

Abstract
Postnatal deficits in brain-derived neurotrophic factor (BDNF) are thought to contribute to pathogenesis of Rett syndrome (RTT), a progressive neurodevelopmental disorder caused by mutations in the gene encoding methyl-CpG-binding protein 2 (MeCP2). In Mecp2-null mice, a model of RTT, BDNF deficits are most pronounced in structures important for autonomic and respiratory control, functions that are severely affected in RTT patients. However, relatively little is known about how these deficits affect neuronal function or how they may be linked to specific RTT endophenotypes. To approach these issues, we analyzed synaptic function in the brainstem nucleus tractus solitarius (nTS), the principal site for integration of primary visceral afferent inputs to central autonomic pathways and a region in which we found markedly reduced levels of BDNF in Mecp2 mutants. Our results demonstrate that the amplitude of spontaneous miniature and evoked EPSCs in nTS neurons is significantly increased in Mecp2-null mice and, accordingly, that mutant cells are more likely than wild- type cells to fire action potentials in response to primary afferent stimulation. These changes occur without any increase in intrinsic neuronal excitability and are unaffected by blockade of inhibitory GABA currents. However, this synaptopathy is associated with decreased BDNF availability in the primary afferent pathway and can be rescued by application of exogenous BDNF. On the basis of these findings, we hypothesize that altered sensory gating in nTS contributes to cardiorespiratory instability in RTT and that nTS is a site at which restoration of normal BDNF signaling could help reestablish normal homeostatic controls.
Introduction
Loss-of-function mutations in the gene encoding methyl-CpG-binding protein 2 (MeCP2) cause most cases of Rett syndrome (RTT), a severe autism spectrum disorder characterized by cognitive, motor, and behavioral deficits as well as marked respiratory arrhythmia and cardiac and gastrointestinal dysautonomia (Chahrour and Zoghbi, 2007). MeCP2 is a transcriptional regulatory protein, and in its absence, large numbers of genes exhibit abnormal increases or decreases in expression. Therefore, a major challenge in understanding the molecular pathogenesis of RTT has been to link dysregulation of specific gene targets of MeCP2 to particular pathophysiological outcomes.
One consequence of MeCP2 loss in mouse models and RTT patients is reduced expression of brain-derived neurotrophic factor (BDNF) after birth (Chang et al., 2006; Wang et al., 2006; Deng et al., 2007). In Mecp2-null mice, the earliest and most dramatic BDNF deficits occur in structures critical for respiratory and autonomic control, including the cranial sensory nodose ganglion and brainstem (Wang et al., 2006). In view of the fact that BDNF normally regulates synaptic function in brainstem respiratory and autonomic cell groups, including the nucleus tractus solitarius [nTS; the principal target of peripheral sensory neurons that convey visceral afferent input to central autonomic pathways (Balkowiec et al., 2000)], preBötzinger complex (Thoby-Brisson et al., 2003), and Kölliker-Fuse nucleus (Kron et al., 2007a,b, 2008), we have hypothesized that reduction of BDNF availability contributes to the disruption of autonomic homeostasis in Mecp2-null mice and, possibly, RTT patients (Ogier and Katz, 2008; Katz et al., 2009). This hypothesis is supported by findings from our laboratory that treatment of Mecp2-null mice with the ampakine CX546 increases BDNF expression and improves RTT-like respiratory deficits (Ogier et al., 2007).
To further define how BDNF deficits associated with loss of MeCP2 may affect respiratory and autonomic network function, the present study first compared the distribution of BDNF protein in the medulla from wild-type and Mecp2-null mice. We then focused on nTS, one of the medullary cell groups exhibiting the largest decline in BDNF levels in the mutants, as a model to examine the effect of reduced BDNF availability on synaptic transmission in Mecp2-null mice. Previous studies in our laboratory showed that BDNF can modulate postsynaptic responses of nTS relay neurons to glutamate by inhibiting postsynaptic AMPA receptors (Balkowiec et al., 2000). Therefore, we hypothesized that nTS neurons in Mecp2-null mice would exhibit exaggerated responses to primary afferent stimulation due to loss of this BDNF-dependent modulation. The present study demonstrates that nTS relay neurons in Mecp2-null mice exhibit significantly larger EPSCs, and are more likely to fire action potentials in response to afferent stimulation, than in wild-type controls. Normal synaptic responses are restored, however, in Mecp2-null slices treated with exogenous BDNF. We propose, therefore, that the loss of BDNF-dependent modulation at primary afferent synapses in nTS may be a critical factor in dysregulation of autonomic homeostasis in Mecp2 mutant mice and, by implication, RTT patients.
Materials and Methods
Animals.
Mecp2tm1-1Jae mice (Chen et al., 2001), developed by Dr. R. Jaenisch (Whitehead Institute, Massachusetts Institute of Technology, Cambridge, MA) and obtained from the Mutant Mouse Regional Resource Center (University of California, Davis, Davis, CA), were maintained on a mixed background (sv129, BALB/c, C57BL/6). Male Mecp2 nulls (Mecp2−/y) were generated by crossing heterozygous Mecp2tm1-1Jae knock-out females with Mecp2tm1-1Jae wild-type males (Mecp2+/y). All experimental procedures were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University and University of Missouri.
Dye labeling.
We used a modification of the technique previously described (Mendelowitz et al., 1992; Kline et al., 2002) to label presynaptic boutons on second-order nTS neurons. Briefly, wild-type adult mice were anesthetized with isoflurane. The vagus nerve was exposed in the neck by a ventral midline excision and isolated from surrounding tissues with Parafilm “M” (Fisher Scientific). Small crystals of the anterograde tracer 4-(4-(didecylamino)styryl)-N-methyl-pyridinium iodide (DiA) (4-Di-10-ASP; Invitrogen) were placed on the isolated intact nerve at the level of the carotid bifurcation and caudal to the nodose ganglion. To prevent dye leakage to surrounding tissues, the region was isolated with a fast hardening silicone elastomer (Kwik-Sil; World Precision Instruments). The animals were then sutured and allowed to recover for a minimum of 3 d. Animals were killed using CO2, then transcardially perfused with PLP fixative (0.075 m l-lysine-monohydrochloride, 0.01 m sodium metaperiodate in 0.1 m sodium phosphate buffer; pH ∼7.4). Brains were removed from the skull, postfixed in PLP fixative for 2 h, and then sectioned with a vibratome (Leica). Forty-five-micrometer sections were processed free-floating for TrkB immunohistochemistry, as described below.
Immunohistochemistry.
In situ detection of BDNF protein was performed on brain tissues collected from three sets of Mecp2 wild-type and null littermate pairs. Mice were perfused transcardially with a 4% paraformaldehyde solution prepared in 0.1 m phosphate buffer, pH 7.4. Brains were removed from the skulls, postfixed, cryoprotected in sucrose, frozen in isopentane, and stored at −80°C. Cryostat-cut sections (25 μm, coronal) were processed free-floating for BDNF immunostaining, using a rabbit polyclonal anti-BDNF antibody (1:1000; Amgen), a biotinylated goat anti-rabbit IgG antibody (1:400; Vector), peroxidase-conjugated avidin-biotin complex (1:150; Vector), and diaminobenzidine (Sigma). In a subset of tissue, sections were incubated with rabbit polyclonal anti-TH (1:200; Pel-Freeze) and visualized using FITC-goat anti-rabbit IgG (ICN/Cappel).
Free-floating sections were also processed for BDNF and TrkB double immunodetection, using a mouse monoclonal anti-BDNF antibody (mAb 9, a generous gift from Prof. Yves-Alain Barde, University of Basel, Basel Switzerland) (Kolbeck et al., 1999) and a chicken polyclonal anti-TrkB antibody (1:100, Promega). An Alexa Fluor 488-conjugated donkey anti-mouse antibody was used for BDNF immunodetection. A biotinyl-tyramide amplification step (PerkinElmer) and Streptavidin-Cy3 (Invitrogen) were used for TrkB immunodetection. Sections were imaged with a laser scanning confocal microscope (Leica TCS SP2 MP, Leica Microsystems) using a 40× oil-immersion objective.
Nissl staining protocol.
Tissue processing was similar to as described above. Twenty-five-micrometer-thick cryostat-cut sections were collected in PBS then mounted onto SuperFrost Plus slides and allowed to dry. Sections were incubated in a 0.5% Cresyl Violet acetate solution for 5 min, quickly rinsed in distilled water then dehydrated in graded ethanol solutions (50, 75, 95, and 100%). Sections were cleared in CitriSolv (Fisher) and mounted under coverslip using Permount (Fisher).
Brainstem slice preparation and electrophysiology.
Brainstem slices containing the nTS were prepared from P35–P48 d mice as previously described (Kline et al., 2005). Mice were anesthetized with isoflurane and decapitated. The brainstem was removed and placed in ice-cold low-calcium, high-magnesium artificial CSF (aCSF) containing the following (in mm): 124 NaCl, 3 KCl, 1.2 NaH2PO4, 1.2 MgSO4, 2 MgCl2, 25 NaHCO3, 11 d-glucose, 0.4 l-ascorbic acid, and 1 CaCl2, saturated with 95% O2–5% CO2, pH 7.4, 300 mOsm. Horizontal slices (∼220 μm) were cut with a stainless steel blade (Campden Instruments) using a vibrating microtome (Leica VT 1000S). Tissue sections were placed in a superfusion chamber (RC-26G, Warner Instruments, volume, ∼234 μl) that contained inlet and outlet ports for aCSF flow. The submerged sections were secured with a nylon mesh and superfused at a flow rate of 3 ml/min with normal recording aCSF at 31–33°C. Recording aCSF contained the following (in mm): 125 NaCl, 3 KCl, 1.2 NaH2PO4, 1.2 MgSO4, 0.4 l-ascorbic acid, 25 NaHCO3, 10 d-glucose, and 2 CaCl2, saturated with 95% O2–5% CO2, pH 7.4, 300 mOsm. All of the recordings were made from cells in the medial nTS.
Neurons in the nTS were visualized using an Olympus microscope (BX51WI, 40× magnification) equipped with differential interface contrast and an infrared-sensitive camera. The pipette was guided using a piezoelectric micromanipulator (PCS-6000; Burleigh). Recording electrodes (8250 glass; 3.5–5.0 MΩ) were filled with a solution containing the following (in mm): 10 NaCl, 130 K+ gluconate, 11 EGTA, 1 CaCl2, 10 HEPES, 1 MgCl2, 2 MgATP, and 0.2 NaGTP, pH 7.3, 295–300 mOsm. Evoked EPSCs and action potentials were generated by placing a concentric bipolar stimulating electrode (Frederick Haer) on tractus solitarius (TS) containing visceral afferents (Andresen and Kunze, 1994; Kline, 2008) and stimulating (0.1 ms duration) at frequencies of 0.5 and 20 Hz with an isolated programmable stimulator (AMPI). In experiments in which we wished to record spontaneous miniature EPSCs (mEPSCs), the extracellular solution contained 1.0 μm tetrodotoxin (TTX) and 10 μm bicuculline methobromide. In all of the voltage-clamp protocols, neurons were voltage clamped at −60 mV in the whole-cell configuration. Data were filtered at 2 kHz and sampled at 10 kHz using pClamp10 software (Molecular Devices). Neurons were rejected if resting membrane was more positive than −45 mV upon initial whole-cell access or if access resistance was not stable throughout recording.
Electrophysiological data were analyzed with Molecular Devices Clampfit, Microsoft Excel, and SigmaPlot software. Each data point for a given trial was an average of 5–20 current sweeps. Synaptic events were compared between control and null mice using an unpaired t test or two-way repeated measures ANOVA (RM ANOVA). Spontaneous miniature EPSC distributions were compared using Kolmogorov–Smirnov tests. p values <0.05 were considered significant. Data were presented as the mean ± SEM.
Results
BDNF protein distribution in the medulla of P35 wild-type mice
To define potential sites at which dysregulation of BDNF expression may influence autonomic and respiratory control, we first compared the brainstem distribution of BDNF protein in P35 wild-type (Mecp2+/y) and Mecp2-null (Mecp2−/y) mice. We chose P35 mice because this is an age at which respiratory function is severely disrupted in Mecp2-null animals (Ogier et al., 2007). In wild-type mice, BDNF protein is detectable in neuronal perikarya and varicose neuronal processes located in diverse cell groups of the medulla oblongata, including cranial sensory and motor nuclei, the medullary reticular formation, the nTS, and the area postrema, a circumventricular organ located in the caudal dorsomedial medulla (Table 1). The highest density of BDNF-positive cell bodies is found in the spinal trigeminal nucleus (pars caudalis and interpolaris) and area postrema, whereas scattered cells are observed in the inferior olive, nTS, prepositus nucleus, and the dorsomedial part of the spinal trigeminal nucleus. BDNF-positive neuronal processes are found at high density in the inferior olive, nTS, dorsal motor nucleus of the vagus nerve, and the dorsomedial part of the principal sensory nucleus of the trigeminal nerve (Table 1). Lower densities of BDNF-stained processes are observed in the medullary reticular formation, gracile nucleus, spinal trigeminal tract and nucleus (pars caudalis and interpolaris), motor nucleus of the glossopharyngeal nerve, area postrema, A1/C1, nucleus ambiguus, ventral spinocerebellar tract, prepositus nucleus, raphe nuclei (obscurus and magnus parts), and lateral paragigantocellularis nucleus.
Table 1.
Distribution of BDNF protein in the medulla oblongata of P35 Mecp2 wild-type mice and comparison to P35 Mecp2-null mice
| Structure | P35 Mecp2 wild type |
P35 Mecp2 null versus wild type |
||
|---|---|---|---|---|
| BDNF-IR cell bodies | BDNF-IR processes | BDNF-IR cell bodies | BDNF-IR processes | |
| Intermediate reticular nucleus | − | ++ | − | ↓↓↓ |
| Gracile nucleus | − | + | − | ↓ |
| Lateral reticular nucleus | − | ++ | − | ↓ |
| Spinal trigeminal tract | − | ++ | − | ↓ |
| Spinal trigeminal nucleus, caudal/interpolar | +++ | ++ | ↓ | ↓ |
| Inferior olive | ++ | +++ | ↓ | ↓↓ |
| Nucleus tractus solitarius | + | +++ | ↓ | ↓↓↓ |
| Motor nucleus of the glossopharyngeal nerve | − | + | − | ↓ |
| Motor nucleus of the vagus nerve | − | ++++ | − | ↓↓↓ |
| Area postrema | +++ | ++ | ↓↓ | ↓↓↓ |
| A1/C1 | − | ++ | − | ↓↓ |
| Raphe obscurus | − | + | − | ↓ |
| Nucleus ambiguus | − | ++ | − | ↓↓ |
| Rostroventrolateral reticular nucleus | − | ++ | − | ↓↓↓ |
| Ventral spinocerebellar tract | − | ++ | − | ↓ |
| Parvocellular reticular nucleus | − | ++ | − | ↓ |
| Prepositus nucleus | ++ | +/++ | ↓↓ | ↓ |
| Raphe magnus | − | ++ | − | ↓ |
| Lateral paragigantocellularis nucleus | − | ++ | − | ↓↓ |
| Principal sensory nucleus of the trigeminal nerve, dorsomedial | − | +++ | − | ↓ |
| Principal sensory nucleus of the trigeminal nerve, ventrolateral | ++ | − | ↓ | − |
For BDNF-immunoreactive (IR) cell bodies: −, no cells observed; +, occasional cells; ++, scattered cells; +++, densely packed cells. For BDNF-IR cell processes: −, no processes observed; +, low density; ++, moderate density; +++, high density; ++++, very high density. For changes in null mice compared to wild types: −, no obvious change; ↓, mild decrease; ↓↓, moderate decrease; ↓↓↓, sharp decrease. Data was collected on three wild-type and three Mecp2-null mice.
Regionally selective deficits in BDNF in P35 Mecp2-null mice
Mecp2-null mice exhibit regionally selective deficits in the intensity of BDNF staining in perikarya and fibers at P35 (Table 1). Dramatic reductions in BDNF fiber staining are observed in autonomic and respiratory-related cell groups, including the dorsal motor nucleus of the vagus nerve (Fig. 1A1,A2,A3), nTS (Fig. 1A1,A2,A3), area postrema (Fig. 1A1,A2,A3), nucleus ambiguus (Fig. 1B1,B2,B3), and medullary reticular nuclei (Fig. 1B1,B2,B3). All other regions exhibit moderate [for example, A1/C1 (Fig. 1C1,C2,C3)] to marginal reductions in the density of BDNF-positive fibers and cell bodies compared to controls.
Figure 1.

BDNF staining intensity in medullary cell groups is markedly decreased in P35 Mecp2-null mice compared to wild-type controls. A1, Representative photomicrograph showing the distribution of tyrosine hydroxylase (TH) protein in the nucleus tractus solitarius region of Mecp2 wild-type mice (Mecp2+/y). A2, A3, BDNF staining in the nucleus tractus solitarius region of Mecp2+/y and null (Mecp2−/y) mice, respectively. B1, Cresyl violet staining in the nucleus ambiguus region of Mecp2+/y mice. B2, B3, BDNF staining in the nucleus ambiguus region of Mecp2+/y and null (Mecp2−/y) mice, respectively. C1, TH protein staining in A1/C1 region of Mecp2 wild-type mice (Mecp2+/y). C2, C3, BDNF staining in the A1/C1 region of Mecp2+/y and null (Mecp2−/y) mice, respectively. A1/C1, A1/C1 catecholaminergic cell group; AP, area postrema; cc, central canal; DMNX, dorsal motor nucleus of the vagus nerve; NA, nucleus ambiguus; RVL, rostroventrolateral reticular nucleus. Scale bar, 200 μm.
Abnormal synaptic function in Mecp2-null mice
To determine whether or not reduced BDNF expression is associated with altered synaptic function, we compared synaptic responses of nTS relay neurons in wild-type and Mecp2-null mice. Although nTS is only one of several brainstem cell groups showing a dramatic reduction in BDNF protein in Mecp2-null mice, it is perhaps the best studied in terms of BDNF responsiveness in normal animals [see Katz (2005) and references therein]. In particular, BDNF can strongly modulate glutamatergic activation of second-order nTS neurons by inhibiting postsynaptic AMPA receptors (Balkowiec et al., 2000). Thus, we hypothesized that nTS second-order neurons in Mecp2-null mice would exhibit abnormally large responses to primary afferent stimulation due to reduced BDNF-dependent modulation.
To define synaptic function in the nTS, we used horizontal brainstem slice preparations from P35–P48 wild-type and Mecp2-null mice to compare EPSCs evoked by stimulation of fibers in the TS, the principal pathway for primary afferent inputs to nTS. We focused in particular on neurons in the medial subnucleus (mnTS), which express TrkB and are densely innervated by BDNF-positive fibers in wild-type animals (Fig. 2), and constitute a critical synaptic site for cardiorespiratory control (Andresen and Kunze, 1994; Subramanian et al., 2007). Only cells receiving monosynaptic input, defined by an SD of EPSC latency (jitter) of <250 μs, were considered for analysis (Doyle and Andresen, 2001; Kline et al., 2002).
Figure 2.
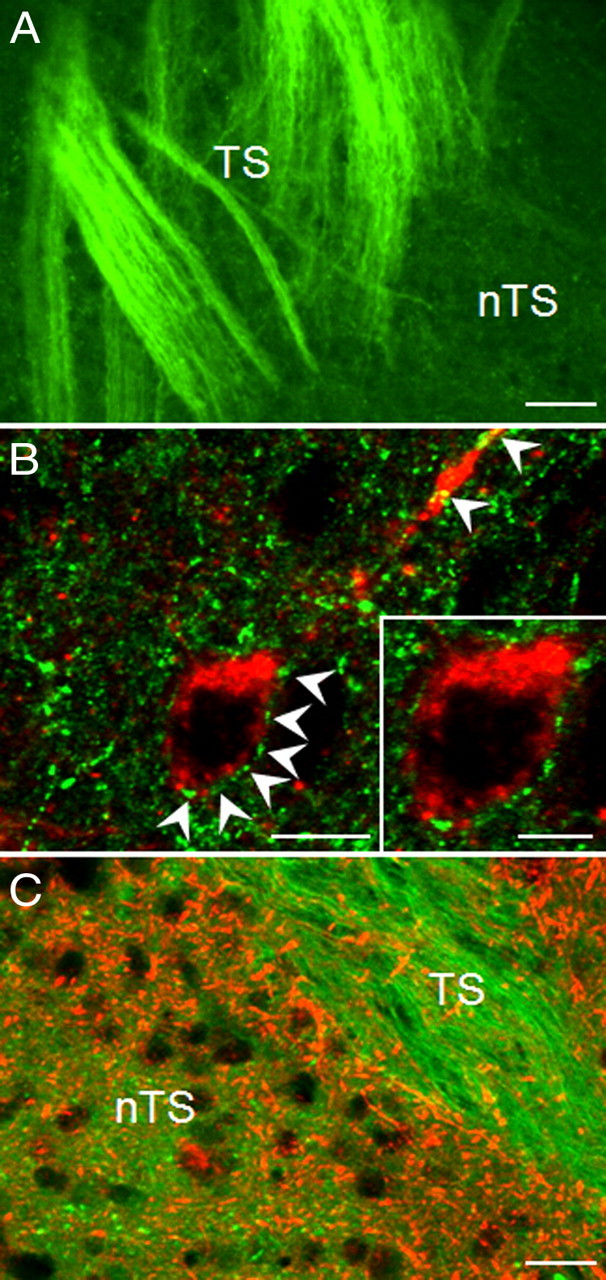
TrkB-positive neurons in the mnTS receive input from BDNF-containing TS fibers. A, Representative picture demonstrating presence of BDNF protein (green) in axons of the TS in the mnTS region of P35 wild-type mice. B, Confocal images of a TrkB-immunoreactive neuron (red) surrounded by BDNF-positive varicosities (green; arrowheads) in the mnTS region. C, DiA-labeled TS axons and varicosities (green) are not immunopositive for TrkB protein (red). Scale bars: 40 μm (A), 10 μm (B), 20 μm (C).
TS stimulation at 0.5 Hz evoked monosynaptic EPSCs (TS-EPSCs) at constant latency in both wild-type and mutant animals (Fig. 3A). However, TS-EPSC amplitude was significantly greater in Mecp2−/y mice than in wild-type controls [wt, 82.3 ± 6.9 pA (n = 18) vs null, 135.8 ± 11.4 pA (n = 13), p < 0.05; t test]. We next asked whether increasing the frequency of TS stimulation would significantly alter afferent processing in wild-type versus null nTS cells. As is characteristic of nTS neurons (Miles, 1986), TS stimulation at 20 Hz for 10 episodes produced TS-EPSC depression following the first evoked event in both groups of mice (Fig. 3B). Although the magnitude of synaptic depression was comparable between Mecp2−/y and Mecp2+/y mice (p > 0.05), the absolute amplitude of TS-EPSCs was increased in Mecp2−/y mice compared to controls, as with 0.5 Hz stimulation. When averaged across the 10 events of the stimulus train, the mean wild-type TS-EPSC amplitude was 49.4 ± 5.0 pA compared to 73.9 ± 6.1 pA in mutants (p < 0.05, t test). The first TS-EPSC averaged 101.6 ± 12.1 pA (n = 22) in wild-type mice and 156.3 ± 15.6 pA (n = 26) in null mice, and EPSC amplitude remained elevated in the mutants throughout the stimulus train (p < 0.05, two-way repeated-measures ANOVA) (Fig. 3C).
Figure 3.
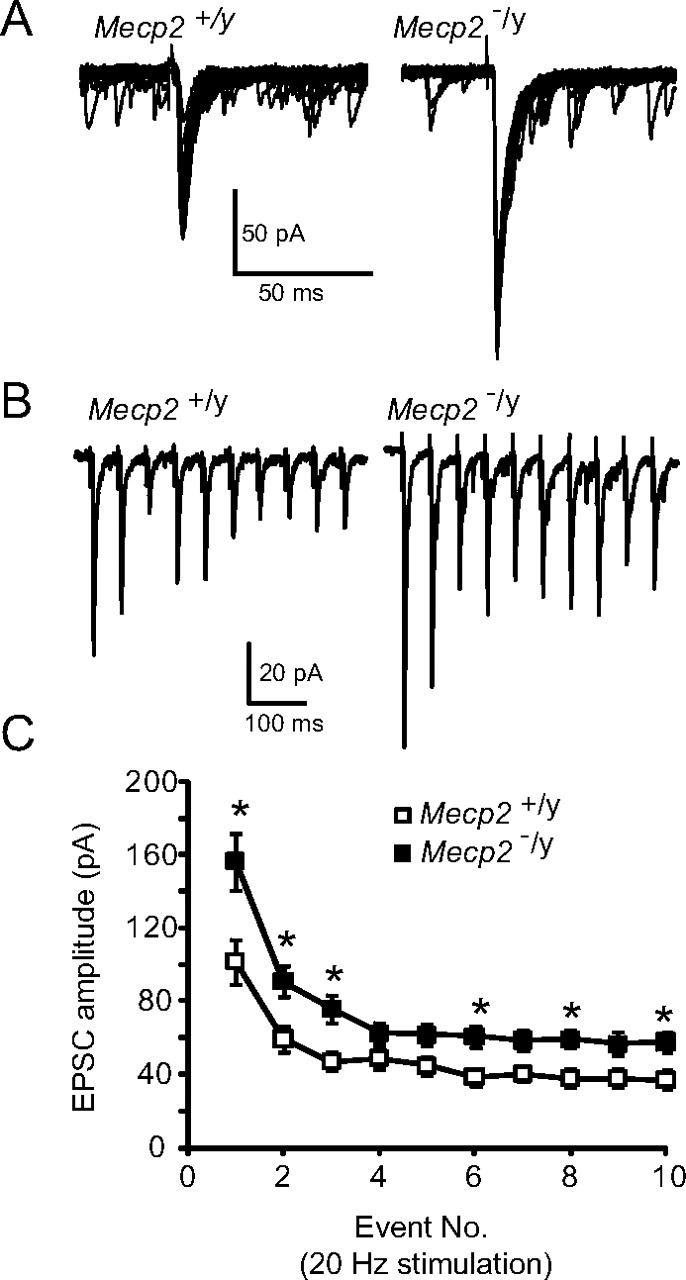
Mecp2-null mice exhibit enhanced solitary tract evoked EPSCs. A, B, Representative tracings of TS evoked EPSCs that were recorded from a wild-type (Mecp2+/y, left) and mutant (Mecp2−/y, right) nTS cell. The TS was stimulated at 0.5 Hz (A, overlay of 20 traces) or 20 Hz (B, average of 5 overlaying traces). C, Average amplitude for each TS-EPSC stimulated at 20 Hz. *p < 0.05, two-way RM ANOVA. Note that TS-EPSC amplitude is higher in Mecp2−/y mice and frequency-dependent depression is observed.
nTS neurons from Mecp2−/y mice exhibit enhanced probability of action potential firing
To examine whether elevated synaptic current amplitude in Mecp2−/y mice is associated with enhanced spiking, we compared the probability of action potential generation in response to TS stimulation in wild-type and null nTS cells. Although TS stimulation at 20 Hz elicited EPSPs in both groups of mice, Mecp2−/y cells were four times more likely to generate an action potential than wild-type controls. Specifically, TS stimulation at resting potential elicited action potential generation in 8 of 10 null cells during the train, but only in 2 of 10 wild-type cells (p < 0.05, z test). This is despite the fact that the resting membrane potential was comparable between the two groups (null, −54 ± 1 mV, n = 10 vs wild type, −57 ± 2 mV, n = 10 each, p > 0.05, t test).
Intrinsic excitability of nTS neurons is comparable in Mecp2−/y and Mecp2+/y mice
To determine whether enhanced EPSC amplitude and action potential firing in Mecp2-null cells are associated with changes in the intrinsic properties of nTS neurons, we compared the electrical properties of monosynaptically driven nTS neurons in wild-type and null mice. Input resistance (wild type, 567 ± 62 MΩ, n = 26 vs null, 559 ± 94 MΩ, n = 26; p > 0.05, t test) and resting membrane potential [−58.3 ± 1.7 mV (n = 31) vs −60.1 ± 1.5 mV (n = 35), respectively; p > 0.05; t test] were not different between cells from wild-type and null mice.
Action potential firing characteristics and dynamic properties of Mecp2−/y and Mecp2+/y were examined by current-induced step depolarization. In monosynaptic cells, current injection (+10–70 pA steps, 500 ms, interval of 2 s) from a constant holding potential of −60 mV evoked action potential discharge in all cells studied. The number of action potentials, as well as the likelihood of generating an action potential, in a given current step was comparable between the two groups (p > 0.05, data not shown). Likewise, the amplitude, afterhyperpolarization, half-width, and threshold, as well as rise time and slope, of the first current-evoked action potential were comparable between both groups (Table 2). Action potential decay time and slope were significantly faster in null mice.
Table 2.
Action potential properties in P35 Mecp2 wild-type and null mice
| Mecp2 wild type | Mecp2 null | |
|---|---|---|
| Peak amplitude (mV) | 67.32 ± 2.38 | 67.58 ± 2.88 |
| Afterhyperpolarization (mV) | −18.93 ± 1.46 | −24.27 ± 2.50 |
| Half-width (ms) | 1.19 ± 0.13 | 0.91 ± 0.07 |
| Threshold (mV) | −34.66 ± 2.32 | −32.22 ± 1.30 |
| Rise time (10–90%, ms) | 0.59 ± 0.07 | 0.51 ± 0.07 |
| Rise slope (10–90%, mV/ms) | 119.44 ± 15.87 | 119.94 ± 11.18 |
| Decay time (90–10%, ms) | 1.02 ± 0.12 | 0.67 ± 0.06* |
| Decay slope (90–10%, mV/ms) | −61.96 ± 5.98 | −91.76 ± 11.10* |
Action potential properties were determined from current-induced depolarization. Data are derived from the initial action potential elicited at threshold.
*p < 0.05, t test. Data are mean ± SEM for wild type (n = 13 cells) and null (n = 15 cells).
Enhanced EPSCs in Mecp2−/y mice are not due to inhibitory shunting
To examine the potential contribution of inhibitory shunting to the EPSC phenotype of Mecp2−/y cells, we recorded EPSCs evoked by stimulation of the TS at 20 Hz during bath application of the GABAA antagonist bicuculline. Application of 10 μm bicuculline did not alter the amplitude of TS-EPSCs at any point in the stimulus train or averaged across the entire train in wild-type (average of 10 events; control, 45.3 ± 4.3 vs bicuculline, 48.8 ± 5.2 pA) or null mice (average of 10 events; control, 59.2 ± 7.0 vs bicuculline, 53.4 ± 7.1 pA) (Fig. 4A,B) (p > 0.05). These data suggest that reduced inhibitory shunting did not occur in either group and is not responsible for elevated EPSC amplitude in null mice.
Figure 4.

Enhanced EPSCs in Mecp2−/y mice are not due to reduced inhibitory shunting. A, Application of 10 μm bicuculline did not alter EPSCs amplitude evoked at 20 Hz in Mecp2+/y mice. Average amplitude for each TS-EPSC stimulated at 20 Hz for aCSF (open diamond) versus bicuculline (closed square). B, Bicuculline (10 μm) did not alter EPSCs amplitude evoked at 20 Hz in Mecp2−/y mice. Average amplitude for each TS-EPSC stimulated at 20 Hz for aCSF (open diamond) versus bicuculline (closed square).
Enhanced miniature EPSCs in Mecp2-null mice
mEPSCs represent the random, action potential-independent release of neurotransmitter and provide information about possible changes in the presynaptic release process. To determine whether mEPSCs were altered by loss of MeCP2, we compared the amplitude and frequency of mEPSCs recorded in the presence of the Na+ channel blocker TTX (1 μm) and bicuculline (10 μm) in wild-type and null mice. Examples of spontaneous miniature activity in a wild-type and null mouse are shown in Figure 5A. Plotting the cumulative probability plots for mEPSC amplitudes demonstrated a significant rightward shift in null mice, indicating an increase in amplitude (p < 0.05; Kolmogorov–Smirnov test) (Fig. 5B) (n = 10 cells each). Similarly, cumulative interevent intervals of mEPSCs demonstrated a significant leftward shift in null mice, indicating an increase in frequency of miniature events (p < 0.05; Kolmogorov–Smirnov test) (Fig. 5C).
Figure 5.
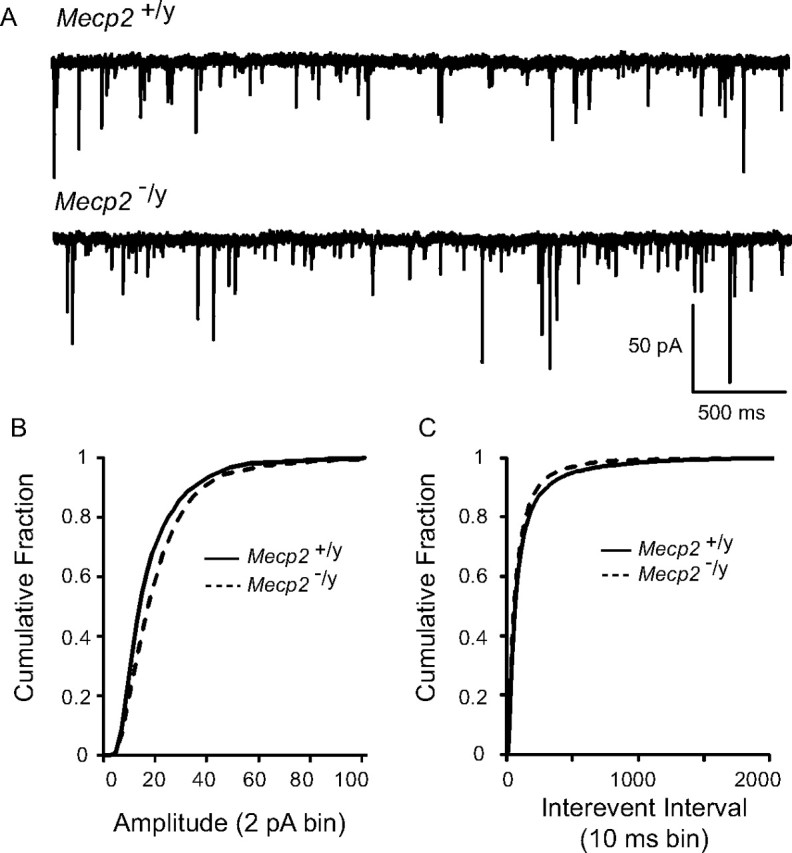
Miniature EPSCs are enhanced in null mice. A, Representative tracings of mEPSCs from a wild-type (top) and null (bottom) mouse nTS cell. Note the increased frequency and amplitude of mEPSCs in null mice. The cell was voltage clamped at −60 mV. B, The cumulative fraction of mEPSC amplitudes (2 pA bin) illustrates a rightward shift in the distribution in null mice. C, The cumulative probability of mEPSC interevent interval distribution (10 ms bin) revealed a significant shift to the left (increased frequency) in null mice.
Exogenous BDNF rescues abnormal EPSC amplitude in Mecp2−/y mice
In view of our previous finding that BDNF can modulate glutamatergic excitation of second-order nTS neurons (Balkowiec et al., 2000), as well as the fact that BDNF is severely depleted in nTS in Mecp2-null mice (present study), we examined whether or not exogenous BDNF could rescue the mutant synaptic phenotype. As shown in Figure 6, bath application of BDNF (100 ng/ml, 15 min) decreased the amplitude of TS-EPSCs evoked by 0.1 or 20 Hz (Fig. 6A) stimulation in Mecp2−/y mice to wild-type values. At 20 Hz, TS-EPSC amplitude averaged across the entire stimulus train decreased from 87.6 ± 9.7 pA (before BDNF) to 41.9 ± 5.1 pA (after BDNF; p < 0.05, paired t test, n = 5). This decrease in average amplitude in the presence of BDNF was reflected in smaller TS-EPSCs throughout the duration of the stimulus train (Fig. 6B) and was partially reversible (Fig. 6C). Moreover, restoration of wild-type TS-EPSC amplitude in null mice during BDNF treatment was accompanied by a significant reduction in the number of action potentials elicited by TS stimulation (20 Hz) (Fig. 6D) (control, 20.5 ± 6.7 vs BDNF, 13.1 ± 6.7, paired t test p < 0.05). This reduction in discharge in the presence of BDNF was not due to alterations in resting membrane potential (control, −53.2 ± 1.97 vs BDNF, −54.9 ± 1.7 mV, p > 0.05, paired t test). Exogenous BDNF also decreased the averaged TS-EPSC amplitude in wild-type mice, albeit to a lesser degree than in mutants (49.8 ± 5.4 pA before BDNF to 41.1 ± 3.7 pA after BDNF; p < 0.05, paired t test, n = 5). In addition to evoked responses, we also compared the effect of exogenous BDNF on miniature EPSCs, which exhibit increased amplitude and frequency in null mice (Fig. 5). As shown in Figure 6E, BDNF induced significant shifts in the cumulative distributions curves for mEPSC size and interevent interval, indicating reductions in amplitude and frequency, respectively (p < 0.05; Kolmogorov–Smirnov test).
Figure 6.
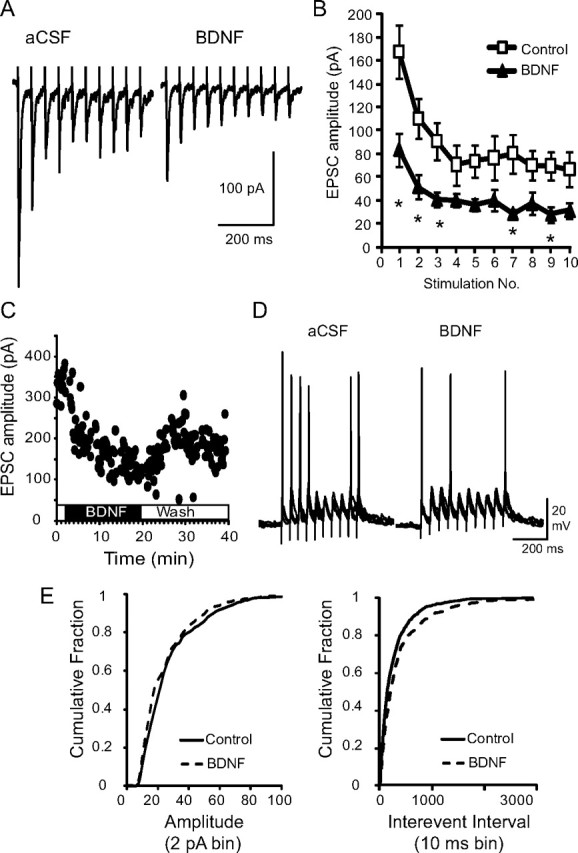
Exogenous BDNF reduces exaggerated postsynaptic responses in null mice. A, Representative raw trace of evoked TS-EPSCs recorded from a Mecp2−/y-null cell during aCSF (left) and following exogenous BDNF (100 ng/ml, 15 min, right). B, Average amplitude for each TS-EPSC stimulated at 20 Hz for aCSF (open square) versus BDNF (closed triangle). *p < 0.05, two-way ANOVA. C, TS-EPSC amplitudes recorded at 0.1 Hz in a null nTS cell in the presence of aCSF, BDNF, and wash out. D, Representative example of TS-evoked discharge in a null nTS cell during aCSF and BDNF (overlap of 5 traces each). Note the decrease in discharge following BDNF. E, In the presence of BDNF, the cumulative fraction of null mEPSC amplitudes (left, 2 pA bin) shifted to the left, while interevent interval distribution (right, 10 ms bin) shifted to the right (decreased frequency).
Discussion
RTT patients and Mecp2 mutant mice exhibit profound disturbances of cardiorespiratory homeostasis that appear to arise, at least in part, from dysregulation of modulatory inputs to brainstem pattern generators and reflex pathways (for review, see Katz et al., 2009). This is illustrated, for example, by the marked state dependence of cardiorespiratory dysfunction in RTT patients (Weese-Mayer et al., 2008) and by recent observations that hypoxic ventilatory reflexes are greatly exaggerated in Mecp2-null mice (Bissonnette and Knopp, 2006; Roux et al., 2008; Voituron et al., 2009). These findings are consistent with observations in other neural systems that the normal balance between synaptic excitation and inhibition is disrupted by loss of MeCP2 function. For example, Dani et al. (2005) observed reduced activity in cortical slices from Mecp2-null mice resulting from a loss of excitatory synaptic drive with no change in intrinsic electrical excitability. In contrast, Medrihan et al. (2008) observed a reduction in IPSCs in the ventrolateral medulla of Mecp2-null mice resulting from reduced levels of GABA and decreased expression of GABA receptor subunits. In hippocampus, on the other hand, both glutamatergic synapse number (Chao et al., 2007) and the strength of basal inhibitory rhythms (Zhang et al., 2008) are decreased in the absence of MeCP2. However, despite markedly abnormal autonomic and respiratory control in RTT, relatively little is known about synaptic deficits caused by loss of MeCP2 in brainstem autonomic reflex pathways.
The present study demonstrates dysregulation of primary afferent synaptic transmission in nTS, a critical site for reflex modulation of autonomic and respiratory function, in Mecp2-null mice. Specifically, we find that the amplitude of spontaneous miniature and evoked EPSCs recorded from second-order nTS neurons is significantly increased in mutants compared to wild-type controls and, accordingly, that mutant cells are more likely to fire action potentials in response to afferent stimulation. These changes occur with no alteration in resting membrane potential, input resistance, or action potential firing in response to step current injections, indicating that they are not due to an increase in the intrinsic excitability of the postsynaptic cells. Moreover, the difference in EPSC amplitude between wild-type and null mice is unaffected by application of bicuculline, which blocks inhibitory GABAergic signaling. Thus, our data indicate that the synaptic phenotype of Mecp2-null nTS neurons most likely reflects either enhanced excitatory synaptic drive and/or decreased inhibition (non-GABAergic) of postsynaptic responses to primary afferent stimulation.
The amplitude of postsynaptic responses to primary afferent stimulation in nTS is normally regulated by interactions between glutamate, released from afferent terminals, and multiple presynaptic and postsynaptic neuromodulators, including BDNF (Balkowiec et al., 2000), dopamine (Kline et al., 2002), GABA (Chen and Bonham, 2005; Bailey et al., 2008), and others [for review, see Andresen and Kunze (1994) and Kline (2008)]. BDNF is synthesized and released by nodose-petrosal cranial sensory ganglion cells (NG), the primary source of peripheral afferent inputs to nTS (Brady et al., 1999; Balkowiec and Katz, 2000), and is also expressed by scattered neurons within nTS itself (Conner et al., 1997; present study). Moreover, BDNF can potently modulate or inhibit glutamatergic excitation of isolated second-order nTS relay neurons through activation of postsynaptic TrkB receptors (Balkowiec et al., 2000). In Mecp2-null mice, BDNF protein levels fall progressively after birth, reaching ∼50% wild-type values in the NG and medulla by the fifth postnatal week (Wang et al., 2006). This BDNF deficit is particularly striking in the neuropil in nTS (Ogier et al., 2007; present study) and is accompanied by a significant reduction in the absolute amount of BDNF released from NG neurons (Wang et al., 2006). Thus, given the inhibitory role of BDNF at primary afferent synapses in nTS (Balkowiec et al., 2000), it seems likely that reduced BDNF availability contributes to the abnormally large synaptic responses we observed in second-order nTS neurons in Mecp2-null mice. However, the fact that mEPSC frequency was slightly increased in mutants compared to controls suggests that enhanced presynaptic glutamate release may also play a role.
The possibility that reduced BDNF availability underlies the mutant synaptic phenotype is further supported by our finding that treatment of nTS slices with exogenous BDNF restores the amplitude of spontaneous and evoked EPSCs to wild-type values and ameliorates the increase in TS-evoked action potential discharge.
Given that TrkB is abundantly expressed by nTS neurons, and virtually undetectable in primary afferent inputs to nTS after birth (Zhuo and Helke, 1996; present study), these effects of BDNF are most likely postsynaptic. This possibility is supported by our previous finding that BDNF inhibits postsynaptic AMPA receptors expressed by second-order nTS relay neurons (Balkowiec et al., 2000). Moreover, the fact that primary afferent fibers do not express TrkB indicates that the reduction in mEPSC frequency in BDNF-treated mutant slices is most likely an indirect effect mediated by TrkB-positive interneurons.
Regardless of the underlying mechanism(s), these results offer a plausible synaptic mechanism to explain why nTS-mediated reflexes are exaggerated in the absence of MeCP2 (Bissonnette and Knopp, 2006; Roux et al., 2008; Voituron, et al., 2009). More generally, increased gain in afferent inputs to cardiorespiratory control networks in the brainstem is thought to have a destabilizing effect on motor output (Dutschmann et al., 2008) and may therefore contribute to the overall instability in respiratory behavior and cardiovascular regulation that are characteristic of RTT (Dutschmann et al., 2008; Katz et al., 2009). In fact, loss of MeCP2 has previously been shown to result in a failure of reflex desensitization to repetitive vagal afferent stimulation in nTS, which, in turn, contributes to highly variable postinspiratory activity and breathing arrhythmia (Stettner et al., 2007). We believe that reduced BDNF-dependent modulation of glutamatergic transmission in nTS, resulting in exaggerated postsynaptic responses to primary afferent stimulation, as described here, could play a key role in generating these disturbances of reflex sensitization and, thereby, cardiorespiratory arrhythmias in RTT.
In addition to cardiorespiratory instability, RTT patients exhibit a significant increase in mean respiratory frequency that is recapitulated in the strain of Mecp2-null mice used in the present study (Ogier et al., 2007). We previously showed that ampakine treatment of these mice restores normal respiratory frequency in association with an increase in BDNF protein levels in the nodose ganglion (Ogier et al., 2007). The present findings suggest that the ability of ampakine treatment to rescue respiratory frequency may be due, at least in part, to restoration of BDNF-dependent modulation at primary afferent synapses in nTS. More generally, the fact that exogenous BDNF reverses the nTS synaptopathy in Mecp2-null mice further supports the possibility that strategies aimed at restoring normal BDNF signaling may be of therapeutic benefit in RTT (Ogier and Katz, 2008; Katz et al., 2009).
Footnotes
This work was supported by a grant to D.M.K. from the National Institute of Neurological Disorders and Stroke. The authors acknowledge the valuable technical assistance of Dr. Qifang Wang.
References
- Andresen MC, Kunze DL. Nucleus tractus solitarius—gateway to neural circulatory control. Annu Rev Physiol. 1994;56:93–116. doi: 10.1146/annurev.ph.56.030194.000521. [DOI] [PubMed] [Google Scholar]
- Bailey TW, Appleyard SM, Jin YH, Andresen MC. Organization and properties of GABAergic neurons in solitary tract nucleus (NTS) J Neurophysiol. 2008;99:1712–1722. doi: 10.1152/jn.00038.2008. [DOI] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkowiec A, Kunze DL, Katz DM. Brain-derived neurotrophic factor acutely inhibits AMPA-mediated currents in developing sensory relay neurons. J Neurosci. 2000;20:1904–1911. doi: 10.1523/JNEUROSCI.20-05-01904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonnette JM, Knopp SJ. Separate respiratory phenotypes in methyl-CpG-binding protein 2 (Mecp2) deficient mice. Pediatr Res. 2006;59:513–518. doi: 10.1203/01.pdr.0000203157.31924.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady R, Zaidi SI, Mayer C, Katz DM. BDNF is a target-derived survival factor for arterial baroreceptor and chemoafferent primary sensory neurons. J Neurosci. 1999;19:2131–2142. doi: 10.1523/JNEUROSCI.19-06-02131.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Bonham AC. Glutamate suppresses GABA release via presynaptic metabotropic glutamate receptors at baroreceptor neurones in rats. J Physiol. 2005;562:535–551. doi: 10.1113/jphysiol.2004.076885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng V, Matagne V, Banine F, Frerking M, Ohliger P, Budden S, Pevsner J, Dissen GA, Sherman LS, Ojeda SR. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum Mol Genet. 2007;16:640–650. doi: 10.1093/hmg/ddm007. [DOI] [PubMed] [Google Scholar]
- Doyle MW, Andresen MC. Reliability of monosynaptic sensory transmission in brain stem neurons in vitro. J Neurophysiol. 2001;85:2213–2223. doi: 10.1152/jn.2001.85.5.2213. [DOI] [PubMed] [Google Scholar]
- Dutschmann M, Mörschel M, Reuter J, Zhang W, Gestreau C, Stettner GM, Kron M. Postnatal emergence of synaptic plasticity associated with dynamic adaptation of the respiratory motor pattern. Respir Physiol Neurobiol. 2008;164:72–79. doi: 10.1016/j.resp.2008.06.013. [DOI] [PubMed] [Google Scholar]
- Katz DM. Regulation of respiratory neuron development by neurotrophic and transcriptional signaling mechanisms. Respir Physiol Neurobiol. 2005;149:99–109. doi: 10.1016/j.resp.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Katz DM, Dutschmann M, Ramirez JM, Hilaire G. Breathing disorders in Rett syndrome: progressive neurochemical dysfunction in the respiratory network after birth. Respir Physiol Neurobiol. 2009;168:101–108. doi: 10.1016/j.resp.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD. Plasticity in glutamatergic NTS neurotransmission. Respir Physiol Neurobiol. 2008;164:105–111. doi: 10.1016/j.resp.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Takacs KN, Ficker E, Kunze DL. Dopamine modulates synaptic transmission in the nucleus of the solitary tract. J Neurophysiol. 2002;88:2736–2744. doi: 10.1152/jn.00224.2002. [DOI] [PubMed] [Google Scholar]
- Kline DD, Buniel MC, Glazebrook P, Peng YJ, Ramirez-Navarro A, Prabhakar NR, Kunze DL. Kv1.1 deletion augments the afferent hypoxic chemosensory pathway and respiration. J Neurosci. 2005;25:3389–3399. doi: 10.1523/JNEUROSCI.4556-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbeck R, Bartke I, Eberle W, Barde YA. Brain-derived neurotrophic factor levels in the nervous system of wild-type and neurotrophin gene mutant mice. J Neurochem. 1999;72:1930–1938. doi: 10.1046/j.1471-4159.1999.0721930.x. [DOI] [PubMed] [Google Scholar]
- Kron M, Mörschel M, Reuter J, Zhang W, Dutschmann M. Developmental changes in brain-derived neurotrophic factor-mediated modulations of synaptic activities in the pontine Kolliker-Fuse nucleus of the rat. J Physiol. 2007a;583:315–327. doi: 10.1113/jphysiol.2007.134726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron M, Zhang W, Dutschmann M. Developmental changes in the BDNF-induced modulation of inhibitory synaptic transmission in the Kolliker-Fuse nucleus of rat. Eur J Neurosci. 2007b;26:3449–3457. doi: 10.1111/j.1460-9568.2007.05960.x. [DOI] [PubMed] [Google Scholar]
- Kron M, Reuter J, Gerhardt E, Manzke T, Zhang W, Dutschmann M. Emergence of brain-derived neurotrophic factor-induced postsynaptic potentiation of NMDA currents during the postnatal maturation of the Kolliker-Fuse nucleus of rat. J Physiol. 2008;586:2331–2343. doi: 10.1113/jphysiol.2007.148916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrihan L, Tantalaki E, Aramuni G, Sargsyan V, Dudanova I, Missler M, Zhang W. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol. 2008;99:112–121. doi: 10.1152/jn.00826.2007. [DOI] [PubMed] [Google Scholar]
- Mendelowitz D, Yang M, Andresen MC, Kunze DL. Localization and retention in vitro of fluorescently labeled aortic baroreceptor terminals on neurons from the nucleus tractus solitarius. Brain Res. 1992;581:339–343. doi: 10.1016/0006-8993(92)90729-s. [DOI] [PubMed] [Google Scholar]
- Miles R. Frequency dependence of synaptic transmission in nucleus of the solitary tract in vitro. J Neurophysiol. 1986;55:1076–1090. doi: 10.1152/jn.1986.55.5.1076. [DOI] [PubMed] [Google Scholar]
- Ogier M, Katz DM. Breathing dysfunction in Rett syndrome: understanding epigenetic regulation of the respiratory network. Respir Physiol Neurobiol. 2008;164:55–63. doi: 10.1016/j.resp.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci. 2007;27:10912–10917. doi: 10.1523/JNEUROSCI.1869-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux JC, Dura E, Villard L. Tyrosine hydroxylase deficit in the chemoafferent and the sympathoadrenergic pathways of the Mecp2 deficient mouse. Neurosci Lett. 2008;447:82–86. doi: 10.1016/j.neulet.2008.09.045. [DOI] [PubMed] [Google Scholar]
- Stettner GM, Huppke P, Brendel C, Richter DW, Gärtner J, Dutschmann M. Breathing dysfunctions associated with impaired control of postinspiratory activity in Mecp2-/y knockout mice. J Physiol. 2007;579:863–876. doi: 10.1113/jphysiol.2006.119966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian HH, Chow CM, Balnave RJ. Identification of different types of respiratory neurones in the dorsal brainstem nucleus tractus solitarius of the rat. Brain Res. 2007;1141:119–132. doi: 10.1016/j.brainres.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Thoby-Brisson M, Cauli B, Champagnat J, Fortin G, Katz DM. Expression of functional tyrosine kinase B receptors by rhythmically active respiratory neurons in the pre-Botzinger complex of neonatal mice. J Neurosci. 2003;23:7685–7689. doi: 10.1523/JNEUROSCI.23-20-07685.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voituron N, Zanella S, Menuet C, Dutschmann M, Hilaire G. Early breathing defects after moderate hypoxia or hypercapnia in a mouse model of Rett syndrome. Respir Physiol Neurobiol. 2009;168:109–118. doi: 10.1016/j.resp.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Wang H, Chan SA, Ogier M, Hellard D, Wang Q, Smith C, Katz DM. Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in Mecp2 null mice. J Neurosci. 2006;26:10911–10915. doi: 10.1523/JNEUROSCI.1810-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weese-Mayer DE, Lieske SP, Boothby CM, Kenny AS, Bennett HL, Ramirez JM. Autonomic dysregulation in young girls with Rett syndrome during nighttime in-home recordings. Pediatr Pulmonol. 2008;43:1045–1060. doi: 10.1002/ppul.20866. [DOI] [PubMed] [Google Scholar]
- Zhang L, He J, Jugloff DG, Eubanks JH. The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus. 2008;18:294–309. doi: 10.1002/hipo.20389. [DOI] [PubMed] [Google Scholar]
- Zhuo H, Helke CJ. Presence and localization of neurotrophin receptor tyrosine kinase (TrkA, TrkB, TrkC) mRNAs in visceral afferent neurons of the nodose and petrosal ganglia. Brain Res Mol Brain Res. 1996;38:63–70. doi: 10.1016/0169-328x(95)00313-h. [DOI] [PubMed] [Google Scholar]