

Abstract
This study investigated the prevalence and the prognostic relevance of the 2 known telomere maintenance mechanisms (TMMs), telomerase activity (TA) and alternative lengthening of telomeres (ALT), in malignant peripheral nerve sheath tumors (MPNST). In 57 specimens from 49 patients with MPNST (35 sporadic, 14 neurofibromatosis type 1-related), TA was determined using the telomeric repeat amplification protocol, and ALT was detected by assaying ALT-associated promyelocytic leukemia bodies (APB) and terminal restriction fragment (TRF) length distribution. TA or ALT (defined on the basis of APB) alone was found in 24.6% or 26.3% of the lesions, respectively, whereas 6 cases (10.5%) were TA+/ALT+. A concordance between APB and TRF results in defining the ALT status was observed in 44 of 57 cases (77.2%; P < .0001). TA was more frequently expressed in samples from patients with neurofibromatosis type 1 than in those with sporadic disease (60% vs 29.4%, P = 0.087). In the overall series, TA proved to be prognostic for 5-year disease-specific death (hazard ratio, 3.78; 95% confidence interval [CI], 1.60–8.95; P = .002), even when adjusted for the presence of neurofibromatosis type 1 (hazard ratio, 4.22; 95% CI, 1.804–9.874; P = .001) and margin status after surgery (hazard ratio, 5.78; 95% CI, 2.19–15.26; P < .001). Conversely, ALT did not significantly affect clinical outcome of MPNST using either APB expression (hazard ratio, 1.25; 95% CI 0.54–2.89; P = 0.605) or TRF distribution (hazard ratio, 0.57; 95% CI, 0.17–1.96; P = .375) as the detection approach. Our results indicate for the first time that both TMMs, TA and ALT, are present in MPNST and differentially affect patient prognosis.
Keywords: alternative lengthening of telomeres, malignant peripheral nerve sheath tumors, schwannoma, telomerase, telomere maintenance mechanisms
Peripheral nerve sheath tumors constitute a group of relatively rare soft tissue neoplasms, which includes benign lesions deriving from specific neural tissues (neurofibromas and schwannomas) and malignant peripheral nerve sheath tumors (MPNSTs) that most commonly involve major nerve trunks.1 MPNSTs are highly aggressive malignancies that account for 3%–10% of all soft tissue sarcomas.2 Approximately 30%–60% of MPNSTs occur in the setting of neurofibromatosis type 1 (NF1), a hereditary tumor syndrome, and are the leading cause of NF1-related mortality.3–6 The remainder of MPNSTs develop sporadically. In patients with NF1, MPNSTs usually arise in the presence of a neurofibroma or, very rarely, a schwannoma.7
Complete surgical excision is the mainstay of MPNST therapy and represents the primary curative modality. Adjuvant chemotherapy and radiation therapy are also often used. However, despite aggressive treatment, local recurrence and metastases are common, leading to a poor prognosis for MPNST patients, with 5-year survival rates in the range of 20%–50%. These findings strongly indicate an urgent need for improved therapeutic approaches able to significantly impact the disease outcome.8
MPNST pathogenesis is poorly understood mainly because of its complex histopathology. A number of molecular and genetic alterations have been found in relation to these tumors.9–12 However, there is no defined molecular signature for MPNST development, although recent work in the field of MPNST molecular pathobiology has identified several altered receptor tyrosine kinase-mediated intracellular signal transduction cascades, posing the possibility of using personalized, targeted therapeutics for the disease.13
The activation of a telomere maintenance mechanism (TMM) is essential for tumor cells to counteract the normal limit on cell proliferation resulting from progressive telomere shortening that normally accompanies each round of cell division.14 Indeed, limitless proliferative potential is a hallmark of cancer cells. There are 2 known TMMs: telomerase15 and alternative lengthening of telomeres (ALT).16 Telomerase is a ribonucleoprotein complex containing an RNA subunit, hTR, that provides the template for the synthesis of telomeric DNA by the catalytic subunit, hTERT.17 Approximately 85%–90% of all tumors express telomerase,18 whereas most of the remaining tumors, mainly those of mesenchymal and neuroepithelial origin,19–26 rely on ALT to maintain their telomeres.
Although the molecular details of ALT are incompletely understood, previous studies have shown that it is consistent with a recombination-dependent DNA replication mechanism.16 Characteristics of ALT cells include an extremely heterogeneous telomere length distribution, ranging from very short to more than 50 kb, and the presence of subnuclear structures termed ALT-associated promyelocytic leukemia (PML) bodies (APBs).16 PML bodies are normal nuclear domains of unknown function that contain the PML protein. In ALT cells, a subset of the PML bodies contains telomeric chromatin (telomeric DNA and the telomere binding proteins TRF-1, TRF-2, TIN2, and RAP1) and other proteins involved in DNA replication, recombination, and repair.16 Another specific feature of ALT mechanisms is the presence of C-circles (self-priming circular telomeric DNA), which may represent molecular intermediates of the ALT mechanism, and whose specificity for assessing ALT activity in biological samples has recently been demonstrated.27
Because the presence of an active TMM is an almost universal feature of cancer and normal cells do not have sufficient levels of TMM activity to counteract telomere shortening, in recent years, TMMs have been suggested as attractive new targets for anti-cancer therapies,28,29 particularly for those tumor types, such as MPNST, that are refractory to conventional therapeutic interventions. In this context, it is important to know whether individual MPNSTs use telomerase or ALT to maintain their telomeres. In a previously published study, telomerase activity was found to be present in approximately 60% of MPNST samples derived from NF1-affected patients.30 However, no information is currently available concerning the presence of ALT in MPNSTs. Taking advantage of a relatively large mono-institutional series of patients with MPNST with long follow-up, we propose to investigate the prevalence of telomerase activity (TA) and ALT and whether they contribute to clinical progression in this disease.
