

Abstract
Aging degrades hematopoietic stem cell (HSC) functions, including stress response; however, the involved molecular pathways are incompletely defined. Murine BM conditionally deleted for One-Twenty-Two-1 (Ott1), is able to maintain lifelong hematopoiesis and has preserved numbers of long-term HSCs, yet cannot repopulate nor sustain itself after transplantation against a competitor even when Ott1 is excised after engraftment. We show, specifically under replicative stress, that Ott1-deleted HSCs have a significant reduction of the G0 cell-cycle fraction associated with self-renewal and undergo early failure. Therefore, Ott1 is required to preserve HSC quiescence during stress but not steady-state hematopoiesis. Reduced tolerance of replicative stress, increased myeloid potential, and greater absolute numbers are mutual characteristics of both Ott1-deleted and aged HSCs, and comparison of their gene expression profiles reveals a shared signature. Ott1-deleted HSCs share multiple aging-associated physiologic changes, including increases in NF-κB activation and DNA damage. Loss of Ott1 causes increased reactive oxygen species; however, antioxidant treatment does not rescue the competitive defect, indicating the existence of additional essential Ott1-dependent HSC pathways. In conclusion, our data establish a requirement for Ott1 in stress hematopoiesis and suggest that Ott1-dependent processes may converge with those affected by aging.
Introduction
The majority of long-term hematopoietic stem cells (LT-HSCs) reside in the G0 phase of the cell cycle, and alterations in pathways that cause aberrant cell-cycle entry or exit typically have a detrimental effect on self-renewal.1,2 The preservation of self-renewal capability is a fundamental requirement for HSCs preventing stem cell exhaustion and BM failure. Aging results in declining HSC self-renewal; however, the responsible affected regulatory pathways remain to be fully identified.3,4
One Twenty Two-1 (OTT1; also known as RNA Binding Motif 15 [RBM15]) plays a broad regulatory role in hematopoiesis. Conditional deletion of Ott1 in adult mice established a requirement for Ott1 in pre-B development and an inhibitory role for myeloid progenitors and megakaryocytes.5 Expansion of the Lin−c-Kit+Sca1+ (LSK) fraction, which contains the HSC population, was observed after Ott1 deletion. Niu et al showed HSCs lacking Ott1 had defects in competitive repopulation.5,6 Embryonic deletion of Ott1 in mice identified nonredundant requirements for organogenesis of the placenta, heart, and spleen.7
OTT1 is the 5′ fusion partner in t(1;22)(p13;q13)–associated infant acute megakaryocytic leukemia.8,9 Translocation of the OTT1 gene into the Megakaryocytic Acute Leukemia, MAL (Megakaryocytic leukemia-1; MKL1) gene produces a fusion transcript encoding a chimeric protein, OTT1-MAL (RBM15-MKL1). OTT1 is related to the widely expressed split ends (spen) family of proteins, the defining features of which are a series of amino-terminal RNA recognition motifs and a Spen Paralog and Ortholog C-terminus (SPOC) domain.10 Ott1 has been implicated in alternative splicing and export of viral RNAs; however, physiologic RNA targets have not been identified.11–13 The SPOC domain has been characterized as having both transcriptional activator and repressor functions, which are histone deacetylase dependent.10 Ott1 contains a RBPJ binding site, and the OTT1-MAL oncoprotein has been shown to cause constitutive activation of gene promoters containing RBPJ consensus sites.14,15
In this report, we demonstrate a cell-autonomous requirement for Ott1 in HSC function for both competitive and noncompetitive repopulation as well as during maintenance in the presence of a competitor. However, Ott1-deficient HSCs are capable of supporting life-long steady-state hematopoiesis in primary animals. We resolve this apparent paradox by showing Ott1 is essential for HSC quiescence during replicative stress but not during the steady state. The inability of Ott1-deficient HSCs to tolerate stress and observed increases in myelopoiesis and LSK populations are similar to changes occurring in aged HSCs.5,16–18 We show the gene expression profile of Ott1-deleted cells are enriched for genes up-regulated during HSC aging, and loss of Ott1 results in physiologic changes similar to aging HSCs, including increases in NF-κB activation and DNA damage.
