

Abstract
Noncovalent association between the von Willebrand factor (VWF) propeptide (VWFpp) and mature VWF aids N-terminal multimerization and protein compartmentalization in storage granules. This association is currently thought to dissipate after secretion into blood. In the present study, we examined this proposition by quantifying the affinity and kinetics of VWFpp binding to mature VWF using surface plasmon resonance and by developing novel anti-VWF D′D3 mAbs. Our results show that the only binding site for VWFpp in mature VWF is in its D′D3 domain. At pH 6.2 and 10mM Ca2+, conditions mimicking intracellular compartments, VWFpp-VWF binding occurs with high affinity (KD = 0.2nM, koff = 8 × 10−5 s−1). Significant, albeit weaker, binding (KD = 25nM, koff = 4 × 10−3 s−1) occurs under physiologic conditions of pH 7.4 and 2.5mM Ca2+. This interaction was also observed in human plasma (KD = 50nM). The addition of recombinant VWFpp in both flow-chamber–based platelet adhesion assays and viscometer-based shear-induced platelet aggregation and activation studies reduced platelet adhesion and activation partially. Anti-D′D3 mAb DD3.1, which blocks VWFpp binding to VWF-D′D3, also abrogated platelet adhesion, as shown by shear-induced platelet aggregation and activation studies. Our data demonstrate that VWFpp binding to mature VWF occurs in the circulation, which can regulate the hemostatic potential of VWF by reducing VWF binding to platelet GpIbα.
Introduction
The multimeric protein von Willebrand factor (VWF) regulates hemostatic processes in blood. By acting as a molecular bridge between extracellular matrix components exposed on the denuded blood vessel wall and platelets in circulation, VWF recruits platelets to sites of vascular injury.1 This macromolecule also participates in coagulation by binding and regulating the half-life of factor VIII (FVIII) in the circulation.
In endothelial cells and megakaryocytes, the 2050-aa mature VWF is produced, along with a relatively large VWF propeptide (VWFpp) that contains 741 amino acids.2 The 2813-aa precursor pre-pro-VWF protein, which includes the signal peptide, consists of 4 homologous units arranged in the following sequence: D1-D2-D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK. A paired dibasic amino acid sequence that is susceptible to proteolysis by furin separates VWFpp (D1-D2) from the remaining domains of mature VWF. After protein synthesis, disulfide bonding between VWF monomers at the C-terminus in the endoplasmic reticulum and N-terminus in the trans-Golgi results in protein multimerization.2,3 Because the acidic Golgi compartment does not favor spontaneous N-terminal disulfide bond formation, the D-domains of VWFpp contain vicinal cysteines (159Cys-Gly-Leu-Cys162 and 521Cys-Gly-Leu-Cys524) that provide the required protein disulfide isomerase or oxidoreductase activity.4 In support of this, mutations in VWFpp result in impaired VWF multimerization and storage,4–8 and the addition of wild-type VWFpp in trans in some of these systems restores multimerization.7–9 The addition of bases such as NH4Cl or chloroquine also abolish VWF multimerization in endothelial cells, likely because of impaired association between the 2 proteins.10 Once synthesized, VWF multimers are cotrafficked with VWFpp into cigar-shaped Weibel-Palade bodies (WPBs) in endothelial cells2,11–14 and α-granules in megakaryocytes,15,16 which are subsequently secreted into circulation in the presence of various proinflammatory or prothrombotic stimuli.
Whereas intracellular calcium levels and pH are important for VWF multimer assembly and storage, current evidence suggests that the pH and calcium conditions prevalent in the circulation (pH 7.4 and 1.5mM Ca2+) do not support VWF binding to VWFpp.17,18 This assertion is partially based on ultracentrifugation studies performed with radiolabeled WPBs in different buffers (pH 6.4 or 7.4 in the presence of Ca2+ or EGTA), which have demonstrated cosegregation of VWF with VWFpp in the centrifugal pellet only under low-pH and high-calcium conditions.19 However, this method lacks time resolution and is better suited to detecting strong binding interactions. Immunofluorescence studies have also shown that endothelial granule secretion results in extracellular patches of VWF from which the propeptide exits rapidly.17–19 In detailed time-course studies, Hannah et al showed that whereas the propeptide dispersed from the endothelial surface with a half-life of 3 seconds after WPB granule release, multimeric VWF persisted on the surface with a longer half-life of 323.5 seconds.18 Whereas this method reports on the association between VWFpp and VWF on the endothelial surfaces, it does not measure VWFpp-VWF binding in the blood.
The present study applies surface plasmon resonance (SPR) to evaluate quantitatively the role of pH and calcium in regulating the affinity and kinetics of VWFpp binding to VWF. The results indicate that, apart from the expected high-affinity interaction at low pH, significant interaction (KD = 25-50nM) also occurs under physiologic conditions. VWFpp binding to VWF was also detected in human plasma. A novel anti-D′D3 mAb (clone DD3.1) blocked this interaction completely, suggesting that VWFpp binds specifically only the D′D3 domain of mature VWF. In shear-induced platelet activation (SIPAct) studies and microfluidic flow-cell–based thrombus formation assays, the addition of either exogenous VWFpp or DD3.1 reduced the extent of platelet adhesion and activation. These processes are initiated by VWF-A1 domain binding to platelet GpIbα. Our results suggest that propeptide binding to VWF likely occurs in the circulation and this interaction may partially regulate VWF function during primary hemostasis. Further, the observation that an anti-D′D3 mAb and VWFpp reduce the function of the VWF-A1 domain reinforces the importance of domain-level architecture in regulating protein function.20
Methods
Abs
All mAbs were mouse IgGs unless otherwise noted. Abs included non-function–blocking anti-VWF mAb AVW-1 directed against the VWF C-terminus (GTI Diagnostics); mAb AVW-3 against the VWF A1-domain (GTI Diagnostics); mAb SZ-123 against the VWF A3-domain (kind gift from Dr Changgeng Ruan, Suzhuo, China)21; 3 anti-VWFpp mAbs 239.2, 239.3, and 242.222,23; and 3 anti-VWF D′D3 domain mAbs, DD3.1, DD3.2, and DD3.3. The last 3 mAbs were generated by fusion of splenocytes from mice immunized with purified human VWF-D′D3 domain expressed in Chinese hamster ovary (CHO) cells (construct described in the next section) with myeloma cells. Polyclonal rabbit anti-VWF Ab was from Dako. Anti-FVIII mAb ESH-4, which blocks VWF binding to FVIII, was from American Diagnostica. Function-blocking anti-GpIbα Ab AK2 was from Millipore.
VWF domain expression and purification
Standard molecular biology methods and primers listed in supplemental Table 1 (available on the Blood Web site; see the Supplemental Materials link at the top of the online article) were used to create lentiviral vectors that encode for VWFpp (VWF 23-745) and individual VWF domains: D′D3 (aa 764-1242), A1 (aa 1243-1480), A2 (aa 1481-1668), and A3 (aa 1671-1875). For protein expression, all constructs were ligated after the VWF signal peptide in the vector pCS-CG (Addgene). Lentivirus and stable CHO cell lines encoding for individual VWF domains (VWFpp, D′D3, A1, A2, or A3) were then established. Individual domains secreted into culture media were purified using poly-histidine affinity and ion-exchange chromatography. Details are provided in supplemental Methods.
