 60, 333–340 10.1002/prot.20512 (doi:10.1002/prot.20512) [DOI] [PubMed] [Google Scholar]
- 286.Wang R., Fang X., Lu Y., Wang S. 2004. The PDBbind database: collection of binding affinities for protein–ligand complexes with known three-dimensional structures. J. Med. Chem. 47, 2977–2980 10.1021/jm030580l (doi:10.1021/jm030580l) [DOI] [PubMed] [Google Scholar]
- 287.Hendlich M., Bergner A., Gunther J., Klebe G. 2003. Relibase: design and development of a database for comprehensive analysis of protein–ligand interactions. J. Mol. Biol. 326, 607–620 10.1016/S0022-2836(02)01408-0 (doi:10.1016/S0022-2836(02)01408-0) [DOI] [PubMed] [Google Scholar]
- 288.Gabanyi M. J. 2011. The structural biology knowledgebase: a portal to protein structures, sequences, functions, and methods. J. Struct. Funct. Genom. 12, 45–54 10.1007/s10969-011-9106-2 (doi:10.1007/s10969-011-9106-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Zhu F., et al. 2010. Update of TTD: therapeutic target database. Nucleic Acids Res. 38, D787–D791 10.1093/nar/gkp1014 (doi:10.1093/nar/gkp1014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Sherry S. T., Ward M. H., Kholodov M., Baker J., Phan L., Smigielski E. M., Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 10.1093/nar/29.1.308 (doi:10.1093/nar/29.1.308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Hindorff L. A., Sethupathy P., Junkins H. A., Ramos E. M., Mehta J. P., Collins F. S., Manolio T. A. 2009. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA 106, 9362–9367 10.1073/pnas.0903103106 (doi:10.1073/pnas.0903103106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Amberger J., Bocchini C., Hamosh A. 2011. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM(R)). Hum. Mutat. 32, 564–567 10.1002/humu.21466 (doi:10.1002/humu.21466) [DOI] [PubMed] [Google Scholar]
- 293.Gamazon E. R., Zhang W., Konkashbaev A., Duan S., Kistner E. O., Nicolae D. L., Dolan M. E., Cox N. J. 2010. SCAN: SNP and copy number annotation. Bioinformatics 26, 259–262 10.1093/bioinformatics/btp644 (doi:10.1093/bioinformatics/btp644) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Mottaz A., David F. P., Veuthey A. L., Yip Y. L. 2010. Easy retrieval of single amino-acid polymorphisms and phenotype information using SwissVar. Bioinformatics 26, 851–852 10.1093/bioinformatics/btq028 (doi:10.1093/bioinformatics/btq028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Ryan M., Diekhans M., Lien S., Liu Y., Karchin R. 2009. LS-SNP/PDB: annotated non-synonymous SNPs mapped to Protein Data Bank structures. Bioinformatics 25, 1431–1432 10.1093/bioinformatics/btp242 (doi:10.1093/bioinformatics/btp242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Yang J. O., Oh S., Ko G., Park S. J., Kim W. Y., Lee B., Lee S. 2011. VnD: a structure-centric database of disease-related SNPs and drugs. Nucleic Acids Res. 39, D939–D944 10.1093/nar/gkq957 (doi:10.1093/nar/gkq957) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Preissner S., et al. 2010. SuperCYP: a comprehensive database on cytochrome P450 enzymes including a tool for analysis of CYP-drug interactions. Nucleic Acids Res. 38, D237–D243 10.1093/nar/gkp970 (doi:10.1093/nar/gkp970) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Thorn C. F., Klein T. E., Altman R. B. 2010. Pharmacogenomics and bioinformatics: PharmGKB. Pharmacogenomics 11, 501–505 10.2217/pgs.10.15 (doi:10.2217/pgs.10.15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Meslamani J., Rognan D., Kellenberger E. 2011. sc-PDB: a database for identifying variations and multiplicity of ‘druggable’ binding sites in proteins. Bioinformatics 27, 1324–1326 10.1093/bioinformatics/btr120 (doi:10.1093/bioinformatics/btr120) [DOI] [PubMed] [Google Scholar]
