

A general approach for targeted gene modification with precise external control and high spatial and temporal resolution will greatly advance investigations in genetics, gene therapy, and developmental biology. However, traditional methods such as homologous recombination[1] and nonhomologous end joining[2] for the introduction and deletion of genomic DNA sequences usually display very low efficiency in vivo, thus limiting their applicability. Recently, the efficiency of these processes has been greatly improved by the ability to site-specifically introduce doublestrand breaks (DSBs) into genomic DNA.[3,4] A family of artificial restriction enzymes, namely zinc-finger nucleases (ZFN), has been developed to sequence-selectively achieve dsDNA scission. ZFNs have since emerged as important and widely recognized tools for the genetic modification of cells, model organisms, and possibly humans to investigate gene function and to treat genetic disorders.[5–9]
Structurally, a ZFN is a chimeric protein containing two domains: an N-terminal zinc-finger domain and a C-terminal nuclease domain. The N-terminal zinc-finger domain usually consists of three to four Cys2His2 “fingers”.[10–13] Each finger recognizes three DNA base-pairs through hydrogen-bonding interactions in the major groove of the DNA. The binding specificities of these fingers to certain DNA sequences can be engineered by selection[14] or modular assembly[15] and subsequent in vivo testing.[16] The C-terminal nuclease domain was evolved from the type IIS restriction enzyme FokI[17] and is dimerized in a tail-to-tail conformation with a second ZFN to introduce a DSB between two recognition sites. ZFN heterodimers recognize a 24 bp composite DNA site which statistically guarantees single occurrence in the genome of targeted cells and organisms.[6]
A potential problem of this design is the dimerization of the nuclease domain without sequence-specific DNA binding, thus potentially leading to nonspecific, off-target cleavage[18] and cellular toxicity.[19] The resulting toxicity has caused considerable problems, and various efforts have been focused on modifying the nuclease domain to diminish these side effects. Miller et al.[20] and Szczepek et al.[21] redesigned dimer interfaces by using structure-based approaches. This has been successfully applied to produce a ZFN-mediated kdr-1 gene knockout in zebrafish embryos.[22] Moreover, destabilizing domains were fused to ZFN proteins to shorten the nuclease half-life, thus reducing the cellular toxicity.[23]
Amid these previous efforts on the modification of nuclease domains, taming the nuclease activity on a molecular level remains an innovative yet unproven concept. We reasoned that by introducing a caged amino acid into the reaction center of the ZFN, its activity should be controllable by UV irradiation, thereby not only providing an opportunity to solve the toxicity problem, but also to control ZFN-mediated gene editing with high spatial and temporal resolution. A light-activated ZFN will enable the conditional generation of gene knock-ins and knock-outs with unprecedented precision. This strategy is based on our previous successes in photochemically controlling β-galactosidase,[24] DNA and RNA polymerases,[25,26] Cre recombinase,[27] protein localization,[28] and kinase activity.[29] In these cases, photocaged amino acids, e.g., ortho-nitrobenzyltyrosine (ONBY, 1), were site-specifically introduced at essential sites of the corresponding proteins to enable the regulation of biological processes with light.[30–34]
Here, we report a light-activatable ZFN, generated through the site-specific incorporation of 1 by cells with an expanded genetic code.[35–37] Furthermore, light-activated gene editing is demonstrated by the activation of luciferase expression in mammalian cells by using a single-strand-annealing (SSA) homologous recombination approach.[38] These findings lay the foundation to achieve spatiotemporal control of sequence-specific genome editing using light as a non-invasive regulatory element.
To identify a tyrosine residue close to the activity center of the ZFN we evaluated several residues in the FokI nuclease domain on the basis of crystallographic data,[17] and selected tyrosine residue Tyr471 close to the putative activity center. In the dimerized and DNA-bound FokI nuclease, the hydroxy group of Y471 points directly into the DNA binding cleft (Figure 1a). Introduction of a sterically demanding ortho-nitrobenzyl group may directly interfere with the protein–DNA interaction and thus render the nuclease catalytically inactive. In addition, the Tyr471 position is located on the protein surface (Figure 1b), thus incorporation of 1 instead of tyrosine will most likely not cause structural perturbation and protein misfolding of the caged ZFN.