Materials and Methods
Study Population
A total of 57 lesions taken from 49 adult patients (median age, 40 years; range, 18–90 years) treated at the Fondazione IRCCS Istituto Nazionale dei Tumori of Milan (INT) from November 1990 through January 2007 were available for analysis. Fourteen patients had NF1 with an associated MPNST, and 35 were classified as having sporadic tumors because they had no clinical signs or family history of NF1. The specimens, stored in the Institutional Tissue Bank, were consecutive with respect to the availability of frozen tissue for TMM studies and adequate clinicopathologic and follow-up information. Six patients developed recurrence and/or metastasis during the follow-up period, and the corresponding 8 lesions were collected and included in the analysis. Patient and tumor characteristics are summarized in Table 1. The median follow-up of the entire group, was 46 months (range, 3–240 months). During the follow-up period, 23 patients died of cancer-related causes. Fourteen schwannoma lesions, obtained from 14 patients who underwent surgery, were included in our research for comparative purposes.
Table 1.
Patient and tumor characteristics
| Characteristic | No. of patients (%) |
|---|---|
| Total | 49 (100%) |
| Gender: | |
| Female | 20 (41.0%) |
| Male | 29 (59.0%) |
| Syndrome: | |
| Sporadic | 34 (69.4%) |
| Neurofibromatosis-1 | 15 (30.6%) |
| Site | |
| Trunk | 17 (34.7%) |
| Extremities | 32 (65.3%) |
| Type of lesion at first presentation | |
| Primary | 42 (85.7%) |
| Recurrence | 4 (8.2%) |
| Metastasis | 3 (6.1%) |
| Size (cm) | |
| <5 | 10 (20.4%) |
| ≥5 | 36 (73.5%) |
| Missing | 3 (6.1%) |
| Grade (FNCLCC): | |
| 1 | 5 (10.2%) |
| 2 | 14 (28.6%) |
| 3 | 23 (46.9%) |
| Missing | 7 (14.3%) |
| Post-surgery treatment | |
| Surgery only | 20 (40.8%) |
| Chemotherapy | 11 (22.4%) |
| Radiotherapy | 11 (22.4%) |
| Chemotherapy + radiotherapy | 5 (10.2%) |
| Missing | 2 (4.1%) |
The study was approved by the Institutional Review Board of INT, and each patient provided written informed consent to donate to the Institute the tissues left over after diagnostic procedures.
Molecular Studies
Normal and tumor tissues were sampled by a pathologist at the time of surgery, flash-frozen in liquid nitrogen, and stored at −80°C. Diagnosis and sampling adequacy were pathologically confirmed on H&E-stained slides. A fragment of 70–100 mg was cut from each lesion and further subdivided for APB detection, protein extraction for TA assay, and DNA extraction for telomere length assessment and array-CGH.
Detection of APB
Frozen sections were cut to 5–7 μm thickness, fixed in 1:1 methanol/acetone, processed to detect APB by combined PML immunofluorescence and telomere fluorescence in situ hybridization,23 and independently scored by 2 observers. Images were captured on a Nikon Eclipse Ti fluorescence microscope using a Volocity 5.3 (Perkin-Elmer) image analysis. APB status was determined according to previously defined criteria.23 The presence of an APB was defined by the localization of a telomeric DNA focus in a nuclear PML body; sections were scored as APB positive if they contained APB in 0.5% or more of tumor cells, and a tumor was considered to be ALT positive when at least one section was APB positive. To avoid false-positive results, an APB was considered to be present only when the telomeric DNA fluorescence within a PML body was more intense than that of telomeres, and a cell was not considered to contain APB if more than 25% of the colocalized foci occurred outside the nucleus. To avoid false-negative results, at least 2000 tumor nuclei were examined, and the assay was repeated in the presence of negative results. An ALT-positive (U2OS) cell line was used as a positive control. To assess whether APB were present in tumor cells and not in admixed stromal cells, we simultaneously performed indirect immunofluorescence for NF1 protein on the frozen sections using a commercial antibody (sc-67; Santa Cruz Biotechnology).31
TA Detection Assay
TA was measured on 0.6 and 6 µg of protein by the telomeric repeat amplification protocol (TRAP)32 with the TRAPeze kit (Intergen Company). A tumor was scored as TA positive when positive TRAP results were obtained for at least one protein concentration. In the case of tumors negative at both protein concentrations, the TRAP assay was repeated to avoid false-negative results.
TRF Analysis
Total DNA was isolated using QuicKpicK genomic DNA kit (BioNobile), digested with a HinfI restriction enzyme (Promega), and further processed by pulsed-field gel electrophoresis, as previously described.33 ALT status was determined by calculating whether the mean, variance, and semi-interquartile range of TRF length distribution were greater than 16 kb, 1000 kb2, and 4 kb, respectively. Tumors were classified as ALT positive when 2 or 3 of these 3 criteria were met for unimodal or bimodal TRF length distributions, respectively. Statistical analysis of TRF length distributions was done with Telometric software.34
Array CGH
The analysis was performed on genomic DNA derived from 31 MPNST (16 NF1-related, 15 sporadic) lesions, using the Agilent's Human Genome CGH Array 44K (Agilent Technologies). Total DNA was isolated with QuicKpicK genomic DNA kit (BioNobile). For each CGH hybridization, 1 μg of genomic DNA from the experimental sample and suitable reference DNA (female XX or male XY, Promega) was digested with AluI and RsaI restriction enzyme (Promega). Labeling reactions were performed using Agilent Genomic DNA Enzymatic Labeling Kit according to the manufacturer's instructions with a modified dNTP pool containing Cy5-dUTP (for the experimental sample) or Cy3-dUTP (for the reference). Labeled samples and references were subsequently filtered by using a Microcon YM-30 column (Millipore). Specific activity was calculated for each sample as a ratio between dyes pmol and μg of DNA; then, experimental and reference targets for each hybridization were pooled and mixed with human Cot-1 DNA (Invitrogen). The samples were loaded in a hybridization chamber, with a clean Gasket Slide and a Microarray Slide (Agilent Technologies). Then hybridization was performed for 24 h at 65°C in a rotating oven. The arrays were then disassembled and washed in specific buffers (Agilent Technologies) according to the manufacturer's instructions; then, slides were dried and scanned using Agilent Scanner (Agilent Technologies). Microarray images were extracted and analyzed using an Agilent Feature Extraction Software. Data from the arrays were analyzed using Agilent CGH Analytic Software 3.4 (Agilent Technologies).