- 300.Aller S. G., et al. 2009. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 10.1126/science.1168750 (doi:10.1126/science.1168750) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Warne T., Moukhametzianov R., Baker J. G., Nehme R., Edwards P. C., Leslie A. G., Schertler G. F., Tate C. G. 2011. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature 469, 241–244 10.1038/nature09746 (doi:10.1038/nature09746) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Rasmussen S. G., et al. 2007. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 450, 383–387 10.1038/nature06325 (doi:10.1038/nature06325) [DOI] [PubMed] [Google Scholar]
- 303.Rutherford K., Le Trong I., Stenkamp R. E., Parson W. W. 2008. Crystal structures of human 108V and 108M catechol O-methyltransferase. J. Mol. Biol. 380, 120–130 10.1016/j.jmb.2008.04.040 (doi:10.1016/j.jmb.2008.04.040) [DOI] [PubMed] [Google Scholar]
- 304.Yano J. K., Hsu M. H., Griffin K. J., Stout C. D., Johnson E. F. 2005. Structures of human microsomal cytochrome P450 2A6 complexed with coumarin and methoxsalen. Nat. Struct. Mol. Biol. 12, 822–823 10.1038/nsmb971 (doi:10.1038/nsmb971) [DOI] [PubMed] [Google Scholar]
- 305.Gay S. C., et al. 2010. Crystal structure of a cytochrome P450 2B6 genetic variant in complex with the inhibitor 4-(4-chlorophenyl)imidazole at 2.0-A resolution. Mol. Pharmacol. 77, 529–538 10.1124/mol.109.062570 (doi:10.1124/mol.109.062570) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Rowland P., et al. 2006. Crystal structure of human cytochrome P450 2D6. J. Biol. Chem. 281, 7614–7622 10.1074/jbc.M511232200 (doi:10.1074/jbc.M511232200) [DOI] [PubMed] [Google Scholar]
- 307.Dobritzsch D., Ricagno S., Schneider G., Schnackerz K. D., Lindqvist Y. 2002. Crystal structure of the productive ternary complex of dihydropyrimidine dehydrogenase with NADPH and 5-iodouracil. Implications for mechanism of inhibition and electron transfer. J. Biol. Chem. 277, 13 155–13 166 [DOI] [PubMed] [Google Scholar]
- 308.Tellez-Sanz R., et al. 2006. Calorimetric and structural studies of the nitric oxide carrier S-nitrosoglutathione bound to human glutathione transferase P1-1. Protein Sci. 15, 1093–1105 10.1110/ps.052055206 (doi:10.1110/ps.052055206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Wu H., et al. 2007. Structural basis of allele variation of human thiopurine-S-methyltransferase. Proteins 67, 198–208 10.1002/prot.21272 (doi:10.1002/prot.21272) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Faratian D., Bown J. L., Smith V. A., Langdon S. P., Harrison D. J. 2010. Cancer systems biology. Meth. Mol. Biol. 662, 245–263 10.1007/978-1-60761-800-3_12 (doi:10.1007/978-1-60761-800-3_12) [DOI] [PubMed] [Google Scholar]
- 311.Prasasya R. D., Vang K. Z., Kreeger P. K. 2011. A multivariate model of ErbB network composition predicts ovarian cancer cell response to canertinib. Biotechnol. Bioeng. 109, 213–224 10.1002/bit.23297 (doi:10.1002/bit.23297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Yang R., Niepel M., Mitchison T. K., Sorger P. K. 2010. Dissecting variability in responses to cancer chemotherapy through systems pharmacology. Clin. Pharmacol. Ther. 88, 34–38 10.1038/clpt.2010.96 (doi:10.1038/clpt.2010.96) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Barabasi A. L., Gulbahce N., Loscalzo J. 2011. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 12, 56–68 10.1038/nrg2918 (doi:10.1038/nrg2918) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.RCSB PDB Statistics [database on the Internet] 2011. RCSB Protein Data Bank. [cited August 6, 2011]. Available from: http://www.pdb.org/pdb/static.do?p=general_information/pdb_statistics/index.html