Silver staining, Western blot, dot blot, and ELISA
The methods used for silver staining, Western blot, dot blot, and ELISA are detailed in supplemental Methods.24–26
SPR
Binding affinity and kinetics were measured using the Dual Channel Surface Plasmon Resonance Spectrometer SPR7000DC (Reichert) and a gold biosensor substrate bearing 10% carboxyl-PEG and 90% PEG mixed in a self-assembled monolayer.27 Carbodiimide coupling was performed to conjugate either 10 μg/mL of anti-VWF mAb AVW-1 or 25 μg/mL of VWF protein domains (VWFpp or D′D3) directly onto the “active channel” of the SPR device. The “reference channel” did not have immobilized ligand. In some cases, 40nM VWF (based on the monomeric VWF molecular mass of 250 kDa) isolated from human plasma cryoprecipitate28 was passed through the channel and captured by the immobilized AVW-1 in the active cell. Various concentrations of analyte (1.9-500nM) were then flown over the sensor at 30 μL/min. Binding measurements at pH 7.4 were performed in HEPES buffer (30mM HEPES, 110mM NaCl, 10mM KCl, and 1mM MgCl2), whereas MES buffer [20mM 2-(N-morpholino)ethanesulfonic acid] was used at lower pH. All binding buffers contain 0.05% Tween-20, with CaCl2 and EDTA being titrated as indicated. SPR data were acquired at 2-second intervals. Zeba Spin Desalting Columns (Thermo-Pierce) were used to match analyte buffer and instrument-running buffer. Binding/association data were typically collected for 5 minutes, after which time, buffer lacking the analyte was perfused for > 7 minutes to obtain dissociation data. HEPES with 1mM EDTA was used for substrate regeneration.
Data processing and model fitting were performed using Scrubber2 (provided by David Myszka, University of Utah, Salt Lake City, UT). Signal from the reference channel was subtracted from active channel to obtain the net response as follows: Response = [Signal]Active_channel− [Signal]Reference_channel. The dissociation constant KD(equilibrium) was calculated by collating response data over the final 20 seconds of the association phase at each analyte concentration, which were then fitted to the single-site or Langmuir-isotherm binding model of Scrubber. Binding on- (kon) and off- (koff) rates were determined by globally fitting kinetic/time-course data at all analyte doses to the simple 1:1 interaction model:
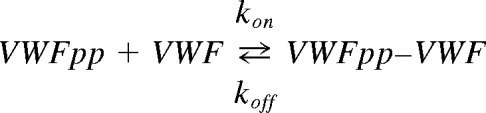 |
Dissociation constant obtained from kinetic data fit, KD(kinetic) (koff /kon), was compared with KD(equilibrium) obtained from near-equilibrium data analysis.
In addition, a model for multivalent binding between VWFpp and VWF was simulated using ClampXP29 to fit kinetic data in MES buffer (pH 6.2) containing 10mM Ca2+ (see supplemental Methods).
SIPAct and SIPA
For SIPAct and shear-induced platelet aggregation (SIPA), blood was drawn from healthy human adult volunteers by venipuncture into either 4% wt/vol sodium citrate, 100μM PPACK (Phe-Pro-Arg-chloromethylketone), or 20 U/mL of heparin following protocols approved by the University at Buffalo Health Sciences Institutional Review Board. Platelet-rich plasma (PRP) and platelet-poor plasma (PPP) were prepared by centrifugation.28
SIPAct.
In some cases, 107 platelets/mL obtained by mixing PRP and PPP were subjected to hydrodynamic shear in a cone-plate viscometer (Thermo-Haake) at a shear rate of 9600 s−1 for 3 minutes in the presence or absence of 50 μg/mL of anti-D′D3 mAbs. In other runs, 3 U/mL of apyrase (Sigma-Aldrich) plus various concentrations of VWFpp (2.5-100 μg/mL) were incubated with blood for 15 minutes in the presence or absence of approximately 100 μg/mL of anti-VWFpp mAb. Blood was then sheared in the viscometer at 3500 s−1 for 3 minutes, after which time 5 μL of sheared sample (either whole blood or platelets in plasma) was withdrawn and incubated with 1 μL of 1:10 diluted CD31 PerCP-eFluor 710 (eBiosciences) and 2 μL of PE-labeled annexin V (BD Biosciences) in HEPES buffer (pH 7.4) containing 5mM CaCl2 for 5 minutes at 37°C. Samples were then analyzed using a FACSCalibur flow cytometer (BD Biosciences). The percent platelet activation was calculated as the single platelets binding more than baseline amounts of fluorescent annexin V.28
SIPA.
PRP labeled with CD31 PerCP-eFluor 710 was diluted in PPP to obtain a platelet count of approximately 108/mL. This solution was preincubated with VWFpp (100 μg/mL), DD3.1 (100 μg/mL), or AVW-3 (20 μg/mL) for 10 minutes at room temperature. The samples were then sheared in a cone-plate viscometer at 9600 s−1 in the presence or absence of agonist (0.5μM ADP or TRAP-6), after which 5 μL samples withdrawn at different time points were read in the flow cytometer for fixed amounts of time. The number of single platelets was measured.30 The percent platelet aggregation was calculated as follows: 100 × (1-singlet platelet number at sampling time point/initial single platelet number at time = 0 s).
Microfluidic flow chamber assay
A 1 × 1-mm patch of a 100-mm tissue-culture Petri dish was incubated with 200 μg/mL of equine tendon collagen (Chrono-log) diluted in 0.02M acetic acid for 1 hour at room temperature.31 The surface was blocked with HEPES buffer containing 1% BSA for 10 minutes. Five milliliters of human blood drawn in PPACK was labeled with 0.625 μg/μL of 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein for 15 minutes at room temperature (0-100 μg/mL of VWFpp or anti-VWF mAb was added in some cases). A custom 1-cm-long microfluidic flow cell made of polydimethylsiloxane with a cross-section of 400 μm width × 100 μm height was vacuum sealed onto the collagen substrate and the apparatus was mounted on the stage of a Zeiss AxioObserver microscope with a Hamamatsu 1394 ORCA camera. Blood was perfused through the flow cell at a wall shear rate of 3000 s−1. Thrombus formation on the collagen substrate was monitored by acquiring images every 10 seconds with an exposure time of 110 milliseconds. Thrombus growth was quantified using ImageJ 1.43u software by measuring the percentage of the collagen-bearing field of view that had immobilized platelets.
Statistics
Data are presented as means ± SD for ≥ 3 experiments. ANOVA was applied for comparison between multiple treatments. P < .05 was considered significant.
Results
The D′D3 domain of mature VWF contains the only binding site for VWF propeptide
While VWFpp has been shown to bind the D′D3 domain of VWF,32,33 it remains to be determined whether this is the only binding site for VWFpp interaction with mature VWF. In the present study, we tested the ability of a panel of mAbs directed against both VWFpp and D′D3 to block this binding. Recombinant VWF domains/variants were also expressed for SPR assays.
VWFpp, individual VWF domains D′D3, A1, A2, and A3, dimeric VWF (ΔPro-VWF), and ΔD′D3-VWF were expressed and purified from CHO cells (Figure 1A). Silver staining (Figure 1B) and Western blot analysis (Figure 1C) confirmed that the isolated domains were > 95% pure and that the molecular mass corresponded to the primary amino acid sequence. All VWF domains were monomeric, without intermolecular disulfide bonds, because protein mobility was not altered by β-mercaptoethanol. ΔPro-VWF and ΔD′D3-VWF were expressed as C-terminal dimers (Figure 1D). Recombinant domain function was confirmed using the FVIII-binding assay for the D′D3 domain, the ristocetin-induced platelet-binding assay for the A1 domain, and the ADAMTS13 cleavage assay for the A2 domain (supplemental Figure 1). The presence of FLAG/His tags did not affect protein function in these studies.
Figure 1.

Expression and purification of individual VWF domains. (A) Mature VWF was purified from human plasma cryoprecipitate. This protein lacks VWFpp because the Arg 763-Ser764 bond is proteolytically cleaved. Propeptide VWFpp (D1-D2 domains) and other domains within the globular section of VWF (D′D3, A1, A2, and A3) were expressed as FLAG- and His-tagged proteins in CHO cells. Amino acid numbers provided are based on pre-pro-VWF. ΔPro-VWF is dimeric, full-length VWF. ΔD′D3-VWF is identical to ΔPro-VWF except it lacks the D′D3 domain. (B) Silver staining of purified VWF domains under reducing (with β-mercaptoethanol) and nonreducing (absence of β-mercaptoethanol) conditions. (C) Western blot of the domains using anti-His Ab under reducing conditions. (D) Western blot of ΔPro-VWF and ΔD′D3-VWF under reducing conditions detected using anti-VWF polyclonal Ab.