Statistical Analysis
The clinical endpoint of this study was disease-specific survival, and the time of its occurrence was computed from the date of surgery to the time of death or censored at the date of the last recorded follow-up for living patients. Survival curves were estimated by means of the Kaplan-Meier product limit method,35 and the Cox proportional hazards model36 was used to calculate hazard ratios (HR) and their 95% confidence interval (95% CI). Fisher's and χ2 exact tests were used to assess the relationship between TMM status and clinico-pathological features. All P values were 2-sided, and values ≤.05 were considered to be statistically significant. The agreement between APB and TRF data was assessed by kappa statistics.
Results
Fifty-seven frozen tumor samples obtained from 49 patients with MPNST were assayed for the presence of TMM (Table 2). TA was detected by the TRAP assay (Fig. 1A), and tumors were defined as ALT+ on the basis of APB presence in at least 10 (0.5%) of 2000 tumor cells (Fig. 1B). Thirty-five lesions (61.4%) expressed at least one TMM. Specifically, 14 lesions (24.6%) were TA+/ALT−, 15 (26.3%) were TA−/ALT+, and 6 (10.5%) were defined as TA+/ALT+ because of the concomitant expression of APB and TA. A consistent fraction of tested samples (22/57, 38.6%) did not express any known TMM.
Table 2.
Telomerase activity and ALT mechanisms in MPNST lesions
| Case | Lesion | TA | APB | TRF length | ||
|---|---|---|---|---|---|---|
| 1 | NF1 | 1 | R | + | − | − |
| 2 | NF1 | 2A | M | + | − | − |
| 2B | M | + | + | − | ||
| 3 | NF1 | 3A | R | + | + | − |
| 3B | R | − | − | − | ||
| 4 | NF1 | 4A | P | + | − | − |
| 4B | R | − | + | − | ||
| 5 | NF1 | 5A | P | − | + | − |
| 5B | R | − | − | − | ||
| 6 | NF1 | 6 | P | + | − | − |
| 7 | NF1 | 7 | P | − | − | − |
| 8 | NF1 | 8 | P | + | − | − |
| 9 | NF1 | 9 | P | − | + | + |
| 10 | NF1 | 10 | P | + | − | − |
| 11 | NF1 | 11 | P | + | − | − |
| 12 | NF1 | 12 | P | + | + | − |
| 13 | NF1 | 13 | P | − | − | − |
| 14 | NF1 | 14 | P | − | − | − |
| 15 | NF1 | 15 | P | − | + | + |
| 16 | sporadic | 16 | P | − | − | − |
| 17 | sporadic | 17 | P | − | + | + |
| 18 | sporadic | 18A | R | + | − | − |
| 18B | R | − | + | − | ||
| 18C | R | − | − | − | ||
| 18D | R | − | − | − | ||
| 19 | sporadic | 19 | R | − | + | − |
| 20 | sporadic | 20 | P | − | − | − |
| 21 | sporadic | 21 | M | + | + | − |
| 22 | sporadic | 22 | P | − | + | − |
| 23 | sporadic | 23A | P | − | − | − |
| 23B | M | − | + | + | ||
| 24 | sporadic | 24 | P | + | + | − |
| 25 | sporadic | 25 | P | − | − | − |
| 26 | sporadic | 26 | M | + | − | − |
| 27 | sporadic | 27 | P | − | + | + |
| 28 | sporadic | 28 | P | − | − | − |
| 29 | sporadic | 29 | P | − | + | + |
| 30 | sporadic | 30 | P | + | − | − |
| 31 | sporadic | 31 | P | − | + | − |
| 32 | sporadic | 32 | P | − | − | − |
| 33 | sporadic | 33 | P | − | + | − |
| 34 | sporadic | 34 | P | + | − | − |
| 35 | sporadic | 35 | P | + | − | − |
| 36 | sporadic | 36 | P | + | + | + |
| 37 | sporadic | 37 | P | − | − | − |
| 38 | sporadic | 38 | P | − | − | − |
| 39 | sporadic | 39 | P | + | − | − |
| 40 | sporadic | 40 | P | − | − | − |
| 41 | sporadic | 41 | P | − | − | − |
| 42 | sporadic | 42 | P | − | + | + |
| 43 | sporadic | 43 | P | − | − | − |
| 44 | sporadic | 44 | P | − | + | − |
| 45 | sporadic | 45 | P | − | − | − |
| 46 | sporadic | 46 | P | + | − | − |
| 47 | sporadic | 47 | P | − | − | − |
| 48 | sporadic | 48 | P | − | − | − |
| 49 | sporadic | 49 | P | − | − | − |
NF1, Neurofibromatosis type 1; P, primary disease; R, recurrent locoregional tumor; M, metastatic lesion; TA, telomerase activity; APB, alternative lengthening of telomeres assessed on the presence of APB (ALT-associated promyelocytic leukaemia nuclear bodies); TRF, alternative lengthening of telomeres assessed on the basis of terminal restriction fragment length distribution.