Figure 1.
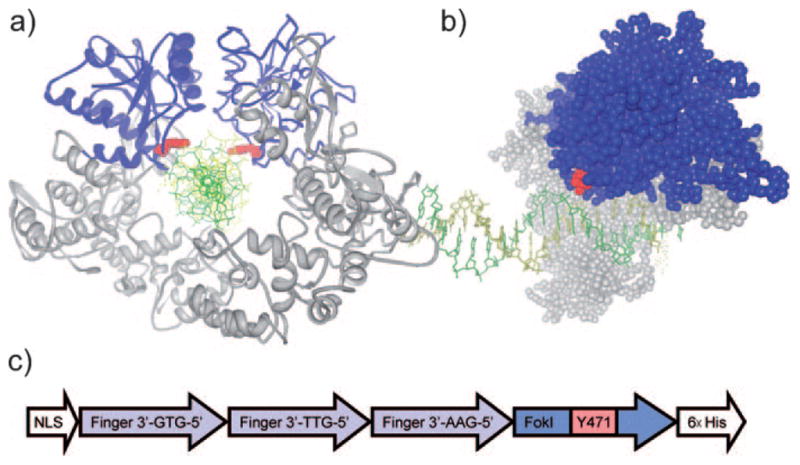
a) Schematic representation of dimerized FokI binding to substrate DNA (PDB: 2FOK). The nuclease and binding domain are shown in blue and gray, respectively, and the DNA helix is shown in green/yellow. The two symmetrical Tyr471 residues are marked in red, and the introduction of an ONB group on the Tyr471-OH may well extend into the DNA binding interface, thus preventing the nuclease domains on both ZFNs from tightly binding to the DNA backbone. b) Side view of DNA-bound FokI nuclease (PDB: 1FOK). Tyr471 is located at the nuclease surface. c) Schematic representation of the zinc-finger nuclease gene expressing the caged ZFN.
To test this hypothesis we cloned a ZFN used for knockout of the zebrafish kdr-1 gene[22] into a pET24d plasmid. The chimeric protein contains an N-terminal nuclear localization signal (NLS), three zinc-finger motifs that recognize a nine-nucleotide DNA sequence (5′-GAAGGTGTG-3′), a FokI nuclease domain, and a C-terminal hexahistidine tag (Figure 1c). The protein was successfully expressed, purified, and tested for its single-strand break (SSB) inducing activity, which indicated that the added C-terminal hexahistidine tag did not interfere with the activity. Thus, a TAG mutation was introduced for direct incorporation of 1 at the Tyr471 position. The resulting pET24d-NLS-ZFNY471TAG was then transformed into chemically competent BL21Gold cells expressing the MjtRNACUA/MjYRSwt or MjYRSONBY pair.[39] Transformed cells were incubated in the presence of 1, protein expression was induced with IPTG, and the caged ZFN was isolated and purified on an Ni-NTA column.[16] The expressed and purified proteins where analyzed by SDS-PAGE (Figure 2). No protein was obtained in the absence of 1, thus indicating a high fidelity of the evolved MjYRSONBY for 1. In the presence of the caged tyrosine 1 (1 mM), a 39 kDa band for caged ZFN was identified which corresponded well to the wild-type ZFN. Moreover, expression levels of caged (1.3 μgmL−1) and wild-type (0.9μgmL−1) ZFN were comparable.
Figure 2.

Structure of the caged tyrosine 1, and Coomassie-stained SDS-PAGE (sodium dodecylsulfate polyacrylamide gel electrophoresis) of the expression of the photocaged ZFN through incorporation of 1 at position Y471. M: NEB broad-range protein marker. WT: wild-type ZFN expressed from pET24d-NLS-ZFNY471TAG plasmid with MjYRSwt (expressed from pEVOL-wt). +1: caged ZFN expressed from pET24d-NLS-ZFNY471TAG and MjYRSONBY (expressed from pEVOL-ONBY) grown in medium supplemented with 1 (1 mM). −1: caged ZFN expressed from pET24dNLS-ZFNY471TAG and pEVOL-ONBY in the absence of 1.