Methods
Mice
Ott1flox/null tg-Mx1-Cre mice were generated as previously described.5 Mice were maintained in a C57BL/6 background. Wild-type C57BL/6 and CD45.1 B6.SJL were obtained from The Jackson Laboratory. Polyinosinic-polycytidilic acid (pIpC; GE Healthcare) treatment was performed at 6 weeks of age with 250 μg injected intraperitoneally every other day for 3 total doses. Excision was consistently greater than 95% as assessed by Southern blot of whole BM and single-colony PCR of myeloid colonies grown in methylcellulose (data not shown). All animal studies were carried out in accordance with and with approval from the University of Massachusetts Medical School Institutional Animal Care and Use Committee.
Transplantation
For competitive repopulation studies, 8 × 105 CD45.2 donor and 2 × 105 helper (CD45.1/CD45.2 heterozygote) cells were injected into tail veins of CD45.1 homozygous mice treated with 1100 rads split-dose γ-irradiation. Peripheral blood chimerism was monitored through nonlethal facial vein bleeding. For noncompetitive transplantation, lethally irradiated mice were injected with the indicated number of donor cells and monitored daily. For competitive maintenance, 5 × 105 Ott1flox/null Mx1-cre or littermate control BM cells were injected with equal numbers of CD45.1/CD45.2 heterozygous competitor marrow into lethally irradiated CD45.1 homozygotes. Mice were analyzed for initial chimerism by facial vein bleed 4 weeks after transplantation, treated with pIpC as described in the previous paragraph. All transplanted mice were maintained on acidified water.
Flow cytometry
Peripheral blood and BM samples were treated with RBC lysis buffer (QIAGEN), then resuspended in PBS with 2% FBS (Invitrogen). Labeling for multiparameter flow cytometry was performed as previously described.5,19 Lin+ antibodies to Ter119, Gr-1, B220, CD19, Il7R, CD3, CD4, and CD8 were obtained from eBioscience. Goat anti–rat-PE and FITC secondary antibodies were from BD Biosciences. Primary antibodies used were anti-CD45.2–FITC, CD45.2-APC, CD45.1-PE, CD45.1-biotin, Mac-1–PE-Cy7, and Gr-1–APC-Cy7 from BD Biosciences and Flt3R-PE, c-Kit–APC-Cy7, Sca-1–PE-Cy7, CD34-APC, CD34-Pacific Blue, CD150-APC, and CD48-biotin from eBioscience. Streptavidin conjugates of AF350 and PECy5.5 were obtained from BD Biosciences. Viable cells were identified through exclusion of 7-aminoactinomycin D (7-AAD; BD Biosciences). Apoptotic and dead/apoptotic cells were identified as annexin V+/7-AAD− and annexin V+/7-AAD+, respectively, using the BD Biosciences Annexin V:PE Apoptosis Detection Kit per the manufacturer's protocol. Cell-cycle analysis was performed using Hoechst 33342 and pyronin Y (Sigma-Aldrich) as described.20 Reactive oxygen species (ROS) measurement of LSK/CD34-labeled cells was performed by incubating cells in 5μM 2′,7′-dichlorfluorescein-diacetate (DCF-DA; Invitrogen) for 30 minutes at 37°C as described previously.19 For γH2AX quantification, LSK/CD34-labeled cells were fixed in 4% paraformaldehyde, permeabilized with methanol/acetone, and stained with anti-γH2AX (Cell Signaling) followed by anti–mouse AF488 secondary antibody before 7-AAD incubation and flow cytometric measurement of the G1 fraction.21 Mitochondrial mass was measured using 50nM Mitotracker Deep Red (Invitrogen) to stain LSK/CD34-labeled cells per the manufacturer's instructions. Flow cytometry was performed on either a BD Biosciences 2-laser 4 color FACSCaliber or 4-laser 11-color LSR II instrument. Data analysis was carried out using CellQuest Version 4.0 (BD Biosciences) or FlowJo Version 7.6.5 (TreeStar) software.
Cell staining and microscopy
For p65 and phospho-p38 mitogen-activated protein kinase (p38Mapk) staining, LSK cells were sorted using a BD Biosciences FACSAria, cytospun onto slides, fixed with 4% paraformaldehyde, permeabilized with 0.25% Triton X-100, and then incubated with anti-p65 (eBioscience) or anti-phospho–p38(Thr180/Tyr182; Millipore) 2 hours at room temperature. After washing, the cells were stained with AlexaFluor-488–conjugated goat anti–rabbit IgG antibody (Cell Signaling) and 4,6-diamidino-2-phenylindole (DAPI) counterstain. Cell images were obtained at room temperature with a Solamere CSU10 Spinning Disk confocal system mounted on a Nikon TE2000-E2 inverted microscope and processed using ImageJ Version 1.32 software (National Institutes of Health). Senescence-associated β-galactosidase (SA–β-Gal) expression was tested using cytospin preparations of LSK cells as described in the previous paragraph. with the Senescence Detection Kit (Abcam) according to the supplier's protocol and scoring positive cells on an Eclipse TS100 inverted microscope (Nikon) with a SPOT Diagnostics camera model 2.2.1 (Diagnostic Instruments Inc).
