

Abstract
T cell triggering through T-cell antigen receptors (TCRs) results in spatial assembly of the receptors on multiple length scales. This assembly is mediated by the T cell actin cytoskeleton, which reorganizes in response to TCR phosphorylation and then induces the coalescence of TCRs into microclusters, followed by their unification into a micrometer-scale structure. The exact outcomes of the association of TCRs with a dynamic and fluctuating actin network across these length scales are not well characterized, but it is clear that weak and transient interactions at the single-molecule level sum to yield significant receptor rearrangements at the plasma membrane. We used the hybrid live cell–nanopatterned supported lipid bilayer system to quantitatively probe the actin–TCR interaction in primary T cells. A specialized tracking algorithm revealed that actin slows as it passes over TCR clusters in a direction-dependent manner with respect to the resistance against TCR motion. We also observed transient actin enrichments at sites corresponding to putative TCR clusters that far exceeded pure stochastic fluctuations and described an image time-autocorrelation analysis method to quantify these accumulations.
Key words: Actin cytoskeleton, T cell receptor cluster, Supported lipid bilayer, Spatial patterning
Introduction
T cells are activated when their T-cell antigen receptors (TCRs) are triggered by interactions with antigen peptide–major histocompatibility complex (pMHC) proteins on antigen-presenting cell (APC) surfaces. T cells exhibit exquisite selectivity and sensitivity, and the physical basis for these attributes within the TCR signaling system has attracted much interest. A significant aspect of TCR triggering is the spatial assembly of receptors on multiple length scales. At the scale of the cell itself, TCRs are visibly transported towards the center of the T-cell–APC interface, and the resulting spatial patterns of TCRs and other molecules are called the immunological synapse (Monks et al., 1998; Grakoui et al., 1999). Physically interfering with these transport processes can induce detectable changes in T cell signaling, such as phosphorylation of immunoreceptor tyrosine-based activation motifs on the TCR complex and intracellular calcium flux (Mossman et al., 2005). On shorter time and length scales, TCRs form microclusters upon engagement with pMHC (Lee et al., 2002; Yokosuka et al., 2005; Dustin et al., 2010). Much of the relevant signaling activity takes place within these TCR microclusters, which function as integrated signaling machines.
Underlying the spatial assembly and transport of TCRs during T cell activation are interactions between the receptors and the actin cytoskeleton (Barda-Saad et al., 2005; DeMond et al., 2008; Gomez and Billadeau, 2008). In response to TCR triggering, the cytoskeleton transitions from a crawling morphology to a pattern of centripetal flow (Ryser et al., 1982; Bunnell et al., 2001; Kaizuka et al., 2007; Burkhardt et al., 2008). Simultaneously, TCR microclusters coalesce and translocate towards the center of the immunological synapse. Both coalescence and translocation are actin-dependent, as disruption of the cytoskeleton with latrunculin prevents both processes (Campi et al., 2005; Varma et al., 2006). Additionally, recent work has shown that actin can affect local TCR–pMHC binding kinetics (Huppa et al., 2010; Huang et al., 2010), illustrating another mechanism for involvement of the cytoskeleton in signaling.
TCR microclusters need not move at the same speed as the retrograde flowing actin (Kaizuka et al., 2007; Yu et al., 2010), nor do they need to move in the same direction. When translocating microclusters encounter physical obstacles imposed using patterned supported membrane substrates, the microclusters are deflected and move at angles to the actin flow with speeds that exhibit a cosine scaling law, which is consistent with dissipative, or frictional, actin-coupling mechanisms (DeMond et al., 2008). In other words, numerous weak interactions sum to yield a net force on the TCR microcluster, but there appears to be no stable or elastic association with the actin network. Such frictional coupling mechanisms appear to extend beyond TCR in the immunological synapse. Differentially clustering the integrin lymphocyte function-associated antigen 1 (LFA-1) using antibodies has been shown to redirect this protein to different positions within the synapse, with the most highly clustered LFA-1 ending up in the center along with TCRs (Hartman et al., 2009).
Although a significant role for actin in the immunological synapse is clear, many physical aspects of actin–TCR interactions are still relegated to the imagination. Preliminary observations of actin engaged in such interactions have been made in Jurkat cells (Kaizuka et al., 2007; Yu et al., 2010), but although these are a tractable T-cell-based model system, they have important differences from primary T cells (Tanimura et al., 2003; Bartelt et al., 2009; Dobson-Belaire et al., 2011). Because their TCR agonist is not known, Jurkat cells must be triggered through artificial antibody-induced crosslinking of their CD3 receptors, which inherently supplants endogenous TCR–TCR associations and has the potential to overstabilize the microcluster. This might add to already-documented differences in membrane protein organization between Jurkat cells and primary T cells (Tanimura et al., 2003). Furthermore, a number of Jurkat cell proteins exhibit levels of phosphorylation unlike those of their primary T cell counterparts, including known actin regulatory molecules such as Pyk2 and Vav1 (Bartelt et al., 2009). These differences raise questions about findings from studies based on Jurkat cell studies and necessitate the ultimate characterization of actin cytoskeleton behavior in primary cells.
In this study, we quantitatively characterized cortical actin motion within antigen-triggered primary T cells to obtain a deeper level of understanding of the elaborate interactions between the cytoskeleton and the plasma membrane during T-cell activation. We employed the hybrid live cell–nanopatterned supported lipid bilayer system, which allows both for assembly of TCR microclusters through interactions with laterally mobile, membrane-tethered pMHC and control of the lateral mobility of these microclusters with nanopatterned barriers in the substrate (Groves et al., 1997; Mossman et al., 2005; Manz and Groves, 2010). Analysis was performed using vector-field identification and tracking algorithms to characterize actin lateral movement and image time-autocorrelation to extract dynamic fluctuations. We observed that the actin cytoskeleton responds to laterally confined TCR clusters by slowing down in their vicinity, a result consistent with a previous report utilizing Jurkat T cells (Yu et al., 2010), and that the slowing is direction-dependent with respect to the resistance against TCR motion. Image time-autocorrelation revealed that the entire actin assembly forms and disassembles en masse. From this observation, we postulate that the primary stability of TCR microclusters originates from the receptors themselves, or other molecules in the microcluster, and that this stability templates assembly of the more dynamic actin, ultimately leading to a linkage to the bulk cytoskeleton.
Results
Immunological synapse formation on supported lipid bilayers and nanopatterned substrates
We visualized the cortical actin cytoskeleton in primary murine T cells during immunological synapse formation by allowing primed T cells to interact with a supported lipid bilayer functionalized with pMHC and intercellular adhesion molecule 1 (ICAM-1) proteins (Fig. 1A). This well-established system simulates the APC surface and effectively triggers T cells while allowing the use of total internal reflection fluorescence (TIRF) microscopy to image events within hundreds of nanometers of the cell–bilayer interface (Mossman and Groves, 2007; Nye and Groves, 2008; Dustin, 2009). An actin-binding probe composed of enhanced green fluorescent protein (EGFP) bound to the calponin homology domain of utrophin (EGFP–UtrCH) enabled observation of F-actin dynamics (Burkel et al., 2007), and key observations made with EGFP–UtrCH were validated using EGFP–β-actin.
Fig. 1.

The supported lipid bilayer allows visualization of the T cell cortical actin cytoskeleton during immunological synapse formation. (A) Schematic outline of the experiment. T cells interact with a supported lipid bilayer functionalized with APC proteins. Centripetal flow of the actin cytoskeleton causes TCR microclusters to coalesce and translocate until they reach a chromium barrier (Cr), where they accumulate. The ICAM-1 APC protein (not shown) is also included and interacts with T cell LFA-1. Ni2+-DOGS lipids (purple circles) and the TIRF illumination beam (cyan) are not labeled. (B–G) TIRF microscopy images of TCRs, the actin cytoskeleton and ICAM-1, as well as bright field and reflection interference contrast microscopy (RICM) images of a cell on a grid-patterned substrate show a typical frustrated immunological synapse. Scale bar: 5 μm.
Experiments in which the spatial configuration of the immunological synapse is physically altered with nanopatterned substrates have proven informative in a variety of studies (Mossman and Groves, 2007; Manz and Groves, 2010). Substrates patterned with grids of continuous metal lines (typically ~100 nm line width, ~10 nm height and 2-5 μm spacing) block diffusive transport of lipids, bilayer-associated protein ligands and their cognate receptors on the plasma membrane between isolated corrals, but they do not impose any effect on lateral diffusion within each corral (Groves et al., 1997; Groves and Boxer, 2002; Mossman et al., 2005; Salaita et al., 2010; Manz and Groves, 2010). Thus, when T cells interact with a protein-functionalized supported lipid bilayer on a nanopatterned substrate, TCR–pMHC microclusters and LFA-1–ICAM-1 assemblies form properly but cannot move outside the corral boundaries. Discontinuous barriers (crosses) that permit lateral diffusion of lipids and bilayer proteins serve as a control for nonspecific interactions of the cell with the metal barrier material. This system reliably reproduces the classic Kupferian synapse in T cells (Fig. 1B–G).
Analysis of cortical actin flow in primary T cells
One effective way of imaging the movement of the actin cytoskeleton is fluorescence speckle microscopy (FSM) (Waterman-Storer et al., 1998). In this methodology, fluorescent probes are incorporated sparsely into the actin cytoskeleton, and intrinsic density fluctuations in their random distribution provide features, or ‘speckles’, whose movement can be tracked. FSM generally does not involve actual imaging of individual fluorophores, though this is possible with modern methods. Stochastic density fluctuations are readily discernable for up to ten fluorophores per diffraction spot. As such, coordinated translational movement of feature patterns mirrors bulk cytoskeletal flow, whereas time evolution of the translating pattern reflects dynamic redistribution of the monomers.
In the experiments described here, we performed FSM using relatively high fluorescent probe densities. This enables better mapping of actual actin density fluctuations (as opposed to stochastic labeling fluctuations) but also requires more sophisticated image analysis. At very low probe densities, speckle features tend to have simpler shapes than they do at higher density. Thus, simple particle tracking algorithms are easily foiled by the complex patterns seen in highly labeled cells. To improve tracking accuracy in these systems, we developed a set of algorithms (see Materials and Methods) that identify features in images based on shape-independent gradient tracking, link them across multiple frames and evaluate their motion. The vector-field algorithm that we employed is a modification of those commonly used in medical imaging applications (Xu and Prince, 1998), which isolate endpoints in the fluorescence intensity gradient of an image as shown in Fig. 2A–C. This method overcomes the tendency of raster-based algorithms to identify multiple false centers in features based on their multiple local fluorescence intensity maxima. Features are then tracked across consecutive frames using a modified nearest-neighbor approach (Fig. 2B,C).
Fig. 2.
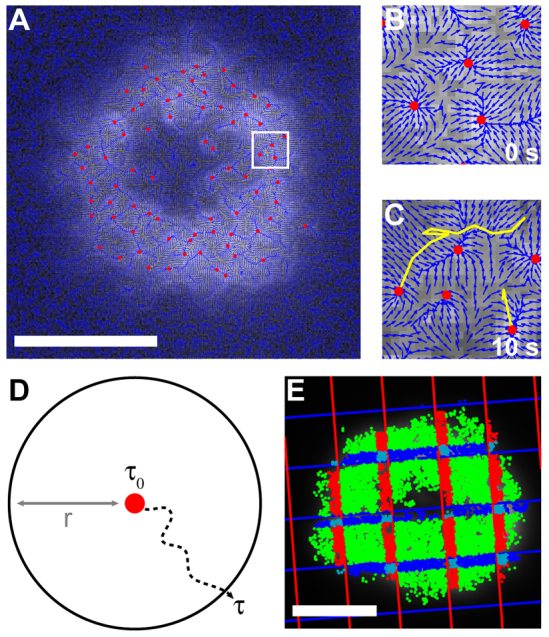
Analysis methods for bulk cytoskeletal flow are distinct from those used to describe speckle motion. (A) A vector-field algorithm identifies the centers of actin features across a whole cell. (B,C) The white square in A is enlarged to clearly show individual vectors in the vector field, and actin features are tracked across multiple frames. New features without tracks are also visible in C. (D) Schematic of the escape time analysis. The time it takes for a feature to travel further than a given escape distance (r) in any direction is its escape time (τ). (E) Escape times are distributed into populations based on each feature's instantaneous position. For example, features within 0.63 μm (10 pixels) of a single pattern boundary and on the opposite side from the cell center are colored dark blue or red, those within 0.63 μm of two boundaries are colored light blue and all other features are colored green. Scale bars: 5 μm.
The most common parameter used for evaluating moving objects is velocity. Although this parameter has been used effectively to describe speckle motion, we found that imaging of the cytoskeleton through an opaque chromium pattern displaced the identified centers of actin features near pattern boundaries and impacted both their apparent speed and direction of travel. We therefore evaluated actin motion in terms of the time it took a feature to travel a certain distance, here referred to as the ‘escape time’ (τ) and the ‘escape distance’ (r) (Fig. 2D). This analysis method, which is demonstrated in supplementary material Figs S1–S5 and explained in detail in the Materials and Methods section, is effectively a determination of mean square displacement, except that the output is in the form of time per (unit distance)2 instead of the inverse. The escape time analysis is not biased by the direction of motion and demonstrably avoids the optical artifacts mentioned above. To test the effects of imaging artifacts, image stacks from cells on unpatterned substrates were overlaid with grid shadow images prior to analysis to simulate the appearance of cells on actual patterned substrates. The escape times in these cells showed no dependence on proximity to pattern boundaries (supplementary material Fig. S6). Escape times were also similar between cells expressing EGFP–UtrCH and EGFP–β-actin (supplementary material Fig. S7).
The primary purpose of the grid experiments was to pin the position of TCR microclusters so that changes in actin movement and fluctuations at these microclusters could be analyzed. Such analysis required binning the escape times of features based on spatial distribution (e.g. Fig. 2E). Escape times were also normalized within each cell to facilitate comparison between different cells in a population, even in the presence of cell-to-cell variability. This normalization allowed statistically robust quantitative data sets to be assembled.
Resistance-dependent slowing of actin at sites of TCR clusters
In the following studies, T cells were allowed to form immunological synapses on substrates nanopatterned with metal grids. Grid barriers effectively trap TCR microclusters, pinning them in place for extended periods of time without otherwise interfering with their assembly. In cells that formed synapses on the edge of a patterned region, large bands of clustered TCR were visible at the pattern boundary. In these partially patterned cells, it was possible to compare escape times of features in regions with and without clustered TCRs that were approximately equidistant from the cell center (Fig. 3A). As anticipated, escape times in a region corresponding to clustered TCRs were higher than those in a region lacking TCRs (Fig. 3B). This can be interpreted as the result of slowing down or of concentration of actin in the immediate vicinity of the TCR cluster.
Fig. 3.

Actin features slow down in TCR-rich regions. (A,B) Two regions of a cell partially on a grid-patterned supported lipid bilayer equidistant from the cell center were selected (yellow and purple rectangles in A) for analysis of actin escape times (B). (C–E) Representative frames from time lapses show cells interacting with a grid-patterned substrate (C), a cross-patterned substrate (D) or an unpatterned substrate overlaid with the grid used for analysis (E). (F–H) Actin features from the cells in C–E were partitioned into four populations and analyzed for escape times. The inset shows that the green population is composed of all steps occurring further than 0.63 μm (10 pixels) from the outside edge of a grid line, the dark blue and red populations are composed of steps occurring within 0.63 μm of an x or y grid line, respectively, and the light blue population is composed of steps occurring within 0.63 μm of both grid lines. Error bars represent s.e.m. Scale bars: 5 μm.
TCR visualization in cells fully on grid patterns is hindered by the overall small size of the structures. Nonetheless, based on the above observations as well as past reports, it is reasonable to assume that TCRs will be generally concentrated along the grid boundaries. With this information, the grids themselves become fiduciary markers of where TCR microclusters are enriched, and statistical analysis of the data can be performed without the need for direct TCR imaging. For this analysis, all observable features in several cells were tracked, and measured escape times from each position in the cell were binned according to their proximity to zero, one vertical, one horizontal or two grid lines (Fig. 3C–H). The results indicate that escape times increased near one grid line and greatly increased near two lines (corresponding to a grid corner) (Fig. 3C,F). Because actin within the T cell can only interact with the metal substrate barriers through TCR–pMHC or possibly also LFA–ICAM interactions (DeMond et al., 2008; Yu et al., 2010), the slowing of actin movement near grids reflects interactions between actin and these proteins on the cytoplasmic side of the T cell membrane. The escape times of all four populations remained essentially constant when cells interacted with control substrates of chromium crosses that did not block long-range TCR transport (Fig. 3D,G) and when cells were not on grids (Fig. 3E,H).
To determine whether actin features are slowed by the mere presence of TCRs or by resistance against their motion from barriers, the directional components of escape times parallel or perpendicular to grid lines were evaluated. These components referred to the amount of time it took a feature to traverse a total vector distance equivalent to the original escape distance in the x or y direction, as defined by the layout of the grid (Fig. 4; supplementary material Figs S4, S5). The components of the escape time perpendicular to a barrier increased in the populations of features near that barrier, whereas components of the escape time parallel to the barrier were essentially unaffected (Fig. 4A–C). Also unaffected by this analysis method were all components in cells interacting with diffusion-permissive substrates patterned with chromium crosses and all components in cells that were not on grids (Fig. 4D–I). These data suggest that active resistance to TCR cluster motion as opposed to the mere presence of TCRs is necessary to induce a slowing of the cortical actin cytoskeleton and it is unlikely that the effect is an artifact of nonspecific cell interactions with chromium.
Fig. 4.
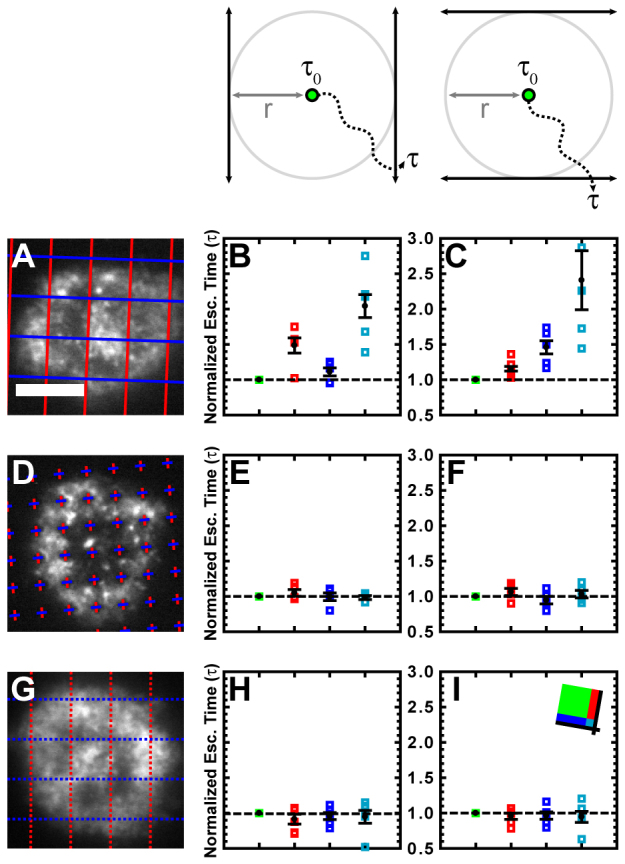
Increased actin escape times correspond to motion perpendicular to TCR diffusion barriers. (A,D,G) Representative frames from time lapses show cells interacting with a grid-patterned substrate (A), a cross-patterned substrate (D) or an unpatterned substrate overlaid with the grid used for analysis (G). (B,E,H) The escape time analysis from Fig. 3 was repeated for the cells in A,D,G, but the escape time was redefined as the time it takes for a feature to move outside of an area identified by boundaries tangential to the original escape area and parallel to the y grid line (schematic above column). This in effect considers only the x component of motion of the feature. (C,F,I) The escape time analysis from Fig. 3 was repeated for the cells in A,D,G, but the escape time was redefined as the time it takes for a heterogeneity to move outside of an area identified by boundaries tangential to the original escape area and parallel to the x grid line (schematic above column). This in effect considers only the y component of motion of the feature. Error bars represent s.e.m. Scale bars: 5 μm.
Coordinated actin fluctuations near TCRs
In time-lapse recordings of EGFP–β-actin, actin accumulations frequently appeared adjacent to substrate barriers (supplementary material Movie 1). These accumulations transiently increased in intensity before dissipating to the background level of fluorescence without translocating past the barrier towards the center of the immunological synapse. Such large-scale fluctuations in total actin density indicate a large degree of coordination in the coupling between actin and T cell surface proteins, including TCRs. Although interactions between actin and TCRs have generally been described as dynamic, the nearly complete dissipation of actin accumulations observed here is somewhat unexpected. At the label densities used in these experiments, the observed fluctuations reflect real dissipation of the actin itself, rather than stochastic fluctuations in fluorophore density. We observed fluorescence intensity accumulations that were two to four times higher than the cell background level that exhibited near 100% variance (complete dissipation). This is more than an order of magnitude beyond what can be expected from stochastic fluctuations (McQuarrie, 2000).
To better characterize the spatiotemporal dynamics of EGFP–β-actin, we developed a numerical approach based on time-autocorrelation of fluorescence intensity as a function of spatial position. This method is distinct from previously developed image correlation spectroscopy (ICS) (Petersen et al., 1993) and spatiotemporal ICS (STICS) (Wiseman et al., 2004; Hebert et al., 2005; Kulkarni et al., 2005; Rossow et al., 2009) in that there is no correlation of the spatial coordinates. Methods like ICS and STICS can be powerful ways to analyze the dynamics of protein clusters in cells, but they are limited in cases where the cluster sizes are heterogeneous. Our approach effectively identifies transient accumulations of arbitrary size with dynamics that are distinct from stochastic fluctuations in the EGFP–β-actin matrix.
In a typical EGFP–β-actin-labeled cell (Fig. 5A–C), the EGFP–β-actin time-average is a weak indication of where and for how long accumulations occur (Fig. 5D). However, the frame-by-frame fluorescence intensity profiles of areas corresponding to accumulations (Fig. 5D,E, blue areas and traces) show discrete large-scale intensity fluctuations, whereas in nearby areas lacking accumulations (Fig. 5D,E, red areas and traces) the fluorescence fluctuates on a much smaller scale around a background value. The time-autocorrelation functions of fluorescence intensity from the respective regions clearly reflect these differences (Fig. 5F). This analysis can be readily applied to the entire image area, as shown in Fig. 5G, where the integral of the autocorrelation function of each image pixel is plotted at the original pixel position. High autocorrelations in areas on the periphery of the cell correspond to extensions and retractions of the lamellipodia (Fig. 5G, white asterisks), whereas locally high autocorrelations are visible at areas adjacent to substrate barriers (Fig. 5G, white arrows and others). These latter areas correspond well to cytoskeletal accumulations visible in the data recordings (supplementary material Movie 1) and to areas that contain a high TCR density prior to time-lapse recording (Fig. 5C).
Fig. 5.
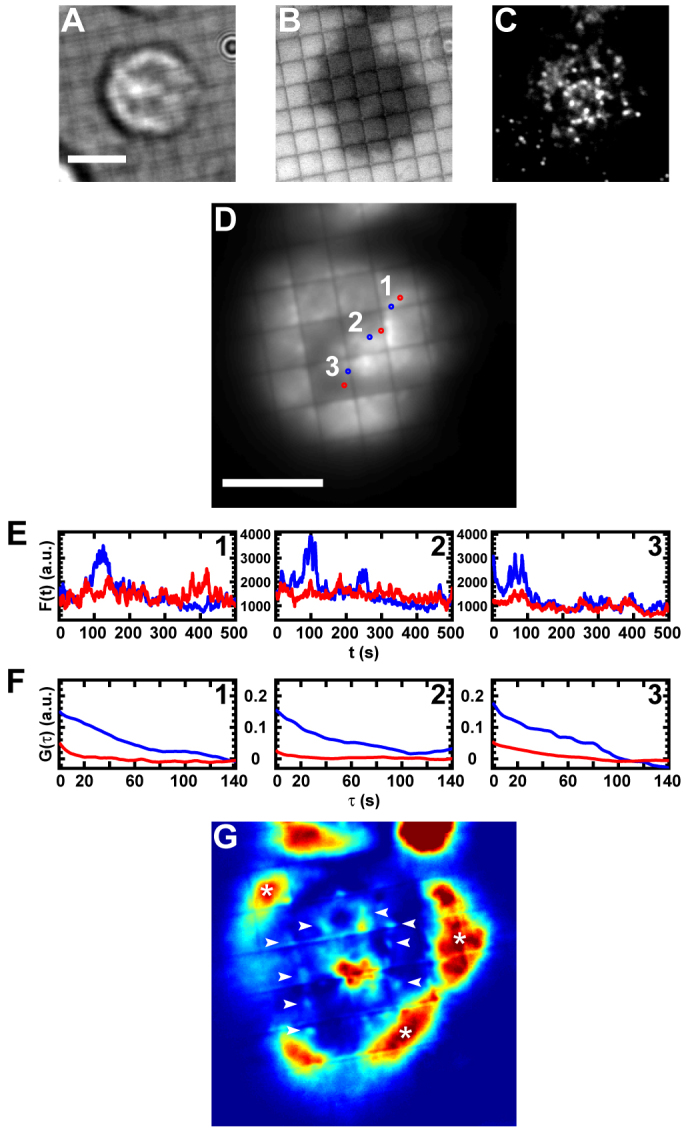
Transient enrichments of the actin cytoskeleton occur at TCR clusters. (A–C) Images taken before cytoskeletal time-lapse recording show the bright field (A), RICM (B) and TCR channel (C) views of an EGFP–β-actin-labeled cell. (D) The entire cytoskeletal time-lapse recording was time-averaged and areas corresponding to actin accumulations (blue regions 1, 2 and 3) and nearby background areas (red regions 1, 2 and 3) selected. (E) Fluorescence intensity profile over time is shown for each pixel within the regions selected in D. (F) Autocorrelation of the results shown in E. (G) The integral of the autocorrelation of the fluorescence intensity profile of every pixel in the cytoskeletal time-lapse image stack is output in the x–y location corresponding to its original pixel and displayed according to a color scale of arbitrary units. The white asterisks correspond to areas of extension and retraction of the lamellipodia and the white arrows highlight areas of high autocorrelation due to putative actin accumulations. Scale bars: 5 μm.
Discussion
The involvement of actin in T cell signaling is well established, but recent studies have shown that beyond acting as a simple transport mechanism, the actin cytoskeleton actively modulates TCR recognition of antigen by altering TCR–pMHC kinetics (Huppa et al., 2010; Huang et al., 2010). To better understand how this modulation occurs, there is a need to move beyond bulk inhibition of the cytoskeleton with latrunculin, cytochalasin and similar compounds by probing specific receptor–actin interactions and using quantitative analysis methods to describe the outcomes. We used the hybrid live cell–nanopatterned supported lipid bilayer system to pin TCR clusters and developed specialized tracking algorithms and an image time-autocorrelation analysis to characterize properties of a mobile and fluctuating cytoskeletal network interacting with these receptor assemblies. Using these tools, we have shown that, in primary T cells, actin slows down and assembles and disassembles en masse in the vicinity of TCR clusters.
The slowing and, to some extent, the overall average increase in density of actin at the sites of pinned TCR clusters are to be expected from a receptor–actin interaction that has previously been shown to be highly dissipative, or frictional (Kaizuka et al., 2007; DeMond et al., 2008; Yu et al., 2010). However, such a coupling does not on its own give rise to the large transient actin enrichments, or fluctuations, that we observe at these sites. We make this argument based on the fact that fluctuations of in a random distribution n objects independent scale with 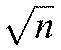 (McQuarrie, 2000). Larger-scale fluctuations are indicative of coordination within the system. Here, proximity to TCR microclusters leads to increases in large cytoskeletal fluctuations, suggesting greater degrees of coordination within the actin network. Taken in the context of studies that have shown microclusters to survive actin disruption (Campi et al., 2005; Varma et al., 2006), this finding demonstrates that TCR microclusters are the more stable element in the TCR–actin interaction and that they template the dynamic actin around themselves.
(McQuarrie, 2000). Larger-scale fluctuations are indicative of coordination within the system. Here, proximity to TCR microclusters leads to increases in large cytoskeletal fluctuations, suggesting greater degrees of coordination within the actin network. Taken in the context of studies that have shown microclusters to survive actin disruption (Campi et al., 2005; Varma et al., 2006), this finding demonstrates that TCR microclusters are the more stable element in the TCR–actin interaction and that they template the dynamic actin around themselves.
Materials and Methods
DNA constructs
A plasmid containing EGFP fused to the calponin homology domain of utrophin (EGFP–UtrCH) was a gift of William Bement, University of Wisconsin, Madison, WI (Burkel et al., 2007). The EGFP–UtrCH gene was amplified using PCR and subcloned into a murine stem cell virus parent-vector-containing puromycin N-acetyltransferase expressed from an internal ribosome entry site (pMSCV-Puro) as previously described (Smith et al., 2011). A retroviral-vector-containing EGFP–β-actin was similarly produced by subcloning the gene from an EGFP–β-actin-containing pCDNA3 vector (Ballestrem et al., 1998).
Retroviral transduction of T cells
AND CD4+ T cells (Kaye et al., 1989) were harvested and retrovirally transduced as previously described (Smith et al., 2011). Briefly, retrovirus-containing cell medium was harvested from cultures of Phoenix retroviral packaging cells transfected with pMSCV-Puro containing the gene of interest. The medium was used to transduce CD4+ T cells isolated from the lymph nodes and spleens of AND × B10.BR mice, primed with 2 μM moth cytochrome c (amino acids 88–103) and stimulated with 50 U/ml of interleukin-2 (IL-2). T cells were transduced 48 and/or 72 hours after isolation, selected using 0.5 μg/ml puromycin from 78 to 120 hours after isolation and used in experiments 168 hours after isolation.
Preparation of T cells for imaging
Imaging experiments were performed as previously described (Smith et al., 2011). Briefly, supported lipid bilayers containing 2 mol% Ni2+-DOGS and 98 mol% DOPC (Avanti Polar Lipids, Alabaster, AL) were formed within FCS2 Closed Chamber Systems (flow cells; Bioptechs, Butler, PA) on piranha-etched glass coverslips or patterned chromium substrates nanofabricated using electron beam lithography as previously described (Salaita et al., 2010). The bilayers were functionalized with polyhistidine-tagged MHC loaded with moth cytochrome c (amino acids 88–103) and with polyhistidine-tagged ICAM-1 proteins, and the flow cells were brought to 37°C. T cells were rinsed with a previously described imaging buffer, (Smith et al., 2011) pelleted, resuspended in a solution of 5 μl Alexa-Fluor-568- or -647-labeled H57 anti-TCR antibody fragment (Fab) diluted in 100 μl imaging buffer and incubated on ice for 20 minutes. Cells were then rinsed with imaging buffer, pelleted, resuspended in pre-warmed imaging buffer and injected into flow cells. The flow cells were kept at 37°C throughout the experiments and all images were acquired within 120 minutes of the injection of the cells.
Microscopy
Cell imaging was performed on an inverted microscope (Nikon Eclipse Ti; Technical Instruments, Burlingame, CA). TIRF microscopy was done using a fiber-coupled Nikon TIRF illuminator with a custom-built laser source (Smith et al., 2011). The 647 nm (RCL-050-640; Crystalaser, Reno, NV), 561 nm (GCL-100-561; Crystalaser, Reno, NV) and 488 nm (Sapphire HP; Coherent, Santa Clara, CA) asers were launched into a single-mode fiber that was connected to the TIRF illuminator. Excitation powers at the sample were on the order of 1 kW/cm2. Images were acquired with an Orca-R2 digital camera (C10600-10B; Hamamatsu Photonics K.K., Japan).
Actin motion analysis
To analyze the motion of the actin cytoskeleton, we developed a set of algorithms for identifying and tracking visibly mobile fluctuations in actin and/or probe density and implemented them in MatLab. Features in the actin distribution are first identified using a supplemented image gradient approach. Each frame of an actin time-lapse recording is convolved with a Gaussian filter to remove low-level imaging noise, and local maxima in the fluorescence intensity image are used to identify candidate features. Because individual features often contain multiple fluorescence intensity maxima, vector fields are created based on fluorescence intensity gradients within the image, and features are assigned locations by positions with converging vectors, which correspond to low local intensity variations. The vector fields also overcome uneven levels of fluorescence across the cell, thus allowing features to be identified consistently in spite of subcellular variations in cytoskeletal density or illumination (Xu and Prince, 1998).
Following identification, nearest neighbor features in consecutive frames are linked to generate actin trajectories as previously described (Yu et al., 2010). Briefly, linking is performed only on features that are visible in consecutive frames within 5 pixels (0.315 μm) of each other. The appearances and disappearances of features are monitored such that if a feature is present in frame t and has no identifiable nearest neighbor in frame t+1, it is defined as disappearing after frame t. Mergers and splits of features are also monitored.
The trajectories of actin features are evaluated using an algorithm that determines the time it takes each feature to move a certain distance. We refer to this time as the ‘escape time’ (τ) to signify that there is no directional bias to the algorithm, and the selected ‘escape distance’ (r=3 pixels=0.189 μm) thus defines a circular boundary around the feature. Note that although the escape distance is below the Rayleigh diffraction limit, it is still possible to localize maxima in the light field with high precision. Our approach is based on centroid analyses used in single molecule localization algorithms (Thompson et al., 2002; Saxton, 2009) except that we are tracking inhomogeneities in the fluorophore density field rather than single objects. This is fundamentally analogous to single fluorophore position identification in standard super-resolution optical techniques such as photoactivated localization microscopy (Betzig et al., 2006) and stochastic optical reconstruction microscopy (Rust et al., 2006). Escape times are determined iteratively for each frame in which a given feature is identified. They are not recorded when a feature disappears before exceeding the escape distance.
Image time-autocorrelation analysis
The time-autocorrelation function of the fluctuating fluorescence intensity was calculated on a pixel-by-pixel basis using the time-lapse image stacks. The time-dependent intensity of each pixel is designated at Ix,y(t), where the x and y subscripts refer to the pixel position. The correlation function was calculated using the fluctuations of the intensity away from time-averaged value:
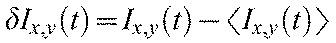 where
where
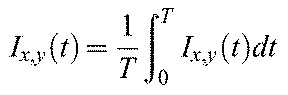
The pixel-by-pixel time-autocorrelation function is then calculated as:
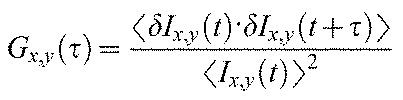
Supplementary Material
Acknowledgements
The authors would like to acknowledge N. Fay and N. Hartman for preparation of key reagents. Patterned substrate fabrication was performed, in part, at the Molecular Foundry, Lawrence Berkeley National Laboratory (LBNL).
Footnotes
Funding
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under contract number DE-AC02-05CH11231. A.S. was partially supported by a National Institutes of Health training grant [grant number T32 GM007232]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.092825/-/DC1
References
- Ballestrem C., Wehrle-Haller B., Imhof B. (1998). Actin dynamics in living mammalian cells. J. Cell Sci. 111, 1649-1658 [DOI] [PubMed] [Google Scholar]
- Barda-Saad M., Braiman A., Titerence R., Bunnell S. C., Barr V. A., Samelson L. E. (2005). Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat. Immunol. 6, 80-89 [DOI] [PubMed] [Google Scholar]
- Bartelt R. R., Cruz-Orcutt N., Collins M., Houtman J. C. D. (2009). Comparison of T cell receptor-induced proximal signaling and downstream functions in immortalized and primary T cells. PLoS ONE 4, e5430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., Hess H. F. (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642-1645 [DOI] [PubMed] [Google Scholar]
- Bunnell S. C., Kapoor V., Trible R. P., Zhang W., Samelson L. E. (2001). Dynamic actin polymerization drives t cell receptor-induced spreading: a role for the signal transduction adaptor LAT. Immunity 14, 315-329 [DOI] [PubMed] [Google Scholar]
- Burkel B. M., von Dassow G., Bement W. M. (2007). Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil. Cytoskeleton 64, 822-832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt J. K., Carrizosa E., Shaffer M. H. (2008). The actin cytoskeleton in T cell activation. Annu. Rev. Immunol. 26, 233-259 [DOI] [PubMed] [Google Scholar]
- Campi G., Varma R., Dustin M. L. (2005). Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J. Exp. Med. 202, 1031-1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMond A. L., Mossman K. D., Starr T., Dustin M. L., Groves J. T. (2008). T cell receptor microcluster transport through molecular mazes reveals mechanism of translocation. Biophys. J. 94, 3286-3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson-Belaire W. N., Cochrane A., Ostrowski M. A., Gray-Owen S. D. (2011). Differential response of primary and immortalized CD4+ T cells to Neisseria gonorrhoeae-induced cytokines determines the effect on HIV-1 replication. PLoS ONE 6, e18133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin M. L. (2009). Supported bilayers at the vanguard of immune cell activation studies. J. Struct. Biol. 168, 152-160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin M. L., Chakraborty A. K., Shaw A. S. (2010). Understanding the structure and function of the immunological synapse. Cold Spring Harbor Perspect. Biol. 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez T. S., Billadeau D. D. (2008). T cell activation and the cytoskeleton: you can't have one without the other. Adv. Immunol. 97, 1-64 [DOI] [PubMed] [Google Scholar]
- Grakoui A., Bromley S. K., Sumen C., Davis M. M., Shaw A. S., Allen P. M., Dustin M. L. (1999). The immunological synapse: a molecular machine controlling T cell activation. Science 285, 221-227 [DOI] [PubMed] [Google Scholar]
- Groves J. T., Boxer S. G. (2002). Micropattern formation in supported lipid membranes. Acc. Chem. Res. 35, 149-157 [DOI] [PubMed] [Google Scholar]
- Groves J. T., Ulman N., Boxer S. G. (1997). Micropatterning fluid lipid bilayers on solid supports. Science 275, 651-653 [DOI] [PubMed] [Google Scholar]
- Hartman N. C., Nye J. A., Groves J. T. (2009). Cluster size regulates protein sorting in the immunological synapse. Proc. Natl. Acad. Sci. USA 106, 12729-12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert B., Costantino S., Wiseman P. W. (2005). Spatiotemporal image correlation spectroscopy (STICS) theory, verification, and application to protein velocity mapping in living CHO cells. Biophys. J. 88, 3601-3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Zarnitsyna V. I., Liu B., Edwards L. J., Jiang N., Evavold B. D., Zhu C. (2010). The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature 464, 932-936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppa J. B., Axmann M., Mörtelmaier M. A., Lillemeier B. F., Newell E. W., Brameshuber M., Klein L. O., Schütz G. J., Davis M. M. (2010). TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature 463, 963-967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaizuka Y., Douglass A. D., Varma R., Dustin M. L., Vale R. D. (2007). Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc. Natl. Acad. Sci. USA 104, 20296-20301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye J., Hsu M.-L., Sauron M.-E., Jameson S. C., Gascoigne N. R. J., Hedrick S. M. (1989). Selective development of CD4+ T cells in transgenic mice expressing a class II MHC-restricted antigen receptor. Nature 341, 746-749 [DOI] [PubMed] [Google Scholar]
- Kulkarni R. P., Wu D. D., Davis M. E., Fraser S. E. (2005). Quantitating intracellular transport of polyplexes by spatio-temporal image correlation spectroscopy. Proc. Natl. Acad. Sci. USA 102, 7523-7528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.-H., Holdorf A. D., Dustin M. L., Chan A. C., Allen P. M., Shaw A. S. (2002). T cell receptor signaling precedes immunological synapse formation. Science 295, 1539-1542 [DOI] [PubMed] [Google Scholar]
- Manz B. N., Groves J. T. (2010). Spatial organization and signal transduction at intercellular junctions. Nat. Rev. Mol. Cell. Biol. 11, 342-352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuarrie D. A. (2000). Statistical Mechanics 1st ed. Sausalito: University Science Books; [Google Scholar]
- Monks C. R. F., Freiberg B. A., Kupfer H., Sciaky N., Kupfer A. (1998). Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 395, 82-86 [DOI] [PubMed] [Google Scholar]
- Mossman K. D., Groves J. T. (2007). Micropatterned supported membranes as tools for quantitative studies of the immunological synapse. Chem. Soc. Rev. 36, 46-54 [DOI] [PubMed] [Google Scholar]
- Mossman K. D., Campi G., Groves J. T., Dustin M. L. (2005). Altered TCR signaling from geometrically repatterned immunological synapses. Science 310, 1191-1193 [DOI] [PubMed] [Google Scholar]
- Nye J. A., Groves J. T. (2008). Kinetic control of histidine-tagged protein surface density on supported lipid bilayers. Langmuir 24, 4145-4149 [DOI] [PubMed] [Google Scholar]
- Petersen N. O., Hoddelius P. L., Wiseman P. W., Seger O., Magnusson K. E. (1993). Quantitation of membrane receptor distributions by image correlation spectroscopy: concept and application. Biophys. J. 65, 1135-1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossow M., Mantulin W. W., Gratton E. (2009). Spatiotemporal image correlation spectroscopy measurements of flow demonstrated in microfluidic channels. J. Biomed. Opt. 14, 024014-024017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust M. J., Bates M., Zhuang X. (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793-796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryser J., Rungger-Brandle E., Chaponnier C., Gabbiani G., Vassalli P. (1982). The area of attachment of cytotoxic T lymphocytes to their target cells shows high motility and polarization of actin, but not myosin. J. Immunol. 128, 1159-1162 [PubMed] [Google Scholar]
- Salaita K., Nair P. M., Petit R. S., Neve R. M., Das D., Gray J. W., Groves J. T. (2010). Restriction of receptor movement alters cellular response: physical force sensing by EphA2. Science 327, 1380-1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton M. J. (2009). Single particle tracking. In Fundamental Concepts in Biophysics, Handbook of Modern Biophysics. pp 1-33 New York: Humana Press; [Google Scholar]
- Smith A. W., Smoligovets A. A., Groves J. T. (2011). Patterned two-photon photoactivation illuminates spatial reorganization in live cells. J. Phys. Chem. A 115, 3867-3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura N., Nagafuku M., Minaki Y., Umeda Y., Hayashi F., Sakakura J., Kato A., Liddicoat D. R., Ogata M., Hamaoka T., et al. (2003). Dynamic changes in the mobility of LAT in aggregated lipid rafts upon T cell activation. J. Cell Biol. 160, 125-135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R. E., Larson D. R., Webb W. W. (2002). Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 82, 2775-2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma R., Campi G., Yokosuka T., Saito T., Dustin M. L. (2006). T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity 25, 117-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer C. M., Desai A., Chloe Bulinski J., Salmon E. D. (1998). Fluorescent speckle microscopy, a method to visualize the dynamics of protein assemblies in living cells. Curr. Biol. 8, 1227-1230 [DOI] [PubMed] [Google Scholar]
- Wiseman P. W., Brown C. M., Webb D. J., Hebert B., Johnson N. L., Squier J. A., Ellisman M. H., Horwitz A. F. (2004). Spatial mapping of integrin interactions and dynamics during cell migration by image correlation microscopy. J. Cell Sci. 117, 5521-5534 [DOI] [PubMed] [Google Scholar]
- Xu C., Prince J. L. (1998). Snakes, shapes, and gradient vector flow. IEEE Trans. Image Process. 7, 359-369 [DOI] [PubMed] [Google Scholar]
- Yokosuka T., Sakata-Sogawa K., Kobayashi W., Hiroshima M., Hashimoto-Tane A., Tokunaga M., Dustin M. L., Saito T. (2005). Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 6, 1253-1262 [DOI] [PubMed] [Google Scholar]
- Yu C.-H., Wu H.-J., Kaizuka Y., Vale R. D., Groves J. T. (2010). Altered actin centripetal retrograde flow in physically restricted immunological synapses. PLoS ONE 5, e11878 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.