We then investigated the light-activation of zinc-finger nuclease activity. A substrate plasmid (pSSA-GAA-GAA) was constructed that contained a 24 bp recognition site that is site-specifically cut by the ZFN enzyme causing a DSB and relieving the DNA from its supercoiled (SC) form. The reaction mixtures were irradiated at 365 nm and the reactions were analyzed by gel electrophoresis. The agarose gels shown in Figure 3 reveal that UV irradiation did not affect wild-type ZFN activity. Gratifyingly, the caged ZFN enzyme was completely inactive before UV irradiation, and approximately 70% of the activity was restored by exposure to UV light of 365 nm for 1 min. By extending the irradiation time from 1 min to 5 or 10 min, complete DSB, comparable to treatment with the wild-type enzyme, was achieved. These results indicate that the site-specifically caged ZFN is completely inactive and that its activity can be restored through UV-light-induced removal of the caging group on Tyr471.
Figure 3.

Light-induced decaging and ZFN activation. The ethidium bromide stained agarose gels show DNA is digested using caged and decaged ZFN. WT: wild-type ZFN. Caged: ZFNONBY471. (–): no ZFN enzyme. 0, 1, 2, 5, 10: UV (365 nm) exposure time in minutes.
The in vitro experiments confirmed that ZFNs can be engineered by unnatural amino acid mutagenesis to be light-activatable. Next, we aimed to demonstrate that these zinc-finger nucleases could be used for gene editing in vivo, such as in mammalian tissue culture.[38] Based on a generalized in vivo strategy to test newly designed ZFNs,[38] the plasmid pSSA-GAA-GAA was used to assay ZFN-mediated SSA recombination intracellularly (Figure 4). If the DSB is introduced by the light-activated ZFN, the two homologous DNA strands will be processed by intracellular exonucleases to create single-stranded regions. The complementary DNA will then be annealed and nonhomologous overhangs will be removed.[40] These events result in the repairing of a previously nonfunctional luciferase gene and lead to the expression of intact luciferase enzyme. As a result, the luciferase activity is proportional to the (light-induced) zinc-finger nuclease activity in vivo.[38]
Figure 4.

General strategy for light-activated DSB induction through a caged ZFN in mammalian cells. A stop codon was introduced into the reporter after the NH region, thereby leading to the expression of truncated protein without luciferase activity. Light-irradiation induces ZFN decaging, followed by a site-specific DSB between two luciferase segments (N-Luc and C-Luc) that share repeated regions (NH and CH). The DNA cleavage enables efficient homologous recombination to produce an active luciferase gene, and the luciferase assay results correspond to the ZFN activity after UV irradiation.
Caged and noncaged ZFNs, together with the luciferase reporter plasmid, were co-transfected into human embryonic kidney (HEK293T) cells. The cells were incubated with the transfection mix, briefly irradiated with light of 365 nm or kept in the dark, and assayed for luciferase activity after 36 h. Without irradiation, HEK293T cells that were transfected with the caged ZFN enzyme only show a basal luciferase expression level identical to the negative control (no enzyme). After a brief (5 min) UV irradiation (365 nm) of the cells, the now decaged and activated ZFN induces an efficient and site-specific DSB, thereby leading to full restoration of the luciferase activity to the level of the wild-type control (Figure 5). These experiments demonstrate that light-activated, sequence-specific gene editing can be achieved with site-specifically caged ZFNs in mammalian cells.
Figure 5.

Light-activation of homologous recombination and gene activation in mammalian cells by ZFN-mediated DSB and subsequent repair of the luciferase reporter gene. The data is normalized to the wild-type zinc-finger nuclease and the background (absence of any zinc-finger nuclease enzyme) was subtracted. The error bars represent standard deviations from three independent experiments. Dark gray bars represent samples kept in the dark and light gray bars represent irradiated samples.
In summary, we have demonstrated that the activity of zinc-finger nucleases (ZFNs) can be photochemically regulated by site-specifically introducing an ortho-nitrobenzyl group at a rationally selected position (Tyr471) close to the activity center of the nuclease. Although the selected tyrosine residue is conserved in FokI and StsI restriction enzymes, its actual catalytic function is unknown. The caged ZFN enzyme was expressed in cells with an expanded genetic code that allowed for the site-specific incorporation of the photocaged tyrosine 1 by using an engineered, orthogonal tRNA/tRNA synthetase pair. Based on structural information, the sterically demanding ONB caging group placed on Tyr471 most likely leads to an unfavorable steric interaction of the nuclease with the DNA substrate, thus preventing the ZFN from cleaving the phosphodiester backbone; importantly, the caged enzyme has no background activity. Nuclease activity can be readily restored through brief irradiation with UV light of 365 nm. An important feature of our design is that the caging of Y471 in the ZFN is fully compatible with additional zinc-finger engineering, such as the design of a tandem array of DNA-binding motifs, dimer interface engineering, enhanced nuclease activity and protein destabilization.[23,41] This highly modular approach will provide a unifying strategy to engineer photocaged ZFNs that target any gene of interest and generate light-activated artificial genome targeting nucleases by using the FokI nuclease domain, such as the most recently emerged transcription-like effector (TALE) fusion proteins.[42–45] Further engineering efforts to overcome protein stability and cellular permeability, such as fusion with maltose binding protein[46] and introduction of a TAT translocation domain,[47] may also be applied to the artificial ZFN design. Overall, this light-activation method lays the groundwork for future studies to enable the manipulation of genomic information with complete sequence specificity and single-cell resolution in multicellular organisms at any time point.
Experimental Section
ZFN expression and in vivo assay: E. coli BL21 cells harboring the plasmids pEVOL-ONBY and pET24d-NLS-ZFNY471TAG were used to inoculate 2 × YT medium (200 mL) supplemented with kanamycin (50 μgmL−1), chloramphenicol (34 μgmL−1), 10M NaOH (100 μL), 1 (1 mM), and ZnCl2 (0.1 mM final concentration). The culture was grown at 37°C for 3 h to reach an OD600 of 0.6. The expression of the caged and wild-type proteins was induced by addition of arabinose (0.02% final concentration) and IPTG (isopropyl β-D-1-thiogalactopyranoside; 0.1 mM final concentration). The cultures were transferred to an 18°C shaker and kept at that temperature for an overnight expression. The cells were harvested and purified according to a slightly modified Ni-NTA (NTA =nickel nitriloacetic acid) resin based protocol,[16] with the addition of 0.2 mM TCEP (tris(2-carboxyethyl)phosphine), 0.1 mM ZnCl2, and 0.1 mM MgCl2 to increase protein stability. The eluted protein was analyzed by SDS-PAGE and Bradford quantification.
HEK293 cells were seeded in polylysine-treated 96-well plates. eluted caged ZFN (0.5 μgμL−1, 40 μL) and the reporter plasmid were co-transfected with Lipofectamine 2000 (Invitrogen). Wild-type ZFN (0.5 μgμL−1) and just elution buffer were also transfected to serve as positive and negative controls, respectively. The cells were incubated with the protein/DNA/Lipofectamine mix for 90 min. The mixture was decanted and replaced with complete DMEM (Dulbecco3s modified eagle medium) containing 10% FBS (fetal bovine serum) and cells were exposed to UV light on a transilluminator (365 nm, 5 min) or kept in the dark. The plates were then incubated for 36 h in a CO2 incubator at 37°C and the luciferase activity was quantified with a BrightGlo assay (Promega) and a luminescence plate reader (Biotek).
Supplementary Material
Footnotes
We gratefully acknowledge financial support from the Beckman Foundation (Beckman Young Investigator Award to A.D.), Research Corporation (Cottrell Scholar Award to A.D.), and the NIH (GM079114). We thank P. G. Schultz (TSRI) for the pEVOL plasmid and D. J. Segal (UC Davis) for the pSSA plasmid.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201101157.
References
- 1.Bibikova M, Carroll D, Segal DJ, Trautman JK, Smith J, Kim YG, Chandrasegaran S. Mol Cell Biol. 2001;21:289. doi: 10.1128/MCB.21.1.289-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bozas A, Beumer KJ, Trautman JK, Carroll D. Genetics. 2009;182:641. doi: 10.1534/genetics.109.101329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright DA, Townsend JA, Winfrey RJ, Irwin PA, Rajagopal J, Lonosky PM, Hall BD, Jondle MD, Voytas DF. Plant J. 2005;44:693. doi: 10.1111/j.1365-313X.2005.02551.x. [DOI] [PubMed] [Google Scholar]
- 4.Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC. Nature. 2005;435:646. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 5.Le Provost F, Lillico S, Passet B, Young R, Whitelaw B, Vilotte JL. Trends Biotechnol. 2010;28:134. doi: 10.1016/j.tibtech.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Katada H, Komiyama M. ChemBioChem. 2009;10:1279. doi: 10.1002/cbic.200900040. [DOI] [PubMed] [Google Scholar]
- 7.Porteus MH. Mol Ther. 2006;13:438. doi: 10.1016/j.ymthe.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Porteus MH, Carroll D. Nat Biotechnol. 2005;23:967. doi: 10.1038/nbt1125. [DOI] [PubMed] [Google Scholar]
- 9.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Nat Rev Genet. 2010;11:636. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 10.Dreier B, Segal DJ, Barbas CF. J Mol Biol. 2000;303:489. doi: 10.1006/jmbi.2000.4133. [DOI] [PubMed] [Google Scholar]
- 11.Dreier B, Beerli RR, Segal DJ, Flippin JD, Barbas CF. J Biol Chem. 2001;276:29466. doi: 10.1074/jbc.M102604200. [DOI] [PubMed] [Google Scholar]
- 12.Dreier B, Fuller RP, Segal DJ, Lund CV, Blancafort P, Huber A, Koksch B, Barbas CF. J Biol Chem. 2005;280:35588. doi: 10.1074/jbc.M506654200. [DOI] [PubMed] [Google Scholar]
- 13.Papworth M, Kolasinska P, Minczuk M. Gene. 2006;366:27. doi: 10.1016/j.gene.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Hurt JA, Thibodeau SA, Hirsh AS, Pabo CO, Joung JK. Proc Natl Acad Sci USA. 2003;100:12271. doi: 10.1073/pnas.2135381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, Curtin SJ, Blackburn JS, Thibodeau-Beganny S, Qi Y, Pierick CJ, Hoffman E, Maeder ML, Khayter C, Reyon D, Dobbs D, Langenau DM, Stupar RM, Giraldez AJ, Voytas DF, Peterson RT, Yeh JRJ, Joung JK. Nat Methods. 2011;8:67. doi: 10.1038/nmeth.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll D, Morton JJ, Beumer KJ, Segal DJ. Nat Protoc. 2006;1:1329. doi: 10.1038/nprot.2006.231. [DOI] [PubMed] [Google Scholar]
- 17.Wah DA, Bitinaite J, Schildkraut I, Aggarwal AK. Proc Natl Acad Sci USA. 1998;95:10564. doi: 10.1073/pnas.95.18.10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Radecke S, Radecke F, Cathomen T, Schwarz K. Mol Ther. 2010;18:743. doi: 10.1038/mt.2009.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bohne J, Cathomen T. Curr Opin Mol Ther. 2008;10:214. [PubMed] [Google Scholar]
- 20.Miller JC, Holmes MC, Wang JB, Guschin DY, Lee YL, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, Gregory PD, Pabo CO, Rebar EJ. Nat Biotechnol. 2007;25:778. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- 21.Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T. Nat Biotechnol. 2007;25:786. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- 22.Meng XD, Noyes MB, Zhu LHJ, Lawson ND, Wolfe SA. Nat Biotechnol. 2008;26:695. doi: 10.1038/nbt1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pruett-Miller SM, Reading DW, Porter SN, Porteus MH. PLOS Genet. 2009;5:e1000376. doi: 10.1371/journal.pgen.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deiters A, Groff D, Ryu YH, Xie JM, Schultz PG. Angew Chem. 2006;118:2794. doi: 10.1002/anie.200600264. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:2728. [Google Scholar]
- 25.Chou CJ, Young DD, Deiters A. Angew Chem. 2009;121:6064. doi: 10.1002/anie.200901115. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:5950. [Google Scholar]
- 26.Chou CJ, Young DD, Deiters A. ChemBioChem. 2010;11:972. doi: 10.1002/cbic.201000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edwards WF, Young DD, Deiters A. ACS Chem Biol. 2009;4:441. doi: 10.1021/cb900041s. [DOI] [PubMed] [Google Scholar]
- 28.Gautier A, Nguyen DP, Lusic H, An WA, Deiters A, Chin JW. J Am Chem Soc. 2010;132:4086. doi: 10.1021/ja910688s. [DOI] [PubMed] [Google Scholar]
- 29.Gautier A, Deiters A, Chin JW. J Am Chem Soc. 2011;133:2124. doi: 10.1021/ja1109979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young DD, Deiters A. Org Biomol Chem. 2007;5:999. doi: 10.1039/b616410m. [DOI] [PubMed] [Google Scholar]
- 31.Deiters A. Curr Opin Chem Biol. 2009;13:678. doi: 10.1016/j.cbpa.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riggsbee CW, Deiters A. Trends Biotechnol. 2010;28:468. doi: 10.1016/j.tibtech.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee HM, Larson DR, Lawrence DS. ACS Chem Biol. 2009;4:409. doi: 10.1021/cb900036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayer G, Heckel A. Angew Chem. 2006;118:5020. doi: 10.1002/anie.200600387. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:4900. [Google Scholar]
- 35.Wu X, Schultz PG. J Am Chem Soc. 2009;131:12497. doi: 10.1021/ja9026067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu CC, Schultz PG. Annu Rev Biochem. 2010;79:413. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Schultz PG. Chem Commun. 2002:1. doi: 10.1039/b108185n. [DOI] [PubMed] [Google Scholar]
- 38.Bhakta MS, Segal DJ. Methods Mol Biol. 2010;649:3. doi: 10.1007/978-1-60761-753-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young TS, Ahmad I, Yin JA, Schultz PG. J Mol Biol. 2010;395:361. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 40.Valerie K, Povirk LF. Oncogene. 2003;22:5792. doi: 10.1038/sj.onc.1206679. [DOI] [PubMed] [Google Scholar]
- 41.Guo J, Gaj T, Barbas CF. J Mol Biol. 2010;400:96. doi: 10.1016/j.jmb.2010.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ. Nat Biotechnol. 2011;29:143. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 43.Li T, Huang S, Jiang WZ, Wright D, Spalding MH, Weeks DP, Yang B. Nucleic Acids Res. 2011;39:359. doi: 10.1093/nar/gkq704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahfouz MM, Li L, Shamimuzzaman M, Wibowo A, Fang X, Zhu JK. Proc Natl Acad Sci USA. 2011;108:2623. doi: 10.1073/pnas.1019533108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Genetics. 2010;110:120717. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cathomen T, Segal DJ, Brondani V, Mueller-Lerch F. Methods Mol Biol. 2008;434:277. doi: 10.1007/978-1-60327-248-3_17. [DOI] [PubMed] [Google Scholar]
- 47.Peitz M, Pfannkuche K, Rajewsky K, Edenhofer F. Proc Natl Acad Sci USA. 2002;99:4489. doi: 10.1073/pnas.032068699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.