- 315.Wang W., et al. 2006. Structural characterization of autoinhibited c-Met kinase produced by coexpression in bacteria with phosphatase. Proc. Natl Acad. Sci. USA 103, 3563–3568 10.1073/pnas.0600048103 (doi:10.1073/pnas.0600048103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Standfuss J., Edwards P. C., D'Antona A., Fransen M., Xie G., Oprian D. D., Schertler G. F. X. 2011. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 471, 656–660 10.1038/nature09795 (doi:10.1038/nature09795) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Nielsen R., Paul J. S., Albrechtsen A., Song Y. S. 2011. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 12, 443–451 10.1038/nrg2986 (doi:10.1038/nrg2986) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Zhang C. C., Kast J. 2010. Applications of current proteomics techniques in modern drug design. Curr. Comput. Aided Drug Des. 6, 147–164 10.2174/157340910791760064 (doi:10.2174/157340910791760064) [DOI] [PubMed] [Google Scholar]
- 319.Mandal S., Moudgil M., Mandal S. K. 2009. Rational drug design. Eur. J. Pharmacol. 625, 90–100 10.1016/j.ejphar.2009.06.065 (doi:10.1016/j.ejphar.2009.06.065) [DOI] [PubMed] [Google Scholar]
- 320.Talele T. T., Khedkar S. A., Rigby A. C. 2010. Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Curr. Top. Med. Chem. 10, 127–241 10.2174/156802610790232251 (doi:10.2174/156802610790232251) [DOI] [PubMed] [Google Scholar]
- 321.Villoutreix B. O., Eudes R., Miteva M. A. 2009. Structure-based virtual ligand screening: recent success stories. Comb. Chem. High Throughput Screen 12, 1000–1016 10.2174/138620709789824682 (doi:10.2174/138620709789824682) [DOI] [PubMed] [Google Scholar]
- 322.Andricopulo A. D., Salum L. B., Abraham D. J. 2009. Structure-based drug design strategies in medicinal chemistry. Curr. Top. Med. Chem. 9, 771–790 10.2174/156802609789207127 (doi:10.2174/156802609789207127) [DOI] [PubMed] [Google Scholar]
- 323.Kalyaanamoorthy S., Chen Y. P. 2011. Structure-based drug design to augment hit discovery. Drug Discov. Today 16, 831–839 10.1016/j.drudis.2011.07.006 (doi:10.1016/j.drudis.2011.07.006) [DOI] [PubMed] [Google Scholar]
- 324.Hubbard R. E. 2011. Structure-based drug discovery and protein targets in the CNS. Neuropharmacology 60, 7–23 10.1016/j.neuropharm.2010.07.016 (doi:10.1016/j.neuropharm.2010.07.016) [DOI] [PubMed] [Google Scholar]
- 325.Cozier G. E., Leese M. P., Lloyd M. D., Baker M. D., Thiyagarajan N., Acharya K. R., Potter K. V. 2010. Structures of human carbonic anhydrase II/inhibitor complexes reveal a second binding site for steroidal and nonsteroidal inhibitors. Biochemistry 49, 3464–3476 10.1021/bi902178w (doi:10.1021/bi902178w) [DOI] [PubMed] [Google Scholar]
- 326.Blackburn E. A., Walkinshaw M. D. 2011. Targeting FKBP isoforms with small-molecule ligands. Curr. Opin. Pharmacol. 11, 365–371 10.1016/j.coph.2011.04.007 (doi:10.1016/j.coph.2011.04.007) [DOI] [PubMed] [Google Scholar]
- 327.Crespan E., Zucca E., Maga G. 2011. Overcoming the drug resistance problem with second-generation tyrosine kinase inhibitors: from enzymology to structural models. Curr. Med. Chem. 18, 2836–2847 10.2174/092986711796150513 (doi:10.2174/092986711796150513) [DOI] [PubMed] [Google Scholar]
- 328.Moriaud F., Richard S. B., Adcock S. A., Chanas-Martin L., Surgand J. S., Ben Jelloul M., Delfaud F. 2011. Identify drug repurposing candidates by mining the protein data bank. Brief Bioinform. 12, 336–340 10.1093/bib/bbr017 (doi:10.1093/bib/bbr017) [DOI] [PubMed] [Google Scholar]
- 329.Ekins S., Williams A. J., Krasowski M. D., Freundlich J. S. 2011. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov. Today 16, 298–310 10.1016/j.drudis.2011.02.016 (doi:10.1016/j.drudis.2011.02.016) [DOI] [PubMed] [Google Scholar]
- 330.Haupt V. J., Schroeder M. 2011. Old friends in new guise: repositioning of known drugs with structural bioinformatics. Brief Bioinform. 12, 312–326 10.1093/bib/bbr011 (doi:10.1093/bib/bbr011) [DOI] [PubMed] [Google Scholar]
- 331.Fitzgerald J. B., Schoeberl B., Nielsen U. B., Sorger P. K. 2006. Systems biology and combination therapy in the quest for clinical efficacy. Nat. Chem. Biol. 2, 458–466 10.1038/nchembio817 (doi:10.1038/nchembio817) [DOI] [PubMed] [Google Scholar]
- 332.Zhang J., et al. 2010. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 463, 501–506 10.1038/nature08675 (doi:10.1038/nature08675) [DOI] [PMC free article] [PubMed] [Google Scholar]