Gene expression and microarray
BM from Ott1 KO and controls 6 weeks after pIpC treatment was labeled as in the preceding paragraph and sorted for LSK cells on a BD Biosciences FACSAria instrument. Total RNA was isolated using an RNeasy microkit (QIAGEN) and treated with RNase-free DNase (QIAGEN). A total of 50 ng of purified total RNA was amplified using the Ovation RNA amplification system Version 2 (Nugen), and biotinylated with the FL-Ovation Biotin Module Version 2 (Nugen), according to the supplied protocol. cDNA was then hybridized to Affymetrix mouse expression array 430A2.0 chips by the Dana-Farber Microarray Core Facility. The raw gene expression values were preprocessed with the robust multiarray analysis algorithm22 using BioConductor Version 2.10 software.23 Gene sets were tested for enrichment for Ott1 knockout versus control using gene set enrichment analysis (GSEA).24 The signal-to-noise ratio was used as the test statistic to rank order the genes for permutation testing. For statistical quantification, permutation testing in GSEA was performed with 2000 permutations by gene set. Microarray data are deposited in the Gene Expression Omnibus accession no. GSE37047.
For quantitative PCR, amplified cDNA from LSK cells as described in “Cell staining and microscopy” was analyzed on an EP Realplex real-time PCR cycler using the QIAGEN Quantifast SYBR Green PCR kit and QuantiTect RT-PCR primers for p16Ink4a, p19Arf, p21Cip, and Gapdh per the manufacturer's protocol. All samples were run in triplicate. Relative expression determined using the ΔΔCT method.
Colony assays
Whole BM was isolated from tibias and femurs of killed mice then treated with RBC lysis buffer (QIAGEN). A total of 20 000 nucleated cells were duplicate plated in M3434 methycellulose (StemCell Technologies) containing erythropoietin, IL-3, IL-6, and SCF and incubated at 37% and 5% CO2 for 10 days. Colonies were scored using an Eclipse TS100 inverted light microscope (Nikon) with a gridded plate and identified by morphology per the manufacturer's instructions.
NAC treatment
Lethally irradiated CD45.1 recipients were injected intravenously with 8 × 105 Ott1flox/null Mx1-cre or littermate control BM cells and 2 × 105 CD45.1/CD45.1 heterozygous competitor cells. After 4 weeks, a nonlethal facial vein bleed was performed, and flow cytometry was used to determine the engraftment percentage via CD45.1/CD45.2 markers on the Mac-1+/Gr-1+ population; then the mice were treated with pIpC 250μg 3 times as described in “Mice.” Cohorts received either 200 mg/kg N-acetyl-cysteine (NAC; Sigma-Aldrich) or saline alone intraperitoneally on a daily basis beginning 3 days before pIpC treatment and continuing until death at 3 weeks after pIpC.
Statistical analysis
Statistical significance was determined by the 2-tailed Student t test and was achieved if P was less than .05. The 5-fluorouracil (5-FU) survival curve was analyzed using a Kaplan-Meier analysis and log-rank test. Error bars represent SD.
Results
HSCs are dependent on Ott1 for engraftment and maintenance
Our previous work demonstrated an increase in the absolute number of LSK cells. Further immunophenotypic characterization was performed to identify which specific subpopulations were altered on Ott1 loss. BM from Ott1flox/null Mx1cre mice approximately 6 weeks after pIpC-mediated excision of the Ott1 allele (hereafter referred to as Ott1 KO) and littermate pIpC-treated controls possessing an intact Ott1 allele was analyzed using 2 alternative markers for HSC identification. LT-HSC populations were identified using LSK/CD150+CD48− or LSK/CD34−/Flk2− staining (Figure 1A).25,26 Ott1 KO and control BM possessed similar numbers of LT-HSCs by both of these criteria. Additional characterization using Flk2 to differentiate short-term HSCs (ST-HSCs) from multipotent progenitors demonstrated a 2-fold increase specifically in ST-HSCs in the Ott1 KO mice.