Fig. 1.

(A) 2 representative specimens of MPNST in which telomerase activity was detected by the TRAP assay using 0.6 and 6 μg of protein. The location of the internal amplification standard (ITAS) is indicated. (B) APB assay in MPNST: combined PML immunofluorescence and telomere fluorescence in situ hybridization in a frozen section of a representative ABP-positive MPNST sample. Indirect immunofluorescence was used for the PML protein (FITC label, green stain); telomere FISH was done using a Cy3 conjugated telomeric peptide nucleic acid probe (red stain); indirect immunofluorescence was used to detect NF1 protein (far-red stain); nuclei were counterstained with 4′,6-diamidino-2-phenylindole, DAPI (blue stain); the foci of telomeric DNA that colocalize with PML represent APBs (white arrows). (C). Terminal restriction fragment (TRF) profiles of 4 representative MPNST specimens.
In ALT+ samples, APBs were observed in a variable but always limited fraction of cells, ranging from 0.5% to 5.7% (mean value, 1.29%). Specifically, the percentage of APB-positive cells ranged from 0.59% to 5.7% (mean value, 1.4%) and from 0.5% to 2.15% (mean value, 0.99%) in TA−/ALT+ and TA+/ALT+ group, respectively. For all 57 samples, results of TRF analysis (Fig. 1C) were also available, and we found concordance between APB and TRF results in defining the ALT status in 44 cases (77.2%; kappa = 0.437; 95% CI, 0.215–0.659; P < .0001). Specifically, 8 lesions (14.0%) were defined as ALT+, and 36 (63.2%) were defined as ALT− with both detection methods, whereas the remaining 13 lesions (22.8%) were defined as ALT+ on the basis of APB expression but did not show a TRF length distribution suggestive of ALT.
In 6 patients, more than 1 lesion was available for TMM investigation. In 3 cases in which both the primary tumor and subsequent lesions were studied, the TA status was concordant (Table 2, cases 2, 5, and 23), and for the other 3 patients, TA status changed during the course of the disease (Table 2, cases 3, 4, and 18). With regard to ALT status, the presence of APB varied in all the patients with metachronous lesions, whereas the TRF phenotype remained stable in 5 of 6 patients (Table 2). The presence of ALT, as determined on the basis of APB presence, was unrelated to gender (P = .966), age (P = .201), tumor location (P = .826), grade (P = .999), size of the lesion (P = 1.000), or NF1 syndrome (P = .964). Such findings were also observed when ALT was determined on the basis of TRF analysis (data not shown). With regard to TA, it did not prove to correlate with gender (P = .298), age (P =.636), tumor location (P = 1.000), grade (P = .603), or size of the lesion (P = .885). However, TA was found to be more frequently expressed in samples from patients with NF1-related MPNST than in those with sporadic disease (60.0% vs 29.4%; P = .087).
With regard to the 14 schwannoma lesions, none of them expressed TA, whereas 2 lesions were scored as ALT+ on the basis of APBs (0.52% and 1.11% APB-positive cells), although they did not show a TRF distribution pattern consistent with ALT.
Array CGH data, obtained in a subset of 31 MPNST samples, both sporadic and NF1-associated, exhibited significant DNA copy number changes, with variations in size, span, and nature of the aberrations on the whole genome (data not shown). In particular, relevant to TMMs, we could detect a copy number gain of hTERT gene on chromosome 5 in 20 (65%) of 31 samples, with a similar percentage in NF1-associated (69%) and sporadic (60%) MPNSTs (Fig. 2A). When considering the prevalence of hTERT aberration in relation to the TMM operating in the tumor, we found a slightly higher frequency of copy number gain of hTERT gene in TA+/ALT− tumors (7/10, 70%) that in TA−/ALT+ tumors (5/9, 56%). In addition, aCGH results for NF1 gene on chromosome 17q11 revealed no significant differences between NF1-associated and sporadic tumors with a copy number loss of NF1 gene in 9 (56%) of 16 NF1-related MPNSTs and in 8 (53%) of 15 sporadic lesions (Fig. 2B).
Fig. 2.
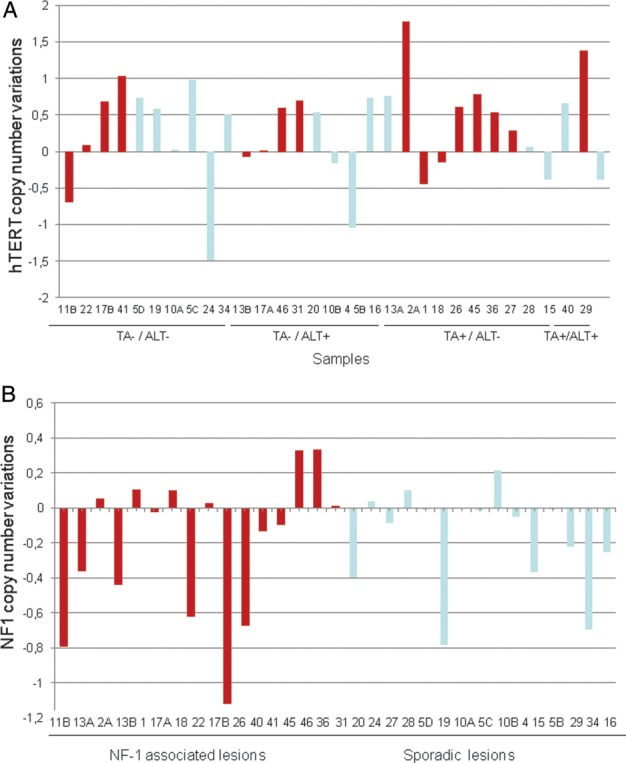
Copy number variation of hTERT gene (A) and NF1 gene (B) in a subset of 31 MPNSTs. Samples arising in the context of NF1 syndrome are highlighted in red.
Clinical outcome was analyzed on the overall series of 49 patients with MPNST. After 5 years of follow-up, 22 patients had died of the disease, and only 1 additional patient died in the 5–10 years interval, which corresponds to survival estimates of 47% and 39%, respectively. From the survival analyses, we found a correlation between the status of microscopic surgical margins and disease-specific survival in univariable analysis (Margin status: positive (negative): HR, 4.11; 95% CI, 1.67–10.10; P = .002) (Supplementary Table S1). Conversely, age, presence of NF1 syndrome, location of the tumor, size, and grade did not show any statistically significant correlation with patients' prognosis (Supplementary Table S1). With regard to relevance of TMMs, for 6 patients who experienced progressive disease (recurrence or metastasis) and whose ALT/TA status changed during the course of the disease, the appearance of any TMM defined the final phenotype (if metachronous lesions were ALT+ and ALT−, the patient would be categorized as ALT+). At 5 years of follow-up, TA proved to be significantly associated with disease-specific mortality. Specifically, results obtained from univariable analysis showed that patients with TA+ tumors had a significantly lower probability of being alive than patients with TA− (HR, 3.78; 95% CI, 1.60–8.95; P = .002) (Fig. 3A). Such findings held true also when adjusted for the presence of NF1 syndrome (HR, 4.22; 95% CI, 1.804–9.847; P = .001) and for the margin status after surgical excision (HR, 5.78; 95% CI, 2.19–15.26; P < .001) (Supplementary Table S1).
Fig. 3.

Disease-specific survival as a function of TA (A) and ALT (B) in patients with MPNST.
Conversely, ALT alone did not prove to be associated with disease-specific mortality either using APB expression (HR, 1.25; 95% CI, 0.54–2.89; P = .605) (Fig. 3B) or TRF distribution analysis (HR, 0.57; 95% CI, 0.17–1.96; P = .375) to define ALT status.
Discussion
This is the first report of a comparative analysis of the expression and clinical relevance of the 2 currently known TMMs, ALT, and TA, in a mono-institutional series of MPNSTs, including both sporadic and NF1-associated tumors. Overall, TA was detected in 35.1% of the lesions, although it was found to be more frequently expressed in samples from patients with NF1-associated MPNST than in those with sporadic disease (60.0% vs 29.4%). This observation is consistent with previous data from Mantripragada et al.,30 who recorded TA in 61% of MPNST samples derived from NF1-affected patients. The authors also showed that all TA+ lesions were high-grade MPNSTs. Conversely, in our case series, we were able to detect TA also in a low-grade sporadic MPNST lesion (out of 5 grade 1 lesions tested). Through a genome–wide high resolution analysis of DNA copy number alterations in NF1-associated MPNSTs and benign tumors, the same research group30 was able to reveal amplification of the hTERT gene in malignant tumors but not in benign lesions. The array CGH results we generated in a subset of 31 MPNSTs showed that such an amplification was also present in sporadic MPNSTs, in a percentage of cases superimposable to that found in NF1-associated tumors (60% vs 69%). The similar genetic alterations that we observed in sporadic and NF1-associated MPNST (as outlined in hTERT and NF1 gene) are not surprising. Indeed, Brekke et al.37 recently suggested that the genetic events leading to MPNST are comparable in NF-related and sporadic tumors. Of note, the observed amplification of hTERT gene is in agreement with gene upregulation in MPNSTs, as previously detected by real-time reverse-transcriptase polymerase chain reaction.38 No clear correlation between hTERT copy number gain and TA was evident, because such a genomic alteration was also found in half of the ALT+ lesions and in a high percentage of TA−/ALT− tumors.
ALT was found in 36.8% of cases when APB expression was used as the detection approach, and the percentage of ALT+ tumors decreased to 14% when the ALT phenotype was determined on the basis of TRF length distribution. The incomplete overlapping of the results obtained with the 2 methods is not surprising. In fact, although the APB assay allows the analysis of individual tumor cells, the TRF pattern could be misleading because of the admixture of normal and tumor cells present in the specimens. However, on the basis of available data, the concordance rate between the 2 assays seems to be dependent on the tumor type. Specifically, the 77.2% concordance rate between the 2 approaches in defining MPNST specimens as ALT+ or ALT− is superimposable to that (77.6%) previously observed in a series of 85 liposarcoma specimens that were comparatively assayed for ALT with the 2 detection methods.39 Conversely, Henson et al.23 reported a complete agreement in the results of the 2 assays in glioblastoma multiforme. The observation that MPNSTs use either TA or ALT to maintain their telomeres suggest that subsets of tumors can undergo different pathways of tumorigenesis. In this context, we previously demonstrated that distinct gene expression signatures can distinguish tumors and cell lines that are TA+ from those that are ALT+.40
As previously reported for other tumor types,19,20,22,24,25 in a few MPNSTs we found a concomitant expression of TA and ALT, confirming the possibility that the 2 TMMs can coexist in the same lesion. However, at present, it is unclear whether TA and ALT can be present in the same tumor cell or whether a given tumor lesion may contain distinct ALT+ and TA+ subpopulations. On the other hand, we found that a significant percentage of MPNSTs (approximately 40%) possessed no apparent TMM despite being informative for the different assays, suggesting that the presence of a constitutively active TMM is not a stringent requirement for a subset of MPNSTs or, alternatively, that these tumors use a mechanism that has not yet been identified. Moreover, such a lack of any known TMM, also previously observed in subsets of other tumor types,19,20,24,25 is in accord with experimental data suggesting that TMM acquisition is not always required for malignant transformation of normal human cells.41 However, the possibility that the lack of TMM expression observed in a high percentage of the tested MPNSTs may be related, at least in part, to the sensitivity of the assays used cannot be excluded.
The activation of a TMM is able to overcome replicative senescence induced by cell division-associated telomere attrition. In a previous study performed in a series of mesenchymal tumors, including 16 MPNSTs, and aimed to understand the contribution of senescence signaling to the biology of these tumors, we used a senescence scoring approach based on expression profiling of well-defined senescence markers as a means to evaluate latent senescence pathways. Specifically, a DNA damage associated signature (DAS) and a modified secretory senescence signature (mSS) were used. Our results indicated that, at an individual tumor level, DAS and mSS scores were not correlated, suggesting that senescence phenotypes may be differentially active during transformation.42
Concerning schwannomas, we did not found evidence of TA in any of the 14 samples tested, in agreement with previous data by Chen et al.,43 who found all 30 of their schwannoma samples to be TA. The authors also characterized the specimens in term of TRF length distribution analysis and could identify 4 lesions having elongated telomeres: 3 of them showed an aggressive clinicopathological behavior. In addition, one patient died of the disease, and one experienced a clinical recurrence. In our case series, no schwannoma lesion could be defined as ALT+ on the basis of TRF results, but 2 lesions were defined as ALT+ based on APB expression. Unfortunately, because no follow-up data are available for these patients, we cannot estimate the possible relevance of the ALT phenotype for the clinical outcome.
With regard to the prognostic relevance of TMMs in MPNST, TA proved to be prognostic for disease-specific survival. Specifically, patients with TA+ tumors had a significantly lower probability of survival 5 years after surgery than did patients with TA− tumors. In addition, TA proved to be a strong prognostic discriminant of increased mortality even when adjusted for the concomitant presence of NF1 syndrome and for margin status after surgical excision. Results available in the literature indicate an association between telomerase activity/expression and poor prognosis in different tumor types, including breast, non-small cell lung cancer, gastric and colorectal cancer, and neuroblastoma,44 although not all the published studies confirmed such an association.
In our series of MPNST patients, ALT failed to significantly affect clinical outcome, independently of the detection method used. Based on available information, the association of ALT with patient prognosis seems to be disease-related. In glioblastoma multiforme, a better survival for patients with a ALT+ tumor was consistently observed in 3 studies in which ALT phenotype was detected by TRF analysis or the presence of APB.19,23,26 On the other hand, in patients with liposarcoma, we found that ALT was a stronger prognostic discriminant of increased mortality than TA in both univariable and multivariable analysis.24 Again, we reported a negligible role for ALT in the prognosis of patients with diffuse malignant peritoneal mesothelioma.25 A tentative explanation for the different outcomes may be that ALT activation results from different sets of tumor-specific genetic changes that are correlated with a better or worse prognosis as a function of the tumor type.
The lack of reliable therapeutic options, other than complete surgery, for MPNST together with our observation that this tumor may use either TA or ALT to maintain telomeres, suggests the opportunity to consider TMMs as new therapeutic targets for the disease. In this context, the characterization of the TMM operating in individual tumors could allow the identification of patients suitable for treatment with anti-telomerase drugs, which are currently used in clinical practice.45 In addition, preclinical evidence indicating that ALT+ tumor models are sensitive to compounds that induce telomere dysfunction by binding to G-quadruplex structures in telomeric DNA and inhibit tumor growth through a mechanism largely independent of the presence of active telomerase,29 would suggest the possibility, in the near future, to treat patients with ALT+ MPNST with these compounds as soon as they reach the clinical setting.
Supplementary Material
Funding
L.V. was supported by a fellowship from the Fondazione Italiana per la Ricerca sul Cancro. The authors also thank the International Union Against Cancer (UICC).
This work was partially supported by grants from the Associazione Italiana per la Ricerca sul Cancro, from the European Community (LSHC-CT-2005-018806), and by funds obtained through a law by the Italian government that allows Italian citizens to allocate a share of their tax payment to support a research or charitable institution of their choice. We wish to thank all those citizens who decided to donate their payment to the IRCCS Fondazione Istituto Nazionale dei Tumori, Milano.
Supplementary Material
Acknowledgments
We thank L. Gioiosa for skilled technical assistance and the Institutional Tissue Bank and Dr. S. Veneroni for sample retrieval.
Conflict of interest statement. The authors declare no potential conflict of interest.
References
- 1.Abreu E, Aubert S, Wavreille G, Gheno R, Canella C, Cotten A. Peripheral tumor and tumor-like neurogenic lesions. Eur J Radiol. doi: 10.1016/j.ejrad.2011.04.036. [published online ahead of print May 10, 2011]. http://dx.doi.org/10.1016/j.ejrad.2011.04.036 . [DOI] [PubMed] [Google Scholar]
- 2.Perrin RG, Guha A. Malignant peripheral nerve sheath tumors. Neurosurg Clin N Am. 2004;15:203–216. doi: 10.1016/j.nec.2004.02.004. doi:10.1016/j.nec.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62(5):1573–1577. [PubMed] [Google Scholar]
- 4.Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311–314. doi: 10.1136/jmg.39.5.311. doi:10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anghileri M, Miceli R, Fiore M, et al. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer. 2006;107(5):1065–1074. doi: 10.1002/cncr.22098. doi:10.1002/cncr.22098. [DOI] [PubMed] [Google Scholar]
- 6.Stucky CC, Johnson KN, Gray RJ, et al. Malignant Peripheral Nerve Sheath Tumors (MPNST): The Mayo Clinic Experience. Ann Surg Oncol. 2012;19(3):878–885. doi: 10.1245/s10434-011-1978-7. [DOI] [PubMed] [Google Scholar]
- 7.Woodruff JM, Selig AM, Crowley K, Allen PW. Schwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumor. Am J Surg Pathol. 1994;18(9):882–895. doi: 10.1097/00000478-199409000-00003. doi:10.1097/00000478-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Grobmyer SR, Reith JD, Shahlaee A, et al. Malignant peripheral nerve sheath tumor: molecular pathogenesis and current management considerations. Journal of Surgical Oncology. 2008;97:340–349. doi: 10.1002/jso.20971. doi:10.1002/jso.20971. [DOI] [PubMed] [Google Scholar]
- 9.Holtkamp N, Atallah I, Okuducu AF, et al. MMP-13 and p53 in the progression of malignant peripheral nerve sheath tumors. Neoplasia. 2007;9(8):671–677. doi: 10.1593/neo.07304. doi:10.1593/neo.07304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birindelli S, Perrone F, Oggionni M, et al. Rb and TP53 pathway alterations in sporadic and NF1-related malignant peripheral nerve sheath tumors. Lab Invest. 2001;81(6):833–844. doi: 10.1038/labinvest.3780293. [DOI] [PubMed] [Google Scholar]
- 11.Upadhyaya M, Kluwe L, Spurlock G, et al. Germline and somatic NF1 gene mutation spectrum in NF1-associated malignant peripheral nerve sheath tumors (MPNSTs) Hum Mutat. 2008;29(1):74–82. doi: 10.1002/humu.20601. doi:10.1002/humu.20601. [DOI] [PubMed] [Google Scholar]
- 12.Mantripragada KK, de Ståhl TD, Patridge C, et al. Genome-wide high-resolution analysis of DNA copy number alterations in NF1-associated malignant peripheral nerve sheath tumors using 32K BAC array. Genes Chromosomes Cancer. 2009;48(10):897–907. doi: 10.1002/gcc.20695. doi:10.1002/gcc.20695. [DOI] [PubMed] [Google Scholar]
- 13.Katz D, Lazar A, Lev D. Malignant peripheral nerve sheath tumour (MPNST): the clinical implications of cellular signalling pathways. Expert Rev Mol Med. 2009;11:e30. doi: 10.1017/S1462399409001227. doi:10.1017/S1462399409001227. [DOI] [PubMed] [Google Scholar]
- 14.Gilson E, Géli V. How telomeres are replicated. Nat Rev Mol Cell Biol. 2007;8(10):825–838. doi: 10.1038/nrm2259. doi:10.1038/nrm2259. [DOI] [PubMed] [Google Scholar]
- 15.Blackburn EH. Telomeres and telomerase: the means to the end (Nobel lecture) Angew Chem Int Ed Engl. 2010;49(41):7405–7421. doi: 10.1002/anie.201002387. doi:10.1002/anie.201002387. [DOI] [PubMed] [Google Scholar]
- 16.Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11(5):319–330. doi: 10.1038/nrg2763. doi:10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- 17.Mitchell M, Gillis A, Futahashi M, Fujiwara H, Skordalakes E. Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat Struct Mol Biol. 2010;17(4):513–518. doi: 10.1038/nsmb.1777. doi:10.1038/nsmb.1777. [DOI] [PubMed] [Google Scholar]
- 18.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33(5):787–791. doi: 10.1016/S0959-8049(97)00062-2. Review doi:10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 19.Hakin-Smith V, Jellinek DA, Levy D, et al. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet. 2003;361(9360):836–838. doi: 10.1016/s0140-6736(03)12681-5. doi:10.1016/S0140-6736(03)12681-5. [DOI] [PubMed] [Google Scholar]
- 20.Ulaner GA, Huang HY, Otero J, et al. Absence of a telomere maintenance mechanism as a favourable prognostic factor in patients with osteosarcoma. Cancer Res. 2003;63:1759–1763. [PubMed] [Google Scholar]
- 21.Ulaner GA, Hoffman AR, Otero J, et al. Divergent patterns of telomere maintenance mechanisms among human sarcomas: sharply contrasting prevalence of the alternative lengthening of telomeres mechanism in Ewing's sarcomas and osteosarcomas. Genes Chromosomes Cancer. 2004;41:155–162. doi: 10.1002/gcc.20074. doi:10.1002/gcc.20074. [DOI] [PubMed] [Google Scholar]
- 22.Johnson JE, Varkonyi RJ, Schwalm J, et al. Multiple mechanisms of telomere maintenance exist in liposarcomas. Clin Cancer Res. 2005;11(15):5347–5355. doi: 10.1158/1078-0432.CCR-05-0684. doi:10.1158/1078-0432.CCR-05-0684. [DOI] [PubMed] [Google Scholar]
- 23.Henson JD, Hannay JA, McCarthy SW, et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res. 2005;11:217–225. [PubMed] [Google Scholar]
- 24.Costa A, Daidone MG, Daprai L, et al. Telomere maintenance mechanisms in liposarcomas: association with histologic subtypes and disease progression. Cancer Res. 2006;66(17):8918. doi: 10.1158/0008-5472.CAN-06-0273. doi:10.1158/0008-5472.CAN-06-0273. [DOI] [PubMed] [Google Scholar]
- 25.Villa R, Daidone MG, Motta R, et al. Multiple mechanisms of telomere maintenance exist and differentially affect clinical outcome in diffuse malignant peritoneal mesothelioma. Clin Cancer Res. 2008;14(13):4134–4140. doi: 10.1158/1078-0432.CCR-08-0099. doi:10.1158/1078-0432.CCR-08-0099. [DOI] [PubMed] [Google Scholar]
- 26.McDonald KL, McDonnell J, Muntoni A, et al. Presence of alternative lengthening of telomeres mechanism in patients with glioblastoma identifies a less aggressive tumor type with longer survival. J Neuropathol Exp Neurol. 2010;69(7):729–736. doi: 10.1097/NEN.0b013e3181e576cf. doi:10.1097/NEN.0b013e3181e576cf. [DOI] [PubMed] [Google Scholar]
- 27.Henson JD, Cao Y, Huschtscha LI, et al. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat Biotechnol. 2009;27(12):1181–1185. doi: 10.1038/nbt.1587. doi:10.1038/nbt.1587. [DOI] [PubMed] [Google Scholar]
- 28.Shay JW, Keith WN. Targeting telomerase for cancer therapeutics. Br J Cancer. 2008;98(4):677–683. doi: 10.1038/sj.bjc.6604209. doi:10.1038/sj.bjc.6604209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Folini M, Venturini L, Cimino-Reale G, Zaffaroni N. Telomeres as targets for anticancer therapies. Expert Opin Ther Targets. 2011;15(5):579–593. doi: 10.1517/14728222.2011.556621. [DOI] [PubMed] [Google Scholar]
- 30.Mantripragada KK, Caley M, Stephens P, et al. Telomerase activity is a biomarker for high grade malignant peripheral nerve sheath tumors in neurofibromatosis type 1 individuals. Genes Chromosomes Cancer. 2008;47(3):238–246. doi: 10.1002/gcc.20525. doi:10.1002/gcc.20525. [DOI] [PubMed] [Google Scholar]
- 31.Malminen M, Peltonen S, Koivunen J, Peltonen J. Functional expression of NF1 tumor suppressor protein: association with keratin intermediate filaments during the early development of human epidermis. BMC Dermatol. 2002;2:10. doi: 10.1186/1471-5945-2-10. doi:10.1186/1471-5945-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. doi:10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 33.Villa R, Folini M, Perego P, et al. Telomerase activity and telomere length in human ovarian cancer and melanoma cell lines: correlation with sensitivity to DNA damaging agents. Int J Oncol. 2000;16(5):995–1002. doi: 10.3892/ijo.16.5.995. [DOI] [PubMed] [Google Scholar]
- 34.Grant JD, Broccoli D, Muquit M, Manion FJ, Tisdall J, Ochs MF. Telometric: a tool providing simplified, reproducible measurements of telomeric DNA from constant field agarose gels. Biotechniques. 2001;31(6):1314–1316. doi: 10.2144/01316bc02. 1318. [DOI] [PubMed] [Google Scholar]
- 35.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. doi:10.2307/2281868. [Google Scholar]
- 36.Cox DR. Regression models and life tables. JR Stat Soc B. 1972;34:187–220. [Google Scholar]
- 37.Brekke HR, Ribeiro FR, Kolberg M, et al. Genomic changes in chromosomes 10, 16, and X in malignant peripheral nerve sheath tumors identify a high-risk patient group. J Clin Oncol. 2010;28(9):1573–1582. doi: 10.1200/JCO.2009.24.8989. doi:10.1200/JCO.2009.24.8989. [DOI] [PubMed] [Google Scholar]
- 38.Lévy P, Vidaud D, Leroy K, et al. Molecular profiling of malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1, based on large-scale real-time RT-PCR. Mol Cancer. 2004;3:20. doi: 10.1186/1476-4598-3-20. doi:10.1186/1476-4598-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Venturini L, Motta R, Gronchi A, Daidone M, Zaffaroni N. Prognostic relevance of ALT-associated markers in liposarcoma: a comparative analysis. BMC Cancer. 2010;10:254. doi: 10.1186/1471-2407-10-254. doi:10.1186/1471-2407-10-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lafferty-Whyte K, Cairney CJ, Will MB, et al. A gene expression signature classifying telomerase and ALT immortalization reveals an hTERT regulatory network and suggests a mesenchymal stem cell origin for ALT. Oncogene. 2009;28(43):3765–3774. doi: 10.1038/onc.2009.238. doi:10.1038/onc.2009.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seger YR, Garcia-Cao M, Piccinin S, et al. Transformation of normal human cells in the absence of telomerase activation. Cancer Cell. 2002;2:401–413. doi: 10.1016/s1535-6108(02)00183-6. doi:10.1016/S1535-6108(02)00183-6. [DOI] [PubMed] [Google Scholar]
- 42.Lafferty-Whyte K, Bilsland A, Cairney CJ, et al. Scoring of senescence signalling in multiple human tumour gene expression datasets, identification of a correlation between senescence score and drug toxicity in the NCI60 panel and a pro-inflammatory signature correlating with survival advantage in peritoneal mesothelioma. BMC Genomics. 2010;11:532. doi: 10.1186/1471-2164-11-532. doi:10.1186/1471-2164-11-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen HJ, Cho CL, Liang CL, Lu K, Lin JW. Implication of telomere length as a proliferation-associated marker in schwannomas. J Surg Oncol. 2002;81(2):93–100. doi: 10.1002/jso.10139. doi:10.1002/jso.10139. [DOI] [PubMed] [Google Scholar]
- 44.Hiyama E, Hiyama K. Telomerase as tumor marker. Cancer Lett. 2003;194(2):221–233. doi: 10.1016/s0304-3835(02)00709-7. Review doi:10.1016/S0304-3835(02)00709-7. [DOI] [PubMed] [Google Scholar]
- 45.Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer. 2008;8(3):167–179. doi: 10.1038/nrc2275. Review doi:10.1038/nrc2275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.