The ability of mAbs directed against VWFpp (239.2, 239.3, and 242.2; Figure 2A) and D′D3 (DD3.1 and DD3.2; Figure 2B) to inhibit multimeric plasma VWF binding to immobilized VWFpp was tested using ELISA, both in MES (pH 6.2) and HEPES (pH 7.4) buffer. All anti-VWFpp mAbs inhibited this molecular interaction, with 239.3 being somewhat superior. Anti-D′D3 mAb DD3.1, but not DD3.2, also blocked VWFpp-VWF binding completely. Dot blots showed that all anti-VWFpp mAbs and clone DD3.1 bound to native protein more readily compared to denatured protein (Figure 2C). Further, mouse mAb DD3.1 did not bind bovine or porcine VWF (Figure 2D). Together with sequence-alignment analysis of human, murine, bovine, and porcine D′D3 (supplemental Figure 2), we conclude that DD3.1 binds a complex 3D epitope that likely includes some of the following residues: L781, M802, K834, T869, K912, H916, K922, K991, N1011, Q1154, F1206, S1217 and V1229.
Figure 2.

Blocking VWF binding to VWFpp. (A) Anti-VWFpp (10 μg/mL) mAbs (239.2, 239.3, and 242.2) was added to microtiter wells bearing immobilized VWFpp for 15 minutes. Next, 2.5 μg/mL of VWF was added in buffer at either low pH (MES buffer, pH 6.2, with 10mM CaCl2) or high pH (HEPES buffer, pH 7.4, with 2.5mM CaCl2). Binding of multimeric VWF to VWFpp was measured using polyclonal anti-VWF Ab. (B) Studies identical to those in panel A were conducted except that 1 μg/mL of anti-D′D3 mAbs (DD3.1 and DD3.2) were incubated with plasma VWF for 15 minutes before the addition of the mixture to wells containing immobilized VWFpp. (C) Fifty nanograms of either native or denatured (heated with β-mercaptoethanol at 95°C for 5 minutes) D′D3 or VWFpp was spotted onto nitrocellulose membrane. After membrane blocking, binding of anti-D′D3 and anti-VWFpp mAbs to these spots was measured using HRP-conjugated goat anti–mouse Ab for detection. (D) Human, porcine, or bovine plasma was immobilized onto wells bearing anti-VWF polyclonal Ab such that equivalent amounts of VWF were captured in each well as determined using HRP-conjugated anti-VWF polyclonal Ab. Binding of anti-D′D3 mAbs to VWF was then measured using HRP-conjugated goat anti–mouse Ab. Neither mAb bound porcine or bovine VWF.
MAbs directed against one section of VWF may alter the function of neighboring domains.20,21 To determine whether D′D3 is the only domain that interacts with VWFpp, SPR studies were performed with other domains located in the globular section of VWF (Figure 3A). VWFpp was covalently coupled to the biosensor surface and individual VWF domains (D′D3, A1, A2, and A3) were passed as analytes in MES buffer containing 10mM Ca2+. Among these, only the D′D3 domain bound VWFpp. Binding between D′D3 and VWFpp was also observed when D′D3 was immobilized on the biosensor and VWFpp was the analyte (Figure 3A inset). To exclude a role of the VWF C-terminal domains (D4-Ck) during the VWF-VWFpp interaction, binding of VWFpp to multimeric plasma VWF (pVWF) and ΔPro-VWF24 was compared with binding to ΔD′D3-VWF (Figure 3B). ΔD′D3-VWF is identical to ΔPro-VWF only it lacks the D′D3 domain. VWFpp binding to pVWF and ΔPro-VWF was specifically blocked by anti-D′D3 mAb DD3.1. VWFpp did not bind ΔD′D3-VWF.
Figure 3.

D′D3 is the only domain in multimeric VWF that binds VWFpp. (A) Eight hundred response unit (RU) VWFpp was covalently coupled onto the SPR biosensor. Each of the VWF domains (D′D3, A1, A2, and A3) at a concentration of 250nM were then injected over this surface in MES buffer (pH 6.2) containing 10mM CaCl2. Inset shows the inverse experiment in which binding of the 250nM VWFpp to 800 RU immobilized D′D3 was measured. Only D′D3 binds VWFpp. (B) Binding of 10 μg/mL of VWF isolated from human plasma cryoprecipitate (multimeric, pVWF), recombinant dimeric ΔPro-VWF, and ΔD′D3-VWF to immobilized VWFpp was measured using ELISA. Binding observed in the case of pVWF and ΔPro-VWF was eliminated when either the D′D3 domain was deleted (ΔD′D3-VWF) or when anti-D′D3 mAb DD3.1 was applied. *P < .05 for pVWF and ΔPro-VWF in the absence of any mAb.
The mAb blocking data and the SPR and ELISA studies show that the D′D3 domain of VWF contains the only binding site for VWFpp in the multimeric protein.
VWFpp binds VWF in blood plasma
Some of the ELISA studies that measured VWFpp binding to VWF (Figure 2) were performed under physiologic conditions of pH 7.4. This suggests that VWFpp and VWF may interact in human blood. To confirm this, VWF from freshly isolated, heparinized, human PPP was immunoprecipitated using anti-VWF Ab onto protein-A/G beads. Coprecipitated VWFpp was then released by chelating calcium and detected using a sandwich ELISA (Figure 4A). The specificity of the measured VWF-VWFpp interaction was confirmed using mAb DD3.1 to block this binding.
Figure 4.
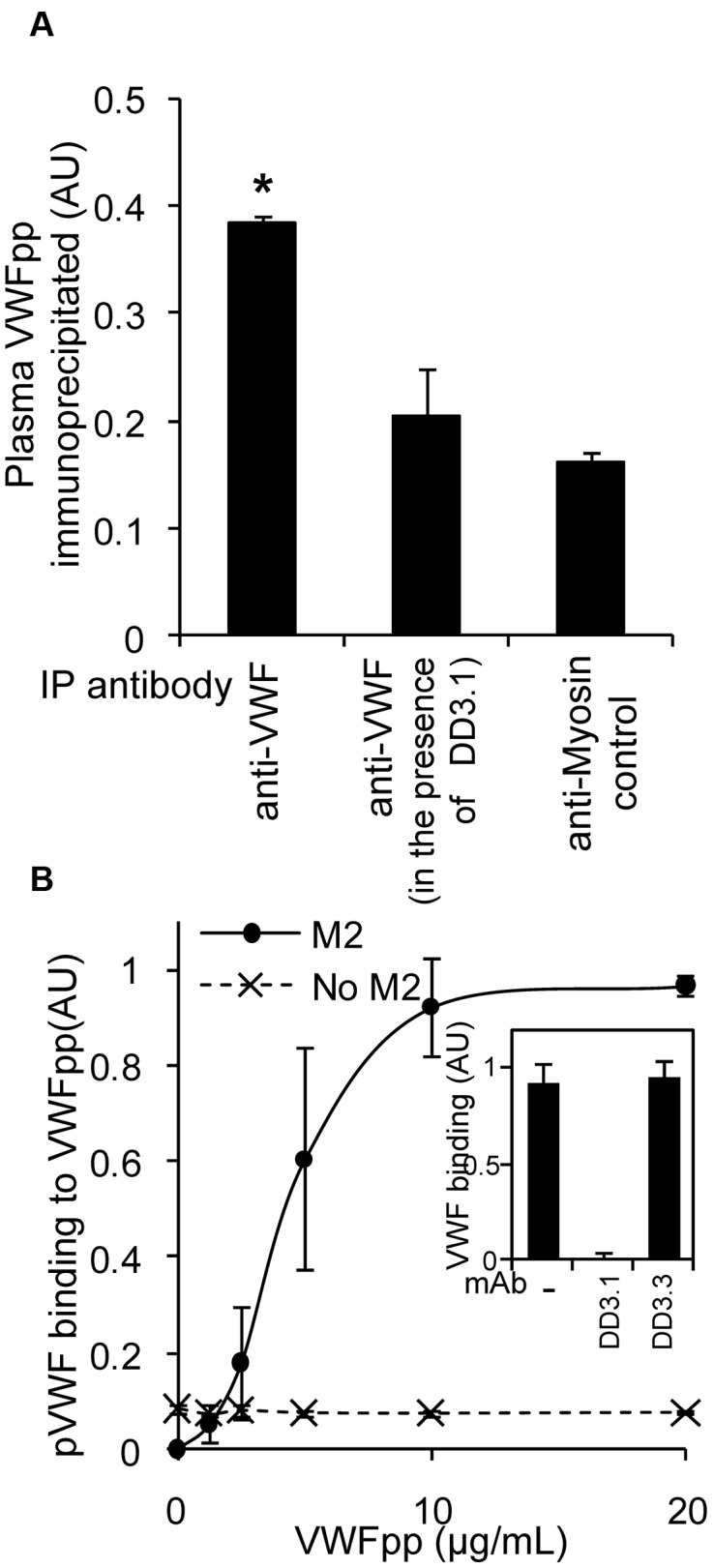
VWFpp and VWF interact in plasma. (A) Anti-VWF Ab (30 μg/mL) or control (antimyosin) was added to immunoprecipitate VWF from 1 mL of heparinized human PPP in the presence or absence of 30 μg/mL of DD3.1 overnight at 4°C. The protein complex was isolated using protein-A/G beads and VWFpp was subsequently released using HEPES buffer containing 5mM EDTA. Released VWFpp was detected with a sandwich ELISA using mAb 242.2 for capture and HRP-conjugated 239.3 for detection. *P < .05 with respect to all other treatments. (B) Studies similar to those in panel A were conducted, only PPP was incubated with 0-20 μg/mL of FLAG-tagged VWFpp for 10 minutes before addition of the mixture to wells bearing anti-FLAG mAb (clone M2). Bound plasma VWF was measured using HRP-conjugated polyclonal anti-VWF Ab. Wells without mAb M2 serve as a negative control. VWF-VWFpp binding in this assay could be blocked by 50 μg/mL of DD3.1, but not by control mAb DD3.3 (panel A, panel B inset).
To quantify the KD of this binding interaction, 0-20 μg/mL (0-250nM) of recombinant FLAG-tagged VWFpp was titrated into PPP (Figure 4B). Binding of VWF to VWFpp was then detected using anti-FLAG mAb bearing wells to immunocapture VWFpp and anti-VWF Ab to detect coprecipitated plasma VWF. VWF bound VWFpp in plasma with a KD of 4 μg/mL (50nM). This interaction was specifically blocked by DD3.1, but not by control mAb DD3.3. These data show that VWFpp binding to mature VWF may be expected in the circulation.
Stronger binding of VWFpp with mature VWF under low-pH and high-calcium conditions compared with physiologic conditions
The affinity and kinetics of VWFpp binding to mature VWF was measured under different buffer conditions using SPR (Figure 5). Human VWF was captured onto the sensor surface via covalently coupled anti-VWF mAb AVW-1. Various concentrations of VWFpp were then injected in 3 different buffers: (1) MES (pH 6.2) with 10mM CaCl2 (Figure 5A,D); (2) HEPES (pH 7.4) with 2.5mM CaCl2 (Figure 5B,E); and (3) HEPES (pH 7.4) with 2.5mM EDTA (Figure 5C). At low pH and high calcium, VWFpp associated almost irreversibly with VWF. This noncovalent binding was reversed within seconds by injecting HEPES buffer containing 1mM EDTA (supplemental Figure 3). VWFpp also bound immobilized VWF at pH 7.4, although the interaction dissipated within 5 minutes into the dissociation phase. No binding occurred when the running buffer was HEPES (pH 7.4) with 2.5mM EDTA. Independent controls showed that buffer salt composition does not affect this molecular interaction (supplemental Figure 4).
Figure 5.

Kinetics and affinity of VWFpp binding to immobilized multimeric VWF. (A) Three hundred twenty response unit (RU) VWF was immobilized on the SPR sensor via anti-VWF mAb. Different concentrations of VWFpp (indicated in nanomolar units in panels A-C) were perfused over the substrate in MES (pH 6.2) binding buffer with 10mM CaCl2. (B) Studies identical to those in panel A except that 920 RU of VWF was immobilized and the running buffer was HEPES (pH 7.4) with 2.5mM CaCl2. (C) 320 RU of VWF was immobilized and running buffer was HEPES with 2.5mM EDTA(pH 7.4). Solid lines in panels A through C represent experimental data and dashed lines are fits to the 1:1 interaction model. Kinetic constants from fits are summarized in Table 1. Equilibrium KD analysis for panels A and B are shown in panels D and E, respectively.
Affinity and kinetic fits of the above data show that the off-rate (7.6 × 10−5 vs 4.4 × 10−3 s−1) and dissociation constants (0.2-3.8nM vs 25.8-30.9nM) were smaller for the lower pH runs compared with runs performed at physiologic pH (Table 1). Further, VWFpp appears to aggregate at low pH. This inference is consistent with the observation that dissociation constants estimated based on equilibrium data and kinetic fits are in reasonable agreement in higher (30.9nM vs 25.8nM) but not lower (0.2nM vs 3.8nM) pH runs. In addition, kinetic fits of experimental data using the 1:1 interaction model (Figure 5A) failed at low pH when VWFpp exceeded 15nM. The number of response units (RUs) of VWFpp binding to multimeric VWF exceeded the maximum theoretically possible amount based on a 1:1 stoichiometric binding model. Overall, both VWF-VWFpp binding and VWFpp aggregation were enhanced at low pH.
Table 1.
Summary of binding constants
| Ligand | Analyte | Buffer | kon, M−1 s−1 × 105 | koff s−1 × 10−5 | KD(kinetics) nM | KD(equilibrium) nM |
|---|---|---|---|---|---|---|
| VWF | VWFpp | HEPES, pH 7.4, 2.5mM Ca2+ | 1.7 | 438 | 25.8 | 30.9 |
| VWF | VWFpp | MES, pH 6.2, 10mM Ca2+ | 3.6 | 7.58 | 0.21 | 3.84 |
| VWF | VWFpp | HEPES, pH 7.4, 2.5mM EDTA | NB | NB | NB | NB |
| VWFpp | VWFpp | HEPES, pH 7.4, 2.5mM Ca2+ | NB | NB | NB | |
| VWFpp | VWFpp | MES, pH 6.2, 10mM Ca2+ | 0.356 | 100.5 | 28.1 | |
| VWFpp | D′D3 | MES, pH 6.2, 10mM Ca2+ | 0.55 | 110 | 20.1 | |
| D′D3 | VWFpp | HEPES, pH 7.4, 2.5mM Ca2+ | 1.76 | 862 | 49 |
NB indicates no binding.
Multivalent binding of VWFpp to proximal D′D3 domains in multimeric VWF augments its binding affinity
Studies were performed with individual VWFpp and D′D3 domains to determine how individual binding events contribute to the overall interaction between the propeptide and multimeric VWF. When VWFpp was immobilized on the sensor surface and the same macromolecule was also the analyte, we observed strong homotypic VWFpp binding (KD = 28nM) in MES (pH 6.2) containing 10mM Ca2+ (supplemental Figure 5A and Table 1). No binding was detected after increasing the pH to 7.4, even in the presence of Ca2+ (data not shown). Therefore, the VWFpp-VWFpp interaction occurs at low but not high pH.
In studies assaying D′D3 binding to VWFpp, higher-affinity binding occurred in MES buffer (pH 6.2) with 10mM Ca2+ (supplemental Figure 5B) compared with runs performed with HEPES buffer (pH 7.4) with 2.5mM Ca2+ (supplemental Figure 5C). KD was 20nM and 49nM, respectively, for these 2 cases (Table 1). The on- and off-rate in the latter case matched the kinetics of VWFpp binding to multimeric VWF at physiologic pH (Figure 5B). Therefore, monovalent VWFpp binding to VWF accounts for the SPR signal at physiologic pH.
The off-rate of VWFpp binding to D′D3 in MES buffer (koff = 110 × 10−5 s−1) was approximately 15-fold higher than that to multimeric VWF (koff = 8 × 10−5 s−1). The “apparent KD” estimated using the simple 1:1 interaction model under low pH conditions also decreased from 20nM in the former case to 0.2nM in the case of the multimer VWF. These differences could be because of the multivalent binding of VWFpp to adjacent D′D3 units located on adjacent VWF monomers in the multimeric protein. We modeled this possibility using a “3 reaction ClampXP model” to fit low pH binding data collected in Figure 5A. This model considered 3 independent processes (supplemental Figure 6): (1) the single site binding of the VWFpp aggregate/cluster to a single D′D3 domain on the immobilized VWF (reaction 1; rate constants for this step are shown in supplemental Figure 5B); (2) The multi-/bivalent binding of VWFpp aggregate to 2 adjacent D′D3 domains located in VWF (reaction 2); and (3) the attachment of previously recruited VWFpp at a single D′D3 site via reaction 1 to additional D′D3 sites on the immobilized protein (reaction 3). The model fit shown in supplemental Figure 6 suggests that the off-rate with which multivalent VWFpp aggregate-VWF interactions are released is likely to be very small (approximately 0 s−1). This binding of VWFpp clusters to proximal D′D3 domains in multimeric VWF is thus likely to be a key feature that enhances the overall affinity of this molecular binding event at low but not high pH. In addition, low pH enhances the affinity of VWFpp binding not only to other VWFpp units, but also to D′D3.
Both VWFpp and anti-D′D3 mAb DD3.1 inhibit VWF-A1 domain function
We also investigated whether the binding of VWFpp to VWF observed under physiologic conditions alters VWF function, with a focus on functions regulated by the D′D3 domain. Some of these experiments measured the effect of VWFpp on SIPAct and SIPA in viscometer-based assays because this is mediated by the VWF A1-domain26,28 and because the D′D3 domain shields A1 function,20 (Figure 6). Other experiments measure platelet recruitment to collagen under shear (Figure 7). The last experiments assay the effect of VWFpp on VWF-D′D3 binding to FVIII (supplemental Figure 8).
Figure 6.
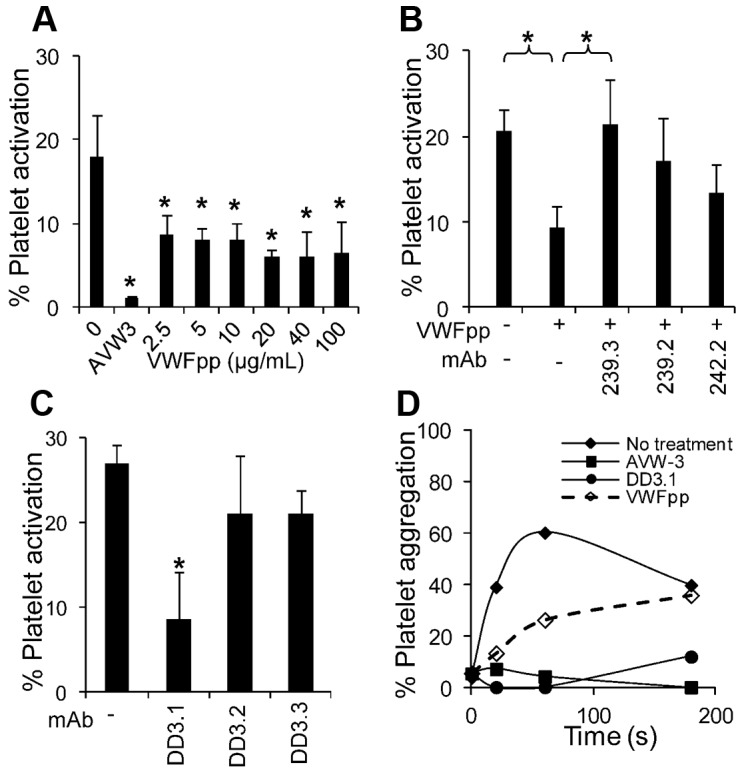
Effect of VWFpp on SIPAct and SIPA. (A) Different concentrations of VWFpp were added to citrated whole blood before the application of shear in a cone-plate viscometer at a shear rate of 3500 s−1 for 3 minutes. Annexin V–PE binding to platelets was used to quantify platelet activation. VWFpp partially inhibits platelet activation (*P < .05 with respect to run without VWFpp). (B) VWFpp (10 μg/mL) was incubated with each of the anti-VWFpp mAbs before the addition of whole blood and application of shear under the conditions described in panel A. 239.3 fully restored platelet-activation levels by countering/blocking the effect of VWFpp (*P < .05). (C) Platelets (107/mL) in plasma were sheared in a viscometer at 9600 s−1 in the presence or absence of anti-D′D3 mAbs. MAb DD3.1, but not other anti-D′D3 mAbs, inhibited platelet activation by approximately 75%. *P < .05 with respect to all other treatments. (D) Approximately 108/mL platelets diluted in plasma were incubated with 100 μg/mL of anti-D′D3 mAb (DD3.1), 20 μg/mL of anti–VWF-A1 domain mAb (AVW-3), or 100 μg/mL of VWFpp for 10 minutes before shear application at 9600 s−1. All reagents reduced platelet aggregation. Data are representative of 3 independent experiments.
Figure 7.

VWFpp inhibits thrombus formation on collagen surfaces. 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein–labeled whole blood was perfused at 3000 s−1 over collagen substrate in the presence of anti-VWF mAbs (SZ-123 and DD3.1) or 5-50 μg/mL of VWFpp. PPACK was used as an anticoagulant. (A) The percent surface area covered with immobilized platelets was quantified. Onset of thrombus formation in the presence of VWFpp was delayed at 2 minutes (*P < .05). (B) Snapshot of the collagen substrate at 2 minutes under the specified conditions.
In the SIPAct assay (Figure 6A), 17% of platelets in citrated whole blood were activated by the application of shear at 3500 s−1 for 3 minutes. Cell activation was completely inhibited by the anti-VWF-A1 mAb AVW-3. Addition of various concentrations of VWFpp (2.5-100 μg/mL) partially inhibited (up to 50%) platelet activation. Inhibition observed after the addition of VWFpp could be reversed by anti-VWFpp mAbs 239.3 (Figure 6B). Further, mAb DD3.1, which inhibits VWF-VWFpp binding, but not non-function-blocking mAbs DD3.2 and DD3.3, reduced SIPAct by approximately 75% (Figure 6C). Partial inhibition of SIPA was also observed after the addition of VWFpp to PRP prepared from PPACK anticoagulated blood (Figure 6D). Whereas DD3.1 and VWFpp were effective at reducing SIPA in the absence of additional stimulus, blocking efficacy decreased after the addition of agonists (ADP and TRAP-6) under high-shear conditions (supplemental Figure 7). The poor inhibition measured is likely because of a more prominent role for GpIIb-IIIa after the addition of stimulus, which results in up to 95% cell aggregation within 20 seconds of shear. Overall, the binding of VWFpp to VWF-D′D3 in solution can alter VWF A1-domain function.
We also monitored thrombus formation in PPACK-anticoagulated whole blood on collagen in a microfluidic chamber at a wall shear rate of 3000 s−1 (Figure 7). Platelet adhesion was VWF dependent because it could be abrogated by SZ-123, an anti–VWF-A3 domain mAb.21 Complete blocking was also achieved by the anti-D′D3 mAb DD3.1, which competes with VWFpp for the same epitope on the D′D3 domain. Similar to the observations in the SIPAct assay, VWFpp (5-50 μg/mL) was partially effective at inhibiting VWF-mediated thrombus formation (Figure 7A). At both 2 and 4 minutes after perfusion of whole blood, the surface coverage of platelets was approximately 60% lower in the presence of VWFpp. Therefore, VWFpp binding to VWF delays the onset of platelet adhesion on collagen surfaces and also slows thrombus growth.
ELISA showed no effect of either VWFpp (up to 40 μg/mL) or mAb DD3.1 on VWF binding to FVIII (supplemental Figure 8). Our data suggest that the VWFpp-VWF interaction may regulate the hemostatic potential of VWF by reducing platelet adhesion and activation.
Discussion
Specific binding of VWFpp to VWF-D′D3
In the present study, the mAb-blocking data, together with SPR and ELISA studies with various VWF variants, demonstrated that VWF-D′D3 harbors the only binding site for VWFpp in mature VWF. VWFpp binding to VWF could be completely blocked using a range of anti-VWFpp mAbs (239.2, 239.3, and 242.2) and also anti-D′D3 mAb DD3.1. Whereas previous studies have demonstrated the transient association of recombinant D3 with VWFpp in the endoplasmic reticulum32 and the in vitro assembly of tubule-like structures on mixing equimolar concentrations of recombinant VWFpp and dimeric D′D3 at low-pH and high-calcium conditions,33 the possibility that other VWF domains can interact with VWFpp was not ruled out. Therefore, the identification of a function-blocking anti-D′D3 mAb DD3.1 that abrogates VWF-VWFpp binding helps to define more precisely the VWFpp-binding epitope on VWF. Further, because DD3.1 and anti-VWFpp mAbs block VWFpp-VWF binding at both low pH (MES buffer) and high pH (HEPES buffer), it may be that alteration of solution pH causes local changes in the structure and charge distribution of the binding pocket, rather than drastically altering binding location.
Whereas the precise D′D3 amino acids that bind VWFpp have not been identified, the data suggest the existence of a complex 3-dimensional binding interface. Mice and humans share approximately 89% homology in the D′D3 sequence, and there is no particular stretch that is distinctly different between the species. Using VWF from additional species, we have narrowed down the binding epitope for DD3.1 to 13 amino acids that are dispersed throughout the D′D3 domain. Among these, F1206 and S1217 lie in close proximity to R1205. Mutation of this D3 residue to histidine (ie, R1205H) results in the loss of pH-dependent binding between VWF and VWFpp.34 Also supporting the notion that the interaction between D′D3 and VWFpp is complex are observations in VWD type 2A suggesting that a variety of mutations dispersed throughout the protein affect multimerization, VWF storage, or both.35,36 These mutations may either participate directly or may destabilize the overall domain structure, thus indirectly affecting the molecular interaction. Finally, using human-canine chimeric constructs, Haberichter et al identified an important role of T869 in the D3 domain and R416 in the D2 domain for cotargeting VWF and VWFpp to WPBs.37 Whether the mAb DD3.1 binds in the proximity of these residues will be determined in the future by undertaking finer epitope-mapping studies. Such identification can enhance the utility of DD3.1 in investigations that assay the role of the VWFpp-VWF interaction on granule storage.
Nature of VWFpp binding to D′D3 over the range of pH and calcium
Low pH and high calcium affected VWFpp more prominently than D′D3. Among these, VWFpp-D′D3 affinity is more dramatically affected by changing pH and is relatively insensitive to changing the calcium concentration (data not shown). Low pH increases the affinity and kinetics of VWFpp for both homotypic binding to other VWFpp and heterotypic interaction with the D′D3 domain. In addition, neither our data nor previous results33 support a possible homotypic interaction between D′D3 units on decreasing pH. In small-angle neuron-scattering studies, we have also observed the aggregation of VWFpp after lowering the pH, but no self-association or structure changes in multimeric VWF (S.N. and L. Porcar, unpublished data, April 2010).
Whereas the D′D3 domain bears the only binding site for VWFpp, the binding avidity of propeptide-multimeric VWF interaction is higher than that of propeptide binding with single D′D3 alone. Whereas VWFpp binds multimeric VWF with subnanomolar apparent affinity (KD approximately 0.2nM), it binds isolated D′D3 with lower affinity (KD approximately 20nM). This experimental observation, along with the Clamp XP model, suggests that clusters of VWFpp formed at low pH bind or link adjacent D′D3 domains on multimeric VWF (supplemental Figure 9). The off-rate of such VWFpp binding to dimeric D′D3 is very low at low pH. We suggest that in cells, VWFpp dimers or aggregates may also be an effective mechanism to bring 2 or more D′D3 domains together. Such aggregation events at low pH, along with the protein disulfide isomerase activity imparted by the Cys-Gly-Leu-Cys epitope of VWFpp may facilitate VWF multimerization in intracellular trans-Golgi compartments.32,33 Consistent with this proposition, the expression of VWFpp in trans8,9 and low pH in Golgi9 are sufficient conditions to allow VWF multimerization and storage.
VWFpp–VWF-D′D3 interaction attenuates VWF-A1 binding to platelet GpIbα
In contrast to the prevailing notion that VWFpp and VWF circulate independently in blood,17,18 we noted that the VWFpp could be coprecipitated with VWF in heparinized human plasma. Supplementing plasma with various concentrations of FLAG-tagged VWFpp resulted in an estimated KD of approximately 50nM; the KD measured using SPR was also 20-30nM under physiologic conditions. The circulatory concentrations of VWF and VWFpp in normal human blood are approximately 10 μg/mL (40nM) and approximately 1 μg/mL (12.5nM), respectively, with protein half-lives being 8-12 and 3-5 hours, respectively.38,39 Based on this, it is to be expected that a fraction of the VWF in circulation would be associated with VWFpp.
The functional interaction between VWFpp and VWF in blood reduced the accessibility of the VWF-A1 domain for platelet GpIbα. In support of this, both VWFpp and anti-D′D3 mAb DD3.1 inhibited the extent of SIPAct and SIPA in viscometer-based studies. Cell activation and aggregation are driven by fluid shear-mediated VWF-A1 binding to the platelet receptor GpIbα and VWF self-association.26,28 In the microfluidics studies, VWFpp and mAb DD3.1 also reduced platelet adhesion and thrombus formation. The A1 domain of immobilized multimeric VWF recruits platelets by engaging GpIbα. In both functional assays, the blocking efficacy of VWFpp was more apparent at early time points because of the transient nature of its interaction with VWF. mAb DD3.1 was more effective compared with VWFpp at blocking cell activation and adhesion at later times because of its approximately 50- to 100-fold higher affinity for the VWF-D′D3 domain. Whereas previous studies suggested that VWFpp may bind collagen,38,39 we did not detect this in our microfluidic and ELISA assays using equine collagen. In addition, neither VWFpp nor the anti-D′D3 mAb DD3.1 affected FVIII binding to VWF. Therefore, whereas uncleaved propeptide in pre-pro-VWF may inhibit FVIII binding to the D′D3 domain,40,41 the D′D3 site recognized by soluble VWFpp is apparently different. Overall, the dominant effect of VWFpp and anti-D′D3 mAb is on the function of the VWF A1 domain.
The consequence of VWFpp-mediated alteration at VWF A1-domain function in normal human physiology and disease requires further examination. In general, the hemostatic function of VWF is regulated by its size in circulation and the availability of VWF-A1–binding sites. These 2 features are not clearly separable, because the binding of VWF to platelet GpIbα under shear via the A1 domain itself promotes ADAMTS13-mediated proteolysis.42 Based on its attenuation of VWF-A1 function, it is possible that VWFpp and VWF-D′D3 can limit platelet adhesion/accumulation in low-pH regions of poorly perfused tissues or in regions of wound repair.19 By reducing VWF binding to GpIbα under hydrodynamic shear conditions, VWFpp may also reduce VWF proteolysis by ADAMTS13. VWFpp may thus play a dual role by enhancing VWF size in normal circulation and by reducing cell adhesion during pathology. Such a regulatory role of VWFpp-VWF binding may be more pronounced during diseases in which the mature VWF half-life in circulation is reduced, either due to autoimmune Abs or to mutations such as those identified in type 1C (Vicenza and other mutations), type 2A, type 2B, or platelet-type pseudo-VWD.43 Under these circumstances, the VWFpp:VWF ratio increases from approximately 1.0 in healthy subjects to 2-11 during disease.22 Finally, VWF/VWFpp mutations that enhance VWFpp-D′D3 binding affinity can enhance the role of VWFpp in regulating hemostasis.
Overall, our results contribute to the growing body of literature demonstrating that interaction between individual VWF domains located in the globular-head section of the protein affect the physiologic function of the multimer.44–46 In particular, our data strengthen the notion that the VWF D′D3 domain functions as a shield to reduce the binding affinity of the VWF-A1 domain for platelet GpIbα.20 By partial regulation of VWF-A1 function, VWFpp may represent yet another control mechanism in the circulation that finely tunes the blood-clotting process.
Supplementary Material
Acknowledgments
The authors thank Nandini Mondal (University of Buffalo, Buffalo, NY) for providing the microfluidic flow cell, Dr Scott L. Diamond (University of Pennsylvania, Philadelphia, PA) for insightful discussions, and Susan Morey (University of Buffalo) for helping to generate the anti-D′D3 mAbs.
This work was supported by the American Heart Association (a predoctoral award to S.R.M.), and the National Institutes of Health (grant HL77258 to S.N., and grants HL33721 and HL81588 to R.R.M.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.R.M. designed and performed the experiments and wrote the manuscript; C.S., K.M.D., and M.M. designed and performed the experiments; K.R.-O. designed the research and supervised the technician on this project; T.E.R. and R.R.M. provided valuable analytical tools or reagents and intellectual input and edited the manuscript; and S.N. designed the research, performed the experiments, coordinated the project activities, and wrote the manuscript.
Conflict-of-interest disclosure: M.M. and T.E.R. are employed by Reichert Inc, manufacturers of the SPR instrumentation used in this manuscript. R.R.M is a consultant to Gen Probe/GTI Diagnostic, CSL Behring and Ocrapharma. The remaining authors declare no competing financial interests.
Correspondence: Sriram Neelamegham, 906 Furnas Hall, State University of New York, Buffalo, NY 14260; e-mail: neel@buffalo.edu.
References
- 1.Ruggeri ZM. Von Willebrand factor: looking back and looking forward. Thromb Haemost. 2007;98(1):55–62. [PubMed] [Google Scholar]
- 2.Wagner DD. Cell biology of von Willebrand factor. Annu Rev Cell Biol. 1990;6:217–246. doi: 10.1146/annurev.cb.06.110190.001245. [DOI] [PubMed] [Google Scholar]
- 3.Valentijn KM, Sadler JE, Valentijn JA, Voorberg J, Eikenboom J. Functional architecture of Weibel-Palade bodies. Blood. 2011;117(19):5033–5043. doi: 10.1182/blood-2010-09-267492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayadas TN, Wagner DD. Vicinal cysteines in the prosequence play a role in von Willebrand factor multimer assembly. Proc Natl Acad Sci U S A. 1992;89(8):3531–3535. doi: 10.1073/pnas.89.8.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haberichter SL, Jozwiak MA, Rosenberg JB, Christopherson PA, Montgomery RR. The von Willebrand factor propeptide (VWFpp) traffics an unrelated protein to storage. Arterioscler Thromb Vasc Biol. 2002;22(6):921–926. doi: 10.1161/01.atv.0000017063.36768.87. [DOI] [PubMed] [Google Scholar]
- 6.Verweij CL, Hart M, Pannekoek H. Expression of variant von Willebrand factor (vWF) cDNA in heterologous cells: requirement of the pro-polypeptide in vWF multimer formation. EMBO J. 1987;6(10):2885–2890. doi: 10.1002/j.1460-2075.1987.tb02591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wise RJ, Pittman DD, Handin RI, Kaufman RJ, Orkin SH. The propeptide of von Willebrand factor independently mediates the assembly of von Willebrand multimers. Cell. 1988;52(2):229–236. doi: 10.1016/0092-8674(88)90511-9. [DOI] [PubMed] [Google Scholar]
- 8.Haberichter SL, Allmann AM, Jozwiak MA, Montgomery RR, Gill JC. Genetic alteration of the D2 domain abolishes von Willebrand factor multimerization and trafficking into storage. J Thromb Haemost. 2009;7(4):641–650. doi: 10.1111/j.1538-7836.2009.03290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayadas TN, Wagner DD. In vitro multimerization of von Willebrand factor is triggered by low pH. Importance of the propolypeptide and free sulfhydryls. J Biol Chem. 1989;264(23):13497–13503. [PubMed] [Google Scholar]
- 10.Wagner DD, Mayadas T, Marder VJ. Initial glycosylation and acidic pH in the Golgi apparatus are required for multimerization of von Willebrand factor. J Cell Biol. 1986;102(4):1320–1324. doi: 10.1083/jcb.102.4.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenberg JB, Haberichter SL, Jozwiak MA, et al. The role of the D1 domain of the von Willebrand factor propeptide in multimerization of VWF. Blood. 2002;100(5):1699–1706. doi: 10.1182/blood-2002-03-0789. [DOI] [PubMed] [Google Scholar]
- 12.Sporn LA, Marder VJ, Wagner DD. Inducible secretion of large, biologically potent von Willebrand factor multimers. Cell. 1986;46(2):185–190. doi: 10.1016/0092-8674(86)90735-x. [DOI] [PubMed] [Google Scholar]
- 13.Weibel ER, Palade GE. New Cytoplasmic Components in Arterial Endothelia. J Cell Biol. 1964;23:101–112. doi: 10.1083/jcb.23.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metcalf DJ, Nightingale TD, Zenner HL, Lui-Roberts WW, Cutler DF. Formation and function of Weibel-Palade bodies. J Cell Sci. 2008;121(pt 1):19–27. doi: 10.1242/jcs.03494. [DOI] [PubMed] [Google Scholar]
- 15.Sporn LA, Chavin SI, Marder VJ, Wagner DD. Biosynthesis of von Willebrand protein by human megakaryocytes. J Clin Invest. 1985;76(3):1102–1106. doi: 10.1172/JCI112064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cramer EM, Meyer D, le Menn R, Breton-Gorius J. Eccentric localization of von Willebrand factor in an internal structure of platelet alpha-granule resembling that of Weibel-Palade bodies. Blood. 1985;66(3):710–713. [PubMed] [Google Scholar]
- 17.Wagner DD, Fay PJ, Sporn LA, Sinha S, Lawrence SO, Marder VJ. Divergent fates of von Willebrand factor and its propolypeptide (von Willebrand antigen II) after secretion from endothelial cells. Proc Natl Acad Sci U S A. 1987;84(7):1955–1959. doi: 10.1073/pnas.84.7.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hannah MJ, Skehel P, Erent M, Knipe L, Ogden D, Carter T. Differential kinetics of cell surface loss of von Willebrand factor and its propolypeptide after secretion from Weibel-Palade bodies in living human endothelial cells. J Biol Chem. 2005;280(24):22827–22830. doi: 10.1074/jbc.M412547200. [DOI] [PubMed] [Google Scholar]
- 19.Vischer UM, Wagner DD. von Willebrand factor proteolytic processing and multimerization precede the formation of Weibel-Palade bodies. Blood. 1994;83(12):3536–3544. [PubMed] [Google Scholar]
- 20.Ulrichts H, Udvardy M, Lenting PJ, et al. Shielding of the A1 domain by the D′D3 domains of von Willebrand factor modulates its interaction with platelet glycoprotein Ib-IX-V. J Biol Chem. 2006;281(8):4699–4707. doi: 10.1074/jbc.M513314200. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y, Dong N, Shen F, et al. Two novel monoclonal antibodies to VWFA3 inhibit VWF-collagen and VWF-platelet interactions. J Thromb Haemost. 2007;5(9):1963–1970. doi: 10.1111/j.1538-7836.2007.02682.x. [DOI] [PubMed] [Google Scholar]
- 22.Haberichter SL, Balistreri M, Christopherson P, et al. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood. 2006;108(10):3344–3351. doi: 10.1182/blood-2006-04-015065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haberichter SL, Castaman G, Budde U, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 VWD (MCMDM-1VWD). Blood. 2008;111(10):4979–4985. doi: 10.1182/blood-2007-09-110940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh I, Shankaran H, Beauharnois ME, Xiao Z, Alexandridis P, Neelamegham S. Solution structure of human von Willebrand factor studied using small angle neutron scattering. J Biol Chem. 2006;281(50):38266–38275. doi: 10.1074/jbc.M607123200. [DOI] [PubMed] [Google Scholar]
- 25.Dayananda KM, Gogia S, Neelamegham S. Escherichia coli-derived von Willebrand factor-A2 domain fluorescence/Forster resonance energy transfer proteins that quantify ADAMTS13 activity. Anal Biochem. 2011;410(2):206–213. doi: 10.1016/j.ab.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shankaran H, Alexandridis P, Neelamegham S. Aspects of hydrodynamic shear regulating shear-induced platelet activation and self-association of von Willebrand factor in suspension. Blood. 2003;101(7):2637–2645. doi: 10.1182/blood-2002-05-1550. [DOI] [PubMed] [Google Scholar]
- 27.Lahiri J, Isaacs L, Tien J, Whitesides GM. A strategy for the generation of surfaces presenting ligands for studies of binding based on an active ester as a common reactive intermediate: a surface plasmon resonance study. Anal Chem. 1999;71(4):777–790. doi: 10.1021/ac980959t. [DOI] [PubMed] [Google Scholar]
- 28.Dayananda KM, Singh I, Mondal N, Neelamegham S. von Willebrand factor self-association on platelet GpIbalpha under hydrodynamic shear: effect on shear-induced platelet activation. Blood. 2010;116(19):3990–3998. doi: 10.1182/blood-2010-02-269266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myszka DG, Morton TA. CLAMP: a biosensor kinetic data analysis program. Trends Biochem Sci. 1998;23(4):149–150. doi: 10.1016/s0968-0004(98)01183-9. [DOI] [PubMed] [Google Scholar]
- 30.Ahmed F, Alexandridis P, Shankaran H, Neelamegham S. The ability of poloxamers to inhibit platelet aggregation depends on their physicochemical properties. Thromb Haemost. 2001;86(6):1532–1539. [PubMed] [Google Scholar]
- 31.Neeves KB, Maloney SF, Fong KP, et al. Microfluidic focal thrombosis model for measuring murine platelet deposition and stability: PAR4 signaling enhances shear-resistance of platelet aggregates. J Thromb Haemost. 2008;6(12):2193–2201. doi: 10.1111/j.1538-7836.2008.03188.x. [DOI] [PubMed] [Google Scholar]
- 32.Purvis AR, Sadler JE. A covalent oxidoreductase intermediate in propeptide-dependent von Willebrand factor multimerization. J Biol Chem. 2004;279(48):49982–49988. doi: 10.1074/jbc.M408727200. [DOI] [PubMed] [Google Scholar]
- 33.Huang RH, Wang Y, Roth R, et al. Assembly of Weibel-Palade body-like tubules from N-terminal domains of von Willebrand factor. Proc Natl Acad Sci U S A. 2008;105(2):482–487. doi: 10.1073/pnas.0710079105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lenting PJ, Westein E, Terraube V, et al. An experimental model to study the in vivo survival of von Willebrand factor. Basic aspects and application to the R1205H mutation. J Biol Chem. 2004;279(13):12102–12109. doi: 10.1074/jbc.M310436200. [DOI] [PubMed] [Google Scholar]
- 35.Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4(10):2103–2114. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 36.Lillicrap D. Genotype/phenotype association in von Willebrand disease: is the glass half full or empty? J Thromb Haemost. 2009;7(Suppl 1):65–70. doi: 10.1111/j.1538-7836.2009.03367.x. [DOI] [PubMed] [Google Scholar]
- 37.Haberichter SL, Jacobi P, Montgomery RR. Critical independent regions in the VWF propeptide and mature VWF that enable normal VWF storage. Blood. 2003;101(4):1384–1391. doi: 10.1182/blood-2002-07-2281. [DOI] [PubMed] [Google Scholar]
- 38.Borchiellini A, Fijnvandraat K, ten Cate JW, et al. Quantitative analysis of von Willebrand factor propeptide release in vivo: effect of experimental endotoxemia and administration of 1-deamino-8-D-arginine vasopressin in humans. Blood. 1996;88(8):2951–2958. [PubMed] [Google Scholar]
- 39.van Mourik JA, Boertjes R, Huisveld IA, et al. von Willebrand factor propeptide in vascular disorders: A tool to distinguish between acute and chronic endothelial cell perturbation. Blood. 1999;94(1):179–185. [PubMed] [Google Scholar]
- 40.Wise RJ, Dorner AJ, Krane M, Pittman DD, Kaufman RJ. The role of von Willebrand factor multimers and propeptide cleavage in binding and stabilization of factor VIII. J Biol Chem. 1991;266(32):21948–21955. [PubMed] [Google Scholar]
- 41.Bendetowicz AV, Morris JA, Wise RJ, Gilbert GE, Kaufman RJ. Binding of factor VIII to von willebrand factor is enabled by cleavage of the von Willebrand factor propeptide and enhanced by formation of disulfide-linked multimers. Blood. 1998;92(2):529–538. [PubMed] [Google Scholar]
- 42.Shim K, Anderson PJ, Tuley EA, Wiswall E, Sadler JE. Platelet-VWF complexes are preferred substrates of ADAMTS13 under fluid shear stress. Blood. 2008;111(2):651–657. doi: 10.1182/blood-2007-05-093021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castaman G, Montgomery RR, Meschengieser SS, Haberichter SL, Woods AI, Lazzari MA. von Willebrand's disease diagnosis and laboratory issues. Haemophilia. 2010;16(suppl 5):67–73. doi: 10.1111/j.1365-2516.2010.02296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin C, Morales LD, Cruz MA. Purified A2 domain of von Willebrand factor binds to the active conformation of von Willebrand factor and blocks the interaction with platelet glycoprotein Ibalpha. J Thromb Haemost. 2007;5(7):1363–1370. doi: 10.1111/j.1538-7836.2007.02536.x. [DOI] [PubMed] [Google Scholar]
- 45.Nishio K, Anderson PJ, Zheng XL, Sadler JE. Binding of platelet glycoprotein Ibalpha to von Willebrand factor domain A1 stimulates the cleavage of the adjacent domain A2 by ADAMTS13. Proc Natl Acad Sci U S A. 2004;101(29):10578–10583. doi: 10.1073/pnas.0402041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao W, Krishnaswamy S, Camire RM, Lenting PJ, Zheng XL. Factor VIII accelerates proteolytic cleavage of von Willebrand factor by ADAMTS13. Proc Natl Acad Sci U S A. 2008;105(21):7416–7421. doi: 10.1073/pnas.0801735105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.