Figure 1.
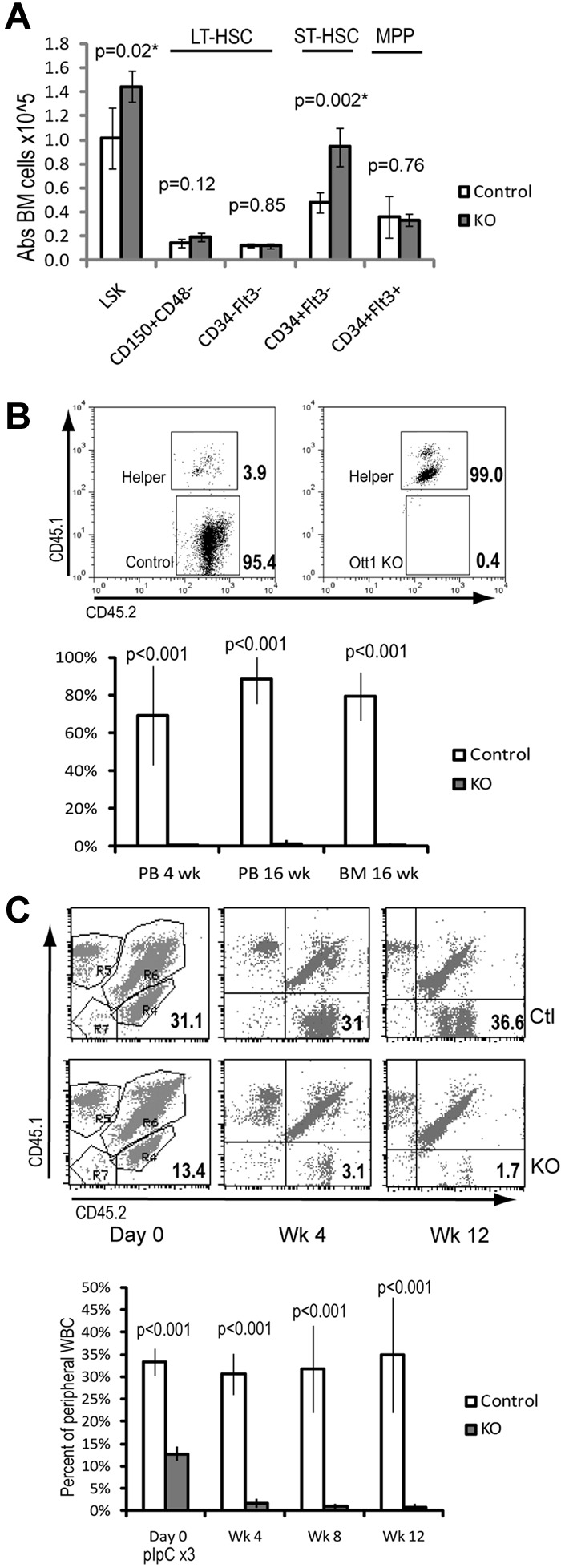
Despite normal numbers of LT-HSCs, Ott1-deleted BM cannot compete with WT BM during engraftment or maintenance. (A) Absolute cell numbers in BM determined by flow cytometry of LSK-gated populations stained with either CD150/CD48 or CD34/Flt3 (Flk2) antibodies. BM was harvested from Ott1flox/null Mx1-cre (KO) or littermates possessing a WT Ott1 allele (control) 6 weeks after pIpC injection. (B) Competitive repopulation of lethally irradiated homozygous CD45.1 recipients transplanted with donor marrow from either Ott1flox/null Mx1-cre (Ott1 KO) or littermates possessing a WT Ott1 allele (control) 6 weeks after pIpC injection. Donor marrow was injected in 4:1 excess with WT CD45.1/45.2 helper marrow. y-axis represents percentage of graft arising from the donor (CD45.2 homozygous) in peripheral blood granulocytes (PB) or BM during the stated posttransplantation interval (n = 4). Flow panels (above) are representative CD45.2-gated PB samples at 16 weeks after transplantation. (C) Maintenance after transplantation. Unexcised BM from Ott1flox/null Mx1-cre (KO) or littermates possessing a WT Ott1 allele (control) was engrafted into lethally irradiated CD45.1 recipients 1:1 with CD45.1/CD45.2 competitor marrow and then excised with pIpC. Percentage is homozygous CD45.2 donor in peripheral blood leukocytes at the stated interval after pIpC (n = 5). Representative flow panels show CD45.2 donor percentage (bottom right quadrant) in PB before and after pIpC. Ctl, top panels; KO, bottom panels. Graphs represent mean values, and error bars represent SD.
Although Ott1 KO mice have similar numbers of LT-HSCs, we sought to establish their functional status by competitive repopulation using donor BM from CD45.2 Ott1 KO mice and littermate controls. BM cells at a ratio of 4:1 (Donor:Competitor) with CD45.1/CD45.2 heterozygous WT competitor were transplanted into lethally irradiated CD45.1 recipients. Flow cytometry of peripheral blood leukocytes was performed every 4 weeks to assess the precentage of engraftment (Figure 1B). At 4 weeks after transplantation, only 0.15% of the peripheral blood granulocytes were derived from the Ott1 KO donor compared with 69% for the control donor marrow. Analysis of BM chimerism at 16 weeks after transplantation demonstrated Ott1 KO BM contributed 1% of total CD45 expressing cells compared with 79.6% of control donor marrow. Analysis of a separate cohort at 2 weeks after transplantation showed less than 1% Ott1 KO cells in the LSK compartment (data not shown). The inability of Ott1-deleted BM to competitively repopulate despite a 4:1 ratio with competitor argues for a severe, cell autonomous defect in HSC function.
Even under noncompetitive conditions, Ott1 KO BM was unable to successfully engraft a lethally irradiated recipient using up to 106 donor cells (supplemental Table 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Transplantation of 5 × 106 cells yielded 3 of 4 surviving mice; however, PCR analysis of the recipients showed the presence of floxed Ott1 alleles indicating engraftment by nonexcised cells (data not shown). Because Ott1 KO BM cannot engraft, the term Ott1 KO “HSCs” in this report refers to an immunophenotypic definition. A defect in HSC function had been previously described using an alternate conditional Ott1 knockout model6; however, we observed a dramatically more severe phenotype in our model under both competitive and noncompetitive conditions. The increased engraftment observed in Niu et al may be the result of expansion of incompletely excised floxed Ott1 alleles in the recipient.
Homing assays were performed using CSFE-labeled BM cells injected into irradiated recipients and in agreement with other studies did not show a significant difference with controls6 (and data not shown). To specifically measure the ability of Ott1 KO HSCs to compete under steady-state conditions, unexcised CD45.2 Ott1flox/null Mx1cre or littermate control BM was transplanted in a 1:1 ratio with WT CD45.1/CD45.2 competitor into lethally irradiated recipients (Figure 1C). After 4 weeks to allow stable engraftment, peripheral blood neutrophils were analyzed to determine baseline chimerism, and mice were then injected with 3 doses of pIpC to excise Ott1. The baseline engraftment for Ott1flox/null Mx1cre is lower than control probably the result of excision by endogenous interferon during transplantation as has been observed in similar experiments.27 The Ott1flox/null Mx1cre graft was rapidly lost after pIpC injection, whereas the control graft retained stable chimerism. Less than 1% Ott1 KO cells were identified in the LSK population on analysis of the BM at 12 weeks (data not shown). The failure of the Ott1flox/null Mx1cre graft is consistent with a physiologic requirement for Ott1 for in HSC function under competitive stress.
Etiologic mechanisms for HSC failure include increases in apoptotic rates, such as in the Forkhead box (FoxO) and Ataxia telangiectasia mutated (Atm) knockouts.19,28 Baseline apoptotic rates in the Ott1 KO as measured by annexin V/7-AAD in LT-HSC and ST-HSC compartments were not significantly different from littermate controls (Figure 2A). Alternatively, increased HSC proliferation and loss of the quiescent, self-renewing population found in G0 leads to stem cell exhaustion.2 Cell-cycle analysis using Hoechst 33342/pyronin Y was performed on BM from Ott1 KO and littermate controls (Figure 2B-C). Ott1 KO LT- and ST-HSC populations had slightly greater G0 populations, but no significant changes in S-G2-M, arguing against baseline loss of quiescence or increased proliferation as an explanation. Finally, to determine whether increased senescence was responsible for the Ott1 KO HSC failure, sorted Ott1 KO LSK cells were stained to detect SA–β-Gal (Figure 2D).29 No evidence of increased SA–β-Gal was observed. In addition, quantitative PCR of gene expression from sorted Ott1 KO LSK cells lacked up-regulation of senescence-associated genes, p16Ink4a, p19Arf, and p21Cip (Figure 2E).30 Niu et al reported reduced c-Myc levels in an alternate Ott1 knockout model with a defect in competitive repopulation.6 Quantitative PCR of sorted Ott1 KO LSK cells demonstrated instead a modest increase in c-Myc levels, suggesting an alternative mechanism underlying the defect (Figure 2E). A possible explanation for this difference is that the Niu et al model used a much larger pIpC dose which would lead to a greater interferon response and interferon is known to suppress c-Myc.31
Figure 2.

Loss of Ott1 does not cause increases in HSC apoptosis, proliferation, or senescence. (A) Flow cytometric measurement of apoptotic (annexin V+/7-AAD−) and dead/apoptotic (annexin V+/7-AAD+) percentage in LT-HSCs (Lin−Sca-1+c-Kit+CD34−) and ST-HSCs (Lin−Sca-1+c-Kit+ CD34+) from BM harvested from Ott1flox/null Mx1-cre (KO) or littermates possessing a WT Ott1 allele (control) 6 weeks after pIpC injection. (B) Cell-cycle analysis of LT-HSCs (Lin−Sca-1+c-Kit+CD34−). KO and control BM obtained as in panel A were stained with Hoechst 33342/pyronin Y, and flow cytometry was performed to quantify G0, G1, and S-G2-M phases. Representative flow panels above of control (left) and KO (right). (C) Cell-cycle analysis as in panel B for ST-HSCs (Lin−Sca-1+c-Kit+CD34+; n = 5, control; n = 4, KO). (D) SA–β-Gal expression in sorted control and Ott1 KO LSK cells. Cytospins of sorted cells were stained for SA–β-Gal and examined under light microscopy. A total of 100 cells were counted from each group, and the experiment was performed in duplicate. No β-Gal–positive cells were observed. Black bars represent 10 μm. (E) Quantitative real-time PCR from total RNA of sorted control and Ott1 KO LSK cells. Bars represent mean of Ott1 KO relative to control (n = 3).Graphs represent mean values, and error bars represent SD.
Long-term hematopoiesis is sustained after in situ deletion of Ott1.
Given the rapid loss of transplanted Ott1-deficient HSCs, a similar reduction in HSCs might be expected over time after in situ deletion. Cohorts of Ott1flox/null Mx1cre and littermate controls underwent pIpC treatment at 6 weeks of age to delete Ott1 and then were followed prospectively. Premature death was not observed in the Ott1 KO arm, and analysis of peripheral blood at approximately 18 months of age showed no significant differences in hematocrit, neutrophil, lymphocyte, or platelet counts (Figure 3A). Analysis of the stem cell compartment demonstrated similar numbers of LT- and ST-HSCs, myeloid progenitors, and total BM cells (Figure 3B). CFU assays from the aged Ott1 KO cohort in methylcellulose demonstrated an approximately 2-fold decrease in colony formation compared with age-matched controls (Figure 3C). Single-colony PCR revealed 100% excision of myeloid colonies, excluding the possibility of outgrowth of an incompletely deleted population (data not shown). The reduction in CFU in light of preserved progenitor numbers demonstrates a loss of clonogenicity with age that was not observed after ST deletion.5 Despite diminished clonogenicity, Ott1 KO animals were able to maintain sufficient hematopoiesis over their life span.
Figure 3.

Mice undergoing Ott1 deletion are able to maintain peripheral blood counts and BM HSC populations older than 18 months but have reduced colony-forming potential. (A) Automated peripheral blood analysis of samples from 18-month-old Ott1flox/null Mx1-cre (KO) or littermates possessing a WT Ott1 allele (control) pIpC-treated at 6 weeks of age. Control, n = 6; KO, n = 5. (B) Absolute numbers of LT-HSCs (Lin−Sca-1+c-Kit+CD34−), ST-HSCs (Lin−Sca-1+c-Kit+CD34+), progenitors (Lin−Sca-1−c-Kit+), and total BM obtained from 18-month-old Ott1 KO and control mice in panel A. (C) Myeloid CFU ability of BM from 18-month-old Ott1 KO or control mice in panel A. Samples plated in duplicate (control, n = 4; Ott1 KO, n = 3). Gran indicates CFU-granulocyte; GM, CFU-granulocyte/monocyte; Mono, CFU-monocyte; and Ery, CFU-erythroