

Abstract
Myosin X (Myo X), also known as MYO10, is an unconventional actin-based motor protein that plays an important role in filopodium formation. Its intra-filopodia movement, an event tightly associated with the function of Myo X, has been extensively studied. However, how the motor activity of Myo X and the direction of its movements are regulated remains largely unknown. In our previous study, we demonstrated that DCC (for ‘deleted in colorectal carcinoma’) and neogenin (neogenin 1, NEO1 or NGN), a family of immunoglobin-domain-containing transmembrane receptors for netrins, interact with Myo X and that DCC is a cargo of Myo X to be delivered to the neurites of cultured neurons. Here, we provide evidence for DCC and neogenin as regulators of Myo X. DCC promotes movement of Myo X along basal actin filaments and enhances Myo-X-mediated basal filopodium elongation. By contrast, neogenin appears to suppress Myo X movement on the basal side, but increases its movement towards the apical and dorsal side of a cell, promoting dorsal filopodium formation and growth. Further studies have demonstrated that DCC, but not neogenin, enhances integrin-mediated tyrosine phosphorylation of focal adhesion kinase and basal F-actin reorganization, providing a cellular mechanism underlying their distinct effects on Myo X. These results thus demonstrate differential regulatory roles on Myo X activity by its cargo proteins, DCC and neogenin, revealing different cellular functions of DCC and neogenin.
Introduction
Myo X (also known as MYO10), an unconventional member of the myosin family, has several distinguished cellular features as compared with other unconventional myosin family proteins. It is primarily localized at the tips of filopodia or the edges of lamellipodia and membrane ruffles (Berg and Cheney, 2002; Berg et al., 2000; Tokuo and Ikebe, 2004; Zhang et al., 2004). It undergoes forward and rearward movements within filopodia and promotes filopodia formation, elongation and sensing, possibly by transporting actin-binding proteins and cell adhesion receptors to the leading edge of the cell (Berg and Cheney, 2002; Tokuo and Ikebe, 2004; Tokuo et al., 2007; Zhang et al., 2004; Zhu et al., 2007). It is widely expressed and implicated in multiple cellular functions in different cell types, including netrin-1-induced neurite outgrowth and growth-cone guidance (Zhu et al., 2007), BMP6-dependent filopodial migration and activation of BMP receptors (Pi et al., 2007), and migration of Xenopus cranial neural crest cells (Hwang et al., 2009; Nie et al., 2009).
Myo X has unique structural features, which provide a basis for its intriguing cellular functions. It contains multiple domains, including a motor domain at its N-terminus, three calmodulin-binding IQ motifs, three PH domains, one myosin tail homology (MyTH) domain and one band 4.1–ezrin–radixin–meosin (FERM) domain (Berg et al., 2000; Yonezawa et al., 2000). Via these domains, Myo X not only binds to membrane lipids, but also interacts with actin-binding proteins (e.g. VASP), microtubules and transmembrane receptors (e.g. integrins and DCC family receptors) (Cox et al., 2002; Isakoff et al., 1998;Plantard et al., 2010; Tokuo and Ikebe, 2004; Umeki et al., 2011; Weber et al., 2004; Zhang et al., 2004; Zhu et al., 2007). In addition to DCC, many Myo-X-binding proteins, such as VASP and integrin, could be candidate cargos of Myo X to be delivered to the dynamic actin-based membrane protrusions, where they promote actin polymerization and cell membrane adhesion and sensing (Tokuo and Ikebe, 2004; Zhang et al., 2004; Zhu et al., 2007). Although many Myo-X-binding proteins have been identified, how the motor activity of Myo X is regulated remains largely unknown.
DCC and neogenin belong to a family of immunoglobulin-domain-containing receptors for the extracellular guidance cue, netrins. Via DCC, netrin-1 promotes axon outgrowth and mediates attractive growth cone guidance of axon projections (Fazeli et al., 1997; Keino-Masu et al., 1996; Kennedy et al., 1994; Serafini et al., 1994; Shekarabi and Kennedy, 2002). Myo X interacts with the DCC intracellular domain via its FERM domain, regulating DCC receptor distribution and function (Zhu et al., 2007). Neogenin also interacts with Myo X in a similar manner to DCC (Zhu et al., 2007). However, the function of neogenin–Myo X interaction remains to be determined.
Here, we present evidence that DCC and neogenin have a role in regulating Myo X activity and cellular distribution. DCC promotes Myo X movement along basal actin filaments and enhances Myo-X-mediated basal filopodia formation and elongation. However, neogenin suppresses Myo X movements on the basal side, but increases its movements towards the intracellular and apical side of cells, promoting dorsal filopodiagrowth. In addition, we showed that DCC promotes, but neogenin inhibits, integrin-dependent focal adhesion kinase (FAK) tyrosine phosphorylation and basal F-actin reorganization, providing a cellular mechanism underlying differential regulation of Myo X activity by DCC and neogenin.
Results
GFP–Myo X motility in NLT cells: filopodia-tip-associated and filopodia-tip-unassociated movements
To understand how Myo X motility is regulated, we took advantage of NLT cells, which are derived from mouse migratory GnRH neurons that express endogenously Myo X and its cargo proteins, DCC and neogenin (data not shown). We first examined the motor activity of GFP–Myo X by time-lapse live imaging analysis as previously described (Berg and Cheney, 2002; Zhu et al., 2007). GFP–Myo X in NLT cells was largely localized at the tip of filopodia (Fig. 1A), consistent with previous reports (Berg and Cheney, 2002; Zhang et al., 2004; Zhu et al., 2007). Analysis of GFP–Myo X motility by time-lapse live imaging revealed three types of Myo X movement. First, a single Myo X punctum was detected at the filopodium tip and its movement was tightly associated with the motility of the filopodium (Fig. 1B,C). We thus named this type of Myo X movement ‘filopodia-tip-associated movement’. Second, several Myo X puncta were detected in a single filopodium, and their movements were unassociated with the filopodium motility (Fig. 1D,E). The filopodia were nearly non-motile, but Myo X exhibited intra-filopodia mobility (Fig. 1D,E). The second type of Myo X movement was named ‘filopodia-tip-unassociated movement’. The third type of Myo X puncta were named ‘non-motile Myo X’ because they appeared to be stable without obvious movement (Fig. 1A; supplementary material Movie 1). In addition, these non-motile puncta were away from cytoplasm, uncoupled with filopodia, and might be result of the remnants of ‘ghost’ filopodia that are disconnected from the cell.
Fig. 1.

GFP–Myo X motility in NLT cells. (A) NLT cell expressing GFP–Myo X. Three types of Myo X movement in a single cell were observed and marked with F (front), T (tail) and S (stable). (B) Time-lapse series of images derived from the front region in A shows movement of GFP–Myo X (left) with corresponding GFP plus phase images (right). A large GFP–Myo X punctum (arrowheads) moves forward and rearward. (C) Plot representing traces of the position of a typical filopodium tip and tip-associated GFP puncta every 20 seconds, obtained from time-lapse images in B. The filopodium undergoes rapid forward (~52 nm/second) and rearward (~39.2 nm/second) extension with a GFP–Myo X punctum presenting at the tip. (D) Time-lapse series of images derived from tail region in A shows movement of GFP–Myo X puncta (left) that are uncoordinated with filopodia movement (GFP phase, right). Although a large GFP punctum (white arrowheads) moves rearward, the filopodium and its tip-associated GFP punctum (red arrowheads) remain stable. (E) Plot representing typical traces of the position of filopodia tip and tip-associated GFP–Myo X punctum every 20 seconds, obtained from time-lapse images in D. The filopodium remains relatively stationary for ~10 minutes, but a punctum of GFP–Myo X undergoes forward (56.5 nm/second) and rearward (36.6 nm/second) movements. (F) Top (upper) and side (bottom) views of a 3D projected image of an NLT cell expressing GFP–Myo X. Anti-Golgin 97 (red), a marker for Golgi, was used for immunostaining. The nuclei (blue) were stained with Topro3. Front (F) and tail (T) regions are indicated. In C and E, the frame-by-frame velocities were obtained from quantitative analysis of 28 filopodia and 40 GFP–Myo X puncta from six GFP–Myo-X-positive cells. The non-motile GFP–Myo X puncta were excluded from the velocity analysis. Mean values are presented. Scale bars: 10 μm.
We analyzed the properties of the first two types of Myo X movement: filopodia-tip-associated and filopodia-tip-unassociated Myo X movement. The tip-associated Myo X puncta appeared to be distributed in a polarized manner and were associated with the leading edge of a moving NLT cell (Fig. 1A; supplementary material Movie 1). This notion was further tested by immunostaining analysis of the fixed cells with a Golgi marker, Golgin-97, an indicator for cell ‘front’ side (Kupfer et al., 1982). Both the tip-associated Myo X and Golgin-97 were on the same side of the cell, ‘facing’ the direction of cell movement (Fig. 1F). In addition, tip-associated Myo X travelled together with filopodia in both directions, with forward and rearward velocities of ~52 and ~39 nm/second, respectively (Fig. 1C; Table 1). The net direction of their movement was forward, or away from the cell body, because the frequency of forward movement was often greater than that of rearward movement. By contrast, the tip-unassociated Myo X puncta were frequently observed to be associated with the filopodia on the ‘rear’ or ‘tail’ region of a moving cell (Fig. 1A,E; supplementary material Movie 1). Those puncta also underwent intra-filopodia movements, but there were more rearward movements (towards the cell body) than forward movements (Fig. 1D,E). The tip-unassociated Myo X seemed to be in association with retracting actin filaments and colocalized with the phase-dense granules (Fig. 1D, white arrows). They travelled at similar rearward (~36 nm/second) and forward (~55 nm/second) speeds as tip-associated Myo X (Table 1), although the net direction appeared to be rearward, or towards the cell body. Taken together, these results suggest that Myo X undergoes different types of movements in a cellular region-specific manner.
Table 1.
GFP–MyoX and filopodium movements

Differential regulation of Myo X motility by DCC and neogenin
We next asked whether DCC, a cargo of Myo X (Zhu et al., 2007), regulates Myo X motor activity. To this end, DCC was coexpressed with GFP–Myo X in NLT cells, and Myo X motility was recorded and analyzed by time-lapse live imaging. Note that Myo X-induced filopodia in cells coexpressing DCC were more elongated than in cells expressing GFP–Myo X alone (Fig. 2A; Table 1), consistent with our previous report (Zhu et al., 2007). In addition, the number of non-motile, stable Myo X puncta was significantly reduced, and both tip-associated and tip-unassociated Myo X movements were observed (Fig. 2A–E). Note that in addition to processive movement, GFP–Myo X puncta in the ‘tail’ filopodia showed diffusion movement in DCC-expressing cells (Fig. 2A,E; supplementary material Movie 2). Quantitative analysis indicated a faster speed of Myo X movements (both tip-associated and tip-unassociated movements, and both forward and rearward intra-filopodia motility) in cells coexpressing DCC as compared with cells expressing Myo X alone (Fig. 2D; Table 1). These results suggest that expression of DCC is sufficient to promote Myo-X-mediated filopodia elongation and increase the speed of Myo X motility.
Fig. 2.

DCC regulation of GFP–Myo X motility in NLT cells. (A) NLT cell coexpressing GFP–Myo X with DCC. The front (F) and tail (T) regions in the cell are marked. The rearward and forward movements are indicated by red and green arrows, respectively. (B) Time-lapse series of images derived from front region in A shows movement of GFP–Myo X (left) with corresponding GFP plus phase images (right). Red arrowheads indicate the tip-associated Myo X puncta. (C) Plot shows typical traces of the position of filopodia tip and tip-associated GFP puncta every 20 seconds, obtained from time-lapse images in B as described for Fig. 1. (D) Comparison of the velocities of GFP–Myo X puncta in the presence and absence of DCC coexpression. Mean + s.d. from three different experiments are shown; *P<0.01. (E) Time-lapse series of images derived from tail region in A. The tip-associated Myo X puncta are marked with red arrowheads, and tip-unassociated Myo X puncta are indicated by white or black arrowheads. (F) Plot shows typical traces of the position of filopodia tip and tip-associated GFP–Myo X puncta every 20 seconds, obtained from time lapse images in D. In C, D and F, the frame-by-frame velocities were obtained by analyzing 24 filopodia and 42 GFP–Myo X puncta from six GFP–Myo X plus DCC-positive cells. Scale bars: 10 μm.
We then ask whether neogenin regulates Myo X motility in a similar manner to DCC, as it is a DCC-related receptor and interacts with Myo X in a similar manner (Zhu et al., 2007). However, cells coexpressing neogenin with GFP–Myo X showed shorter filopodia and exhibit a non-polarized cell morphology (Fig. 3A; Table 1). Both tip-associated Myo X and non-motile, stable Myo X puncta were reduced in number, and most GFP–Myo X puncta appeared to be filopodia-tip-unassociated, with an average of 5–10 puncta per filopodium (Fig. 3A,B). Those Myo X puncta in DCC-expressing cells also travelled at a faster speed rearward, but not forward, compared with Myo X alone (Fig. 3D; Table 1; supplementary material Movie 3). In addition, GFP–Myo X was largely distributed intracellularly or in the perinuclear region as a diffuse pattern (Fig. 3A). These results suggest that expression of neogenin might prevent Myo-X-induced filopodia elongation by inhibiting tip-associated Myo X movement, revealing a different regulatory role on Myo X motility to that of DCC.
Fig. 3.

Neogenin regulation of GFP–Myo X motility in NLT cells. (A) NLT cell coexpressing GFP–Myo X with neogenin. The tail (T)-like region in the cell is marked. The rearward and forward movements are indicated by red and green arrows, respectively. (B) Time-lapse series of images derived from tail region in A. The tip-associated and unassociated Myo X puncta are marked by red and white arrowheads, respectively. (C) Plot of time-lapse images in B shows typical traces of the position of filopodia tip and tip-associated GFP–Myo X puncta every 20 seconds. (D) Comparison of the velocities of GFP–Myo X puncta in the presence and absence of neogenin coexpression. Mean + s.d. from three different experiments are shown; *P<0.01. In C and D, 14 filopodia and 35 GFP–Myo X puncta from four GFP–Myo X plus neogenin-positive cells were analyzed. Scale bars: 10 μm.
Differential regulation of Myo X cellular distribution by DCC and neogenin
To understand why Myo X movements were differentially regulated, we examined whether Myo X subcellular distribution is affected by DCC and neogenin. 3D imaging analysis of NLT cells expressing GFP–Myo X demonstrated that GFP–Myo X puncta were largely distributed at basal side of the cell periphery (Fig. 4A,B). Coexpression of DCC showed little effect on the distribution of GFP–Myo X, as most Myo X puncta were also distributed at the basal side of cell peripheral membrane protrusions (Fig. 4A,B). However, coexpression of neogenin altered Myo X distribution. Neogenin–Myo X was colocalized as puncta largely inside the cell (Fig. 4A,B). Many short and small finger-like neogenin- and Myo-X-positive protrusions were observed on the apical side of the cell surface (Fig. 4A). These results suggest that neogenin was capable of changing the direction of Myo X movement towards the inside and/or apical cell membrane, a different direction to that of Myo X with or without DCC expression. This view was further verified by quantification of the GFP fluorescence intensity in the basal and apical membranes (Fig. 4C). In cells coexpressing DCC, the GFP–Myo X was mainly distributed at the basal side of the cell periphery, with low signal detected inside the cell or at the apical membrane (Fig. 4B,C). By contrast, strong GFP–Myo X signal was detected not only at the basal membrane, but also on the apical surface of cells coexpressing neogenin (Fig. 4B,C). In addition, GFP–Myo X puncta were distributed around the whole cell surface in neogenin coexpressing cells, whereas it was asymmetrically distributed at the basal side of cells expressing GFP–Myo X alone or with DCC (Fig. 4A). Cell spreading (based on the cell surface area at the basal side) was also reduced in NLT cells coexpressing neogenin, but not DCC (Fig. 4D). These results provide additional support for the differential regulation of Myo X activity by DCC and neogenin. DCC promotes basal filopodia elongation and tip-associated Myo X movement, and neogenin prevents basal tip-associated movement, but increases Myo X movement towards the cell body and apical membrane and thus promotes dorsal filopodia growth.
Fig. 4.

Differential regulation of GFP–Myo X distribution by DCC and neogenin. (A) 3D confocal imaging analysis of NLT cells expressing GFP–Myo X in the absence or presence of DCC or neogenin. The three rows show projected views from the points of apical (top), basal (bottom), and a Z-axis (side) of the same cell. Arrows indicate the apical sides of cells. Scale bars: 20 μm. (B) Quantification analysis of GFP–Myo X puncta distribution in cell periphery or cell inside (intracellular regions). Mean + s.d. from three different experiments are shown. (C) Quantification of fluorescence intensity at the apical and basal focal planes of NLT cells expressing GFP–Myo X with DCC or neogenin. (D) Quantification of cell spreading (basal area) in NLT cells expressing GFP–Myo X in the presence or absence of DCC or neogenin. The data were normalized against NLT cells expressing control GFP (5126±441.4 μm2). Mean + s.d. (n=5–10 cells) are shown; *P<0.01.
We then determined whether DCC and/or neogenin expression in NLT cells is necessary for the regulation of Myo X movement. To this end, microRNA (miRNA) constructs of DCC and neogenin (both GFP and DsRed) were generated and tested for their effects on DCC and neogenin expression. Transfection of miRNA-DCC or miRNA-neogenin, but not control (a scrambled miRNA), in HEK293 cells led to a reduction in the expression of FLAG–DCC or FLAG–neogenin, respectively (supplementary material Fig. S1). NLT cells were then co-transfected with GFP–Myo X and scrambled miRNA, miRNA-DCC or miRNA-neogenin (indicated by DsRed). In control cells, GFP–Myo X puncta were distributed at the basal side of cell periphery (Fig. 5A), exhibiting three types of movements (tip-associated, tip-unassociated and non-motile) as described above. However, in cells coexpressing miRNA-DCC, but not miRNA-neogenin, fractions of GFP–Myo X puncta were detected towards the apical side of cells (Fig. 5A,B). The tip-associated Myo X puncta were reduced in number (Fig. 5C) and the speeds of Myo X movement (tip-associated, both forward and rearward) were decreased in cells depleted of DCC expression (Fig. 5D). These phenotypes resemble to certain degree that of NLT cells coexpressing neogenin with Myo X: both exhibited an increase in apical Myo X distribution and a decrease in filopodia-tip-associated Myo X movements. These results suggest that DCC is not only sufficient but also necessary for keeping Myo X at the basal side of the cell periphery, implying that the ratio of DCC to neogenin in a cell might be crucial for Myo X association with the basal filopodium tip and its asymmetrical distribution.
Fig. 5.

Requirement of DCC, but not neogenin, expression for the basal distribution of GFP–Myo X. (A) 3D confocal imaging analysis of NLT cells expressing GFP–Myo X in the absence or presence of miRNA-DCC. The front (F), putative front (“F”), and stable (S) regions are marked. The two rows show projected views from the points of apical (top) and a Z-axis (side) of the same cell. The apical Myo X puncta are marked with white arrows, and the basal Myo X puncta is indicated by a green arrow. Scale bars: 20 μm. (B,C) Quantification of GFP–Myo X puncta distribution at the ‘apical’/non-basal region (B) and at the basal filopodia tip (C). The GFP puncta that are away from the basal side based on the Z-axis view were counted as ‘apical’ GFP puncta. Mean + s.d. from three different experiments is shown. (D) Comparison of the velocities of GFP–Myo X puncta in the presence or absence of miR-DCC or miR-neogenin. The velocities were analyzed as described in Figs 1, 2 and 3. 25 filopodia and 32 GFP–Myo X puncta from four GFP–Myo X-positive cells were analyzed. Mean + s.d. from three different experiments are shown; *P<0.01.
Differential regulation of F-actin remodeling by DCC and neogenin
Actin filaments are essential ‘tracks’ for Myo X movement (Nagy et al., 2008). We thus asked whether DCC–Myo X or neogenin–Myo X complexes differentially regulate F-actin remodeling in NLT cells. F-actin structures in NLT cells expressing GFP–Myo X in the presence of DCC or neogenin were revealed by phalloidin staining. As shown in Fig. 6A, F-actin-containing stress fibers and filopodia were observed in cells expressing Myo X alone. Compared with untransfected cells, no difference was seen in the number and organization of stress fibers, but an increase in filopodia was observed in cells expressing GFP–Myo X (Fig. 6A), consistent with previous reports (Berg and Cheney, 2002; Bohil et al., 2006; Zhu et al., 2007). Remarkably, coexpression of DCC further enhanced the length and number of filopodia and enriched stress-fiber-like F-actin at the cell periphery, but not in the central region (Fig. 6B,D). Coexpression of neogenin, on the other hand, decreased the number of stress fibers and shortened the length of filopodia significantly (Fig. 6C,D). We also examined the effect of DCC and neogenin on microtubules by co-immunostaining using antibodies against detyrosinated tubulin (data not shown) and tyrosinated tubulin, and little difference was observed on microtubules in these cells (Fig. 6A–D). These results suggest that DCC–Myo X and neogenin–Myo X complexes appeared to have different regulatory effects on F-actin remodeling.
Fig. 6.

Altered basal F-actin filaments, but not microtubules, in NLT cells coexpressing GFP–Myo X with DCC or neogenin. (A–C) Phalloidin and tyrosinated tubulin (Tyr-tubulin) staining of F-actin filaments and microtubules, respectively, in NLT cells expressing GFP–Myo X alone (A), GFP–Myo X plus DCC (B) or GFP–Myo X plus neogenin (C). The front (“F”) and tail (“T”) regions are marked. The white arrow in A indicates the tip of a filopodium. Scale bars: 10 μm. (D) Quantification of F-actin structures (stress fibers and filopodia) and Tyr-tubulin intensity. A typical stress fiber is a long thick actin bundle that spans across the cell body and is distributed along the basal side of the cell. A single NLT cell that contains >10 stress fibers was considered as a stress fiber-positive cell. About 90% of NLT cells or NLT cells expressing GFP–Myo X alone in a normal culture condition are stress fiber-positive cells. Filopodia were defined as protrusions that are thin (less than 200 nm diameter) and long (more than 0.75 μm) measured horizontally from the cell body margin to the tip of a filopodium. The average length of a filopodium was measured and normalized based on NLT cells expressing GFP–Myo X alone (10.11±07 μm). The average fluorescence intensity (5951 pixels) of Tyr-tubulin per cell (on projected images) was also quantified by ImageJ software. Mean + s.d. from three different experiments are shown; *P<0.01.
We next asked whether expression of DCC or neogenin alone in NLT cells had any effect on F-actin remodeling. Cells expressing GFP vector showed no difference in F-actin to that of untransfected cells, which exhibited predominant stress fibers, as shown by phalloidin staining (Fig. 7A,C). Expression of DCC and neogenin in NLT cells increased cell periphery lamellipodia-like structures and membrane ruffles at the cell periphery, respectively, without significant effect on stress fibers (Fig. 7A,C). Note that both DCC and neogenin were co-stained with phalloidin at the lamellipodia and ruffle membranes, respectively (Fig. 7A). These results suggest that both DCC and neogenin might be sufficient to induce F-actin remodeling and increase cell periphery and/or cortical actin polymerization. Because lamellipodia, but not membrane ruffles, require cell attachment to the extracellular matrix (Geiger et al., 2001; Ridley et al., 2003), these results imply that neogenin, but not DCC, plays a negative role in cell adhesion.
Fig. 7.

Reorganization of F-actin structures in NLT cells expressing DCC or neogenin. NLT cells expressing GFP, FLAG-DCC, or FLAG-neogenin were fixed and subjected to staining analysis using indicated antibodies. F-actin structures were revealed by phalloidin staining, and cells expressing GFP, DCC or neogenin were indicated by GFP or anti-FLAG immunostaining. (A) Projected views of confocal imaging analysis. (B) Images at the apical and basal focal planes of the GFP- and neogenin-expressing cells. The focal plane of the side view at the yellow vertical line region is also included in the bottom panel. The colocalization of neogenin with phalloidin is indicated by a yellow arrow. (C) Quantification of different F-actin structures (stress fibers, lamellipodia and ruffles). (D) Quantification of F-actin staining at the basal and apical focal planes of the cells expressing GFP, DCC or neogenin. Stress fibers were defined as described for Fig. 6D. Lamellipodia were defined as surface-attached sheet-like phalloidin-stained membrane protrusions. About 10% of NLT cells expressing GFP control at normal culture condition (low density) show lamellipodia. Membrane ruffles are thick actin filaments that are frequently associated with apical side of leading edge of motile cells. About 5% of NLT cells expressing GFP control at normal culture condition (low density) show membrane ruffles. In C and D, the mean + s.d. from three different experiments (10 cells per experiment) are shown.
We next determined whether DCC and/or neogenin are required for F-actin remodeling in NLT cells by expressing miRNA-DCC or miRNA-neogenin (indicated by GFP or DsRed). F-actin structures were revealed by either phalloidin staining or by co-transfection with CFP–actin or GFP–actin. In cells transfected with scrambled miRNA, normal stress fibers and cell periphery actin filaments were observed in most (~90%) cells (Fig. 8A,B). Remarkably, many cells expressing miRNA-DCC (~60%), but not miRNA-neogenin, showed significant reduction in the number of stress fibers and disorganized cell periphery actin filaments (Fig. 8A,B). These results suggest that DCC, but not neogenin, might be necessary for the basal F-actin organization in NLT cells.
Fig. 8.
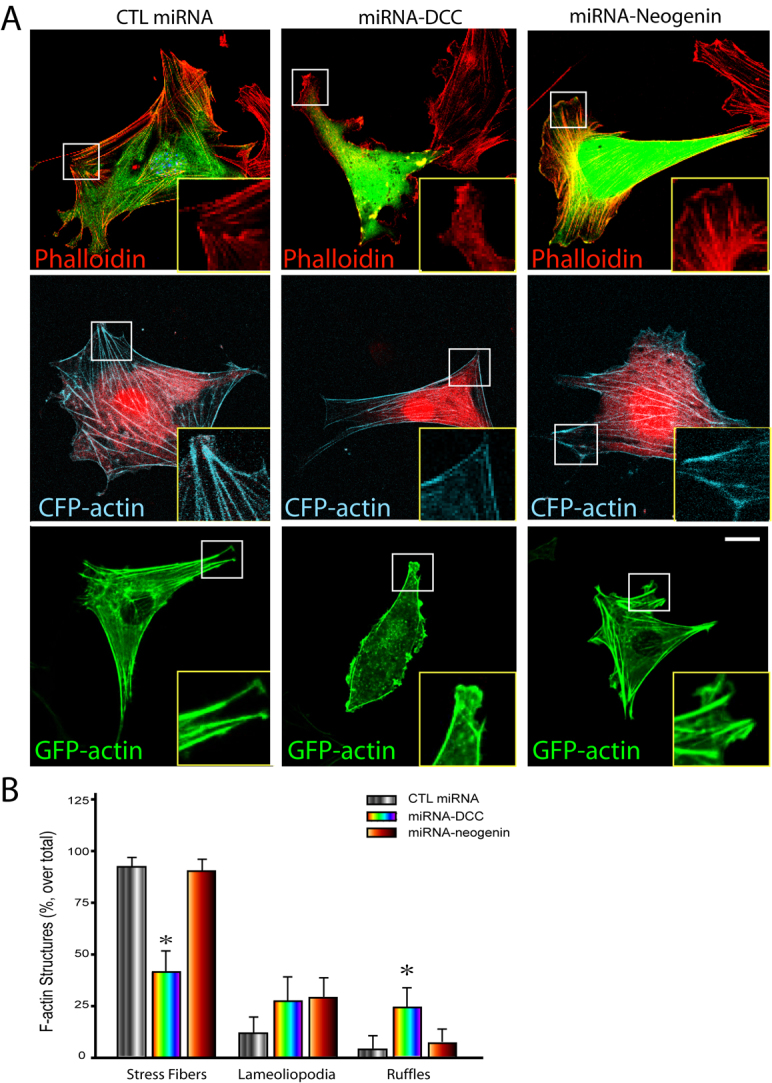
Requirement of DCC, but not neogenin, expression for the basal F-actin organization in NLT cells. (A) Analysis of F-actin filaments in NLT cells expressing scramble miRNA, miRNA-DCC, and miRNA-neogenin. F-actin filaments were revealed by phalloidin staining (top row), coexpressing CFP–actin (middle row) or GFP–actin (bottom row). Insets show amplified images of the boxed areas. Scale bar: 10 μm. (B) Quantification of F-actin structures (stress fibers, lamellipodia and ruffles) in cells expressing scramble miRNA, miRNA-DCC, and miRNA-neogenin as described in the legend of Fig. 7D. Mean + s.d. from three different experiments (10 cells per experiment) are shown; *P<0.01.
Differential regulation of integrin-dependent FAK tyrosine phosphorylation by DCC and neogenin
The reduced number of basal stress fibers in NLT cells transfected with miRNA-DCC suggested DCC regulation of integrin-mediated signaling. FAK tyrosine 397 phosphorylation is induced upon cell attachment to the extracellular matrix (Martin et al., 2002) and phosphorylated tyrosine 397 (Y397-P) is therefore used as a integrin signaling ‘reporter’. It also interacts with both DCC and neogenin (Ren et al., 2004). We thus assessed the role of DCC and neogenin on FAK Y397-P in NLT cells in response to cell adhesion. NLT cells expressing DCC or neogenin were plated onto collagen-coated coverslips for 4 hours. Cells were then fixed and subjected to immunostaining analysis using antibodies against Y397-P. As shown in Fig. 9A, cells expressing DCC showed an increase of Y397-P compared with untransfected cells. By contrast, expression of neogenin decreased the Y397-P signal significantly (Fig. 9A,B). These results suggest that DCC promotes, but neogenin inhibits, integrin-mediated FAK tyrosine phosphorylation. Supporting this view is the observation that a reduced Y397-P signal was found in cells expressing miRNA-DCC, but not miRNA-neogenin (Fig. 9C,D).
Fig. 9.

Differential regulation of FAK tyrosine 397 phosphorylation by DCC and neogenin. (A) Immunostaining analysis of FAK phosphorylated tyrosine 397 (pY397) in NLT cells expressing FLAG–DCC or FLAG–neogenin. Arrows indicate the cells expressing DCC or neogenin, and stars marked the untransfected cells. (B) Quantification analysis of the relative FAK Y397-P fluorescence intensity in cells expressing DCC or neogenin. (C) Immunostaining analysis of FAK Y397-P in NLT cells expressing scramble miRNA, miRNA-DCC, or miRNA-neogenin. Arrows indicate the cells expressing miRNA-DCC or miRNA-neogenin. (D) Quantification of the relative FAK Y397-P fluorescence intensity in cells transfected with miRNA-DCC or miRNA-neogenin. In A–D, NLT cells transiently transfected with indicated plasmids were trypsinized and re-plated onto collagen-coated coverslips for 4 hours. Cells were then fixed and subjected to immunostaining analysis. In B and D, the average fluorescence intensity of Y397-P per cell was quantified by NIH ImageJ software and normalized against untransfected cells (1221 pixels). The mean ± s.d. from three different experiments (10 cells per experiment) are shown; *P<0.01, #P<0.01. Scale bars: 10 μm.
Discussion
In this study, we have shown an asymmetrical localization and a polarized intra-filopodia movement of Myo X in NLT cells. Myo X cargo proteins, DCC and neogenin, are not only delivered to the different destinations of a cell, they also play an important role in regulating the activity of Myo X. DCC promotes Myo X movement along the intra-filopodia actin filaments at the basal side of a cell. Neogenin, by contrast, changes the direction of Myo X movement towards the intracellular and dorsal side of a cell. A possible mechanism underlying this differential regulation of Myo X might involve their differential regulation of integrin signaling and integrin-induced basal F-actin reorganization (Fig. 10).
Fig. 10.

A working model of differential regulation of Myo X movements by DCC and neogenin. The DCC–Myo X complex appears to promote integrin-induced FAK phosphorylation (e.g. pY397) and basal filopodia elongation. However, a neogenin–Myo X complex suppresses integrin signalling, but increases dorsal filopodia growth.
The filopodia-tip-associated and filopodia-tip-unassociated Myo X movements in NLT cells are similar to those described in Hela cells (Berg and Cheney, 2002). However, the velocity of Myo X movements in NLT cells appears to be slower than in Hela cells (Berg and Cheney, 2002). In addition, a significant number of GFP–Myo X puncta in NLT cells are in a relatively stationary state and have no obvious connection with filopodia or the cell membrane, which is not described in Hela cells. The exact cellular functions of the three types of Myo X movement are unclear. It is noteworthy that the different types of Myo X movement appear to be in association with cell polarity: the filopodia-tip-associated Myo X is largely distributed at the ‘front’ side of a moving cell, and the tip-unassociated Myo X and the stationary Myo X puncta are frequently found at the ‘tail’ side of the cell. The tip-associated Myo X movement correlates well with its function in filopodia formation and extension at the front side, and the tip-unassociated Myo X movement seems to be associated with the tail retraction. We thus speculate that tip-associated Myo X plays an important role in the transport of cell adhesion receptors (such as integrins and DCC) and actin remodeling proteins (e.g., VASP) to the leading edge of a moving cell, and that the tip-unassociated Myo X carries ‘tail’-associated cargo proteins and/or lipids. This speculation remains to be further investigated.
Myo X motility has been extensively studied; however, mechanisms underlying its regulation remain largely unknown. We found that DCC and neogenin, both cargos of Myo X, regulate Myo X movements, which suggests that Myo X cargos act as regulators and guide Myo X movement to different destinations. DCC enhances Myo-X-mediated basal filopodia extension and promotes the velocity of Myo X movements, both tip-associated and tip-unassociated, along actin filaments at the basal side. Neogenin, however, appears to promote Myo X movement towards the intracellular and dorsal or apical direction. In addition, neogenin inhibits filopodia-tip-associated Myo X movements, decreases filopodia formation and extension, and reduces cell spreading. These results suggest that DCC-Myo X plays a role in basal filopodia formation and extension, but neogenin-Myo X is involved in dorsal filopodia formation and extension. These observations also show that the ratio of DCC to neogenin might be crucial for the moving direction of Myo X. When the level of DCC over neogenin is dominant, Myo X moves along the basal actin filaments, promoting basal filopodia formation and extension. On the other hand, if the ratio of DCC to neogenin is decreased (by overexpression of neogenin or depletion of DCC), Myo X tip-associated movement along the basal actin filaments is reduced and its dorsal movement is enhanced. This view is in agreement with a previous report (Bohil et al., 2006) that proposes a reciprocal relationship between dorsal filopodia formation and basal cell spreading.
The mechanisms underlying the differential regulation of Myo X by DCC and neogenin remain largely unclear. We propose three working models for their differential regulation. First, DCC, but not neogenin, promotes integrin signaling and integrin-mediated basal F-actin organization, which might be essential for its stimulatory effect on Myo X activity. This model is in line with the observations that overexpression of DCC, but not neogenin, enhances integrin-dependent FAK tyrosine 397 phosphorylation and F-actin organization at the basal side of cells (Figs 7, 9), and suppression of DCC expression, but not neogenin, reduces integrin signaling and F-actin based filaments (Figs 8, 9). Neogenin, on the other hand, appears to have a negative role in integrin signaling, but promotes Myo-X-mediated dorsal filopodia growth.
Second, DCC, but not neogenin, promotes phosphoinositide 3-kinase (PI3K) signaling and phosphatidylinositol (3,4,5)-trisphosphate [PtdIns(3,4,5)P3] production, another potential mechanism underlying DCC stimulation of filopodia-tip-associated Myo X movement. This hypothesis is in light of recent reports that the binding of the second PH domain of Myo X to PtdIns(3,4,5)P3 enhances Myo X association with the filopodia tips and promotes its motor activity (Plantard et al., 2010; Umeki et al., 2011).
Third, DCC, but not neogenin, promotes Myo X dimerization. Several reports based on the in vivo and in vitro motility assay have suggested that Myo X forms a dimer in the cell and moves processively toward the barbed ends of actin filaments (Sun et al., 2010; Watanabe et al., 2010). Regulation of Myo X dimerization might affect its motor activity (Tokuo et al., 2007). It is postulated that the putative coiled-coil domain plays a role in the dimerization of Myo X (Sousa and Cheney, 2005). However, this putative coiled-coil domain of Myo X is highly charged and unable to dimerize (Knight et al., 2005). Thus, the dimerization of Myo X might require other cellular components or cargos of Myo X (Hirano et al., 2011; Umeki et al., 2011). One hypothesis is that Myo X might dimerize upon its cargo binding, just like Myo VI (Yu et al., 2009). Interestingly, DCC, but not neogenin, is known to be dimerized via its intracellular P3 domain (data not shown), and this P3 domain also interacts with the FERM domain of Myo X (Zhu et al., 2007). In light of these observations, we speculate that DCC, but not neogenin, stimulates Myo X dimerization, thus promoting Myo X motor activity and inducing filopodia formation and extension.
Materials and Methods
Reagents
Monoclonal antibodies anti-FLAG (cat#F3165), anti-Myc (cat#9E10) and anti-Golgin 97 (cat# A21270) were purchased from Sigma (St. Louis, MO). Goat anti-DCC polyclonal antibody (cat#sc-6535) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Alexa-Fluor-568–phalloidin (cat#A12380), nuclear counter staining TO-PRO-3 iodide (cat#T3605) and Lipofectamine 2000 (cat#11668-019) were obtained from Molecular Probes (Invitrogen, Carlsbad, CA). Anti-mouse and anti-goat secondary fluorescence-conjugated antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). A stable HEK293 cell line expressing human netrin-1 was used as described previously (Ren et al., 2004; Xie et al., 2005; Zhu et al., 2007). Unless otherwise indicated, ~200 ng/ml human netrin-1 was used for stimulation.
Expression vectors
The plasmids expressing FLAG-tagged mouse neogenin and human DCC were used as previously described (Ren et al., 2004). The cDNA of mouse Myo X was subcloned into mammalian expression vector (pEGFP-C1) fused with GFP at the N-terminus, as described previously (Zhu et al., 2007). The pDsRed-C1 mammalian expression vector was obtained from Clontech Laboratories (Mountain View, CA). The miRNA expression vectors were generated by the BLOCK-iT lentiviral miRNA Expression System (Invitrogen) according to the manufacturer's instruction as previously described (Zhu et al., 2007). In addition, we also generated the scramble RFP-miR, RFP-miR-DCC and RFP-miR-neogenin expression vectors by replacing GFP with RFP in the EmGFP-miR and EmGFP-miR-DCC and EmGFP-miR-neogenin plasmids. The knockdown efficiency of newly generated plasmids was verified by western blot and sequencing analysis. The sequences for the DCC miRNA and neogenin miRNA constructs were as follows:
mouse DCC, 5′-GCTGAGAACAATCTGTAACCTTGGAGTTTTGGCCAC-TGACTGACTCCAAGGTCAGATTGTTCT-3′ and 5′-CCTGAGAACAATCTG-ACCTTGGAGTCAGTCAGTGGCCAAAACTCCAAGGTTACAGATTGTTCT-3′; mouse neogenin, 5′-TGCTGTAATGAAGCGAGTAGAGACCAGTTTTGG-CCACTGACTGACTGGTCTCTTCGCTTCAT-3′ and 5′-CCTGTAATGAAG-CGAAGAGACCAGTCAGTCAGTGGCCAAAACTGGTCTCTACTCGCTTCA-TTA-3′. The authenticity of all constructs was verified by DNA sequence.
Cell culture and transfection
NLT cells were grown in DMEM supplemented with 10% FBS, 100U/ml penicillin G and 100 μg/ml streptomysin (Invitrogen, GIBCO, Carlsbad, CA). Cells were cultured on glass coverslips precoated with 10 μg/ml collagen. For imaging experiments, 50–70% confluent NLT cells in 12-well plates were transfected with 1.6 μg of the indicated plasmids using 3 μl Lipofectamine 2000 in DMEM without FBS and antibiotics. For the co-transfection experiments, the molar ratio of GFP–Myo X to FLAG–DCC or FLAG–neogenin was 1:3 to ensure the expression of DCC and neogenin in GFP-positive cells. For western blot analysis, 48 hours after transfection, cells were lysed in modified RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM sodium chloride, 1% NP-40, 0.25% sodium deoxycholate and proteinase inhibitors). Lysates were separated by SDS-PAGE. The gel was transferred to vinylidene difluoride membrane and probed with the indicated antibodies.
Immunostaining, confocal imaging and quantification analysis
Cells were growing on the coverslip and fixed with 4% paraformaldehyde, 0.1% Triton X-100 in PBS for 15 minutes at room temperature. Fixed cells were blocked in TBS with 5% BSA at 37°C for 1 hour, then incubated with primary antibodies at 4°C for overnight. Cells were then washed and incubated with goat anti-rabbit or anti-mouse secondary antibodies coupled to Alexa Fluor 488 or Alexa Fluor 594 supplemented with 1 μM TO-PRO-3 for 50 minutes at room temperature. Finally, cells were mounted with FluorSave mounting medium (Invitrogen) to inhibit photobleaching. Images were acquired with a 63× NA 1.4 objective at a resolution of 1024×1024 pixels of the LSM510 META multiphoton confocal system (Zeiss). Image projection was developed by using corresponding LSM510 software. 3D images were developed using Volocity 8.0 software. For fluorescence intensity quantification, the projected Z-stack images were analyzed by ImageJ software (NIH, Bethesda, MD). Cells were selected randomly and the displayed data are representative of three experiments. Statistical evaluations for these and all other quantifications were performed with the software Graph Pad Prism version 4.0.
Live cell time-lapse imaging and quantification analysis
Transfected cells were grown on Delta T dishes (Biotechs) in DMEM supplemented with 10% FBS and antibiotics. For visualizing GFP–Myo X movement, the medium was replaced with F10 medium and sealed with a large coverslip. The dish was then placed in a temperature control system (Biotechs) that maintained a temperature of 37°C. Time-lapse intervals were 20 seconds, and cells were imaged over periods of 25–40 minutes. Image sequences were collected using a LSM510 META microscope with a 63× 1.4 N.A. objective. Movie files were created using LSM image examiner software. Distance plots were generated using the Trackpoints program of the Volocity 8.0 software. For velocity quantification, ImageJ and Microsoft Excel software were used to analyze 25–40 filopodia and/or 30–50 GFP–Myo X puncta from four to six different transfected cells that had a comparable level of the GFP fluorescence. The data were pooled and presented as mean ± s.d. from at least three independent experiments. The tip of a filopodium was estimated with phase images from the time-lapse series, and the puncta of GFP–Myo X were tracked by estimating the center of the fluorescent puncta.
Supplementary Material
Acknowledgements
We thank members of the Mei and Xiong laboratories for technical advice, helpful discussions and comments on the manuscript.
Footnotes
Funding
This study was supported by grant from the National Institutes of Health [grant number NS060648 to W.-C. X]. Deposited in PubMed for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.094946/-/DC1
References
- Berg J. S., Cheney R. E. (2002). Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nat. Cell Biol. 4, 246-250 [DOI] [PubMed] [Google Scholar]
- Berg J. S., Derfler B. H., Pennisi C. M., Corey D. P., Cheney R. E. (2000). Myosin-X, a novel myosin with pleckstrin homology domains, associates with regions of dynamic actin. J. Cell Sci. 113 Pt 19, 3439-3451 [DOI] [PubMed] [Google Scholar]
- Bohil A. B., Robertson B. W., Cheney R. E. (2006). Myosin-X is a molecular motor that functions in filopodia formation. Proc. Natl. Acad. Sci. USA 103, 12411-12416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D., Berg J. S., Cammer M., Chinegwundoh J. O., Dale B. M., Cheney R. E., Greenberg S. (2002). Myosin X is a downstream effector of PI(3)K during phagocytosis. Nat. Cell Biol. 4, 469-477 [DOI] [PubMed] [Google Scholar]
- Fazeli A., Dickinson S. L., Hermiston M. L., Tighe R. V., Steen R. G., Small C. G., Stoeckli E. T., Keino-Masu K., Masu M., Rayburn H., et al. (1997). Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature 386, 796-804 [DOI] [PubMed] [Google Scholar]
- Geiger B., Bershadsky A., Pankov R., Yamada K. M. (2001). Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2, 793-805 [DOI] [PubMed] [Google Scholar]
- Hirano Y., Hatano T., Takahashi A., Toriyama M., Inagaki N., Hakoshima T. (2011). Structural basis of cargo recognition by the myosin-X MyTH4-FERM domain. EMBO J. 30, 2734-2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang Y. S., Luo T., Xu Y., Sargent T. D. (2009). Myosin-X is required for cranial neural crest cell migration in Xenopus laevis. Dev. Dyn. 238, 2522-2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakoff S. J., Cardozo T., Andreev J., Li Z., Ferguson K. M., Abagyan R., Lemmon M. A., Aronheim A., Skolnik E. Y. (1998). Identification and analysis of PH domain-containing targets of phosphatidylinositol 3-kinase using a novel in vivo assay in yeast. EMBO J. 17, 5374-5387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keino-Masu K., Masu M., Hinck L., Leonardo E. D., Chan S. S., Culotti J. G., Tessier-Lavigne M. (1996). Deleted in Colorectal Cancer (DCC) encodes a netrin receptor. Cell 87, 175-185 [DOI] [PubMed] [Google Scholar]
- Kennedy T. E., Serafini T., de la Torre J. R., Tessier-Lavigne M. (1994). Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell 78, 425-435 [DOI] [PubMed] [Google Scholar]
- Knight P. J., Thirumurugan K., Xu Y., Wang F., Kalverda A. P., Stafford W. F., 3rd, Sellers J. R., Peckham M. (2005). The predicted coiled-coil domain of myosin 10 forms a novel elongated domain that lengthens the head. J. Biol. Chem. 280, 34702-34708 [DOI] [PubMed] [Google Scholar]
- Kupfer A., Louvard D., Singer S. J. (1982). Polarization of the Golgi apparatus and the microtubule-organizing center in cultured fibroblasts at the edge of an experimental wound. Proc. Natl. Acad. Sci. USA 79, 2603-2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K. H., Boerner S. A., Parsons J. T. (2002). Regulation of focal adhesion targeting and inhibitory functions of the FAK related protein FRNK using a novel estrogen receptor “switch”. Cell Motil. Cytoskeleton 51, 76-88 [DOI] [PubMed] [Google Scholar]
- Nagy S., Ricca B. L., Norstrom M. F., Courson D. S., Brawley C. M., Smithback P. A., Rock R. S. (2008). A myosin motor that selects bundled actin for motility. Proc. Natl. Acad. Sci. USA 105, 9616-9620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie S., Kee Y., Bronner-Fraser M. (2009). Myosin-X is critical for migratory ability of Xenopus cranial neural crest cells. Dev. Biol. 335, 132-142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi X., Ren R., Kelley R., Zhang C., Moser M., Bohil A. B., Divito M., Cheney R. E., Patterson C. (2007). Sequential roles for myosin-X in BMP6-dependent filopodial extension, migration, and activation of BMP receptors. J. Cell Biol. 179, 1569-1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantard L., Arjonen A., Lock J. G., Nurani G., Ivaska J., Stromblad S. (2010). PtdIns(3,4,5)P is a regulator of myosin-X localization and filopodia formation. J. Cell Sci. 123, 3525-3534 [DOI] [PubMed] [Google Scholar]
- Ren X. R., Ming G. L., Xie Y., Hong Y., Sun D. M., Zhao Z. Q., Feng Z., Wang Q., Shim S., Chen Z. F., et al. (2004). Focal adhesion kinase in netrin-1 signaling. Nat. Neurosci. 7, 1204-1212 [DOI] [PubMed] [Google Scholar]
- Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R. (2003). Cell migration: integrating signals from front to back. Science 302, 1704-1709 [DOI] [PubMed] [Google Scholar]
- Serafini T., Kennedy T. E., Galko M. J., Mirzayan C., Jessell T. M., Tessier-Lavigne M. (1994). The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell 78, 409-424 [DOI] [PubMed] [Google Scholar]
- Shekarabi M., Kennedy T. E. (2002). The netrin-1 receptor DCC promotes filopodia formation and cell spreading by activating Cdc42 and Rac1. Mol. Cell. Neurosci. 19, 1-17 [DOI] [PubMed] [Google Scholar]
- Sousa A. D., Cheney R. E. (2005). Myosin-X: a molecular motor at the cell's fingertips. Trends Cell Biol. 15, 533-539 [DOI] [PubMed] [Google Scholar]
- Sun Y., Sato O., Ruhnow F., Arsenault M. E., Ikebe M., Goldman Y. E. (2010). Single-molecule stepping and structural dynamics of myosin X. Nat. Struct. Mol. Biol. 17, 485-491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuo H., Ikebe M. (2004). Myosin X transports Mena/VASP to the tip of filopodia. Biochem. Biophys. Res. Commun. 319, 214-220 [DOI] [PubMed] [Google Scholar]
- Tokuo H., Mabuchi K., Ikebe M. (2007). The motor activity of myosin-X promotes actin fiber convergence at the cell periphery to initiate filopodia formation. J. Cell Biol. 179, 229-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeki N., Jung H. S., Sakai T., Sato O., Ikebe R., Ikebe M. (2011). Phospholipid-dependent regulation of the motor activity of myosin X. Nat. Struc. Mol. Biol. 18, 783-788 [DOI] [PubMed] [Google Scholar]
- Watanabe T. M., Tokuo H., Gonda K., Higuchi H., Ikebe M. (2010). Myosin-X induces filopodia by multiple elongation mechanism. J. Biol. Chem. 285, 19605-19614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber K. L., Sokac A. M., Berg J. S., Cheney R. E., Bement W. M. (2004). A microtubule-binding myosin required for nuclear anchoring and spindle assembly. Nature 431, 325-329 [DOI] [PubMed] [Google Scholar]
- Xie Y., Ding Y. Q., Hong Y., Feng Z., Navarre S., Xi C. X., Zhu X. J., Wang C. L., Ackerman S. L., Kozlowski D., et al. (2005). Phosphatidylinositol transfer protein-alpha in netrin-1-induced PLC signalling and neurite outgrowth. Nat. Cell Biol. 7, 1124-1132 [DOI] [PubMed] [Google Scholar]
- Yonezawa S., Kimura A., Koshiba S., Masaki S., Ono T., Hanai A., Sonta S., Kageyama T., Takahashi T., Moriyama A. (2000). Mouse myosin X: molecular architecture and tissue expression as revealed by northern blot and in situ hybridization analyses. Biochem. Biophys. Res. Commun. 271, 526-533 [DOI] [PubMed] [Google Scholar]
- Yu C., Feng W., Wei Z., Miyanoiri Y., Wen W., Zhao Y., Zhang M. (2009). Myosin VI undergoes cargo-mediated dimerization. Cell. 138, 537-548 [DOI] [PubMed] [Google Scholar]
- Zhang H., Berg J. S., Li Z., Wang Y., Lang P., Sousa A. D., Bhaskar A., Cheney R. E., Stromblad S. (2004). Myosin-X provides a motor-based link between integrins and the cytoskeleton. Nat. Cell Biol. 6, 523-531 [DOI] [PubMed] [Google Scholar]
- Zhu X. J., Wang C. Z., Dai P. G., Xie Y., Song N. N., Liu Y., Du Q. S., Mei L., Ding Y. Q., Xiong W. C. (2007). Myosin X regulates netrin receptors and functions in axonal path-finding. Nat. Cell Biol. 9, 184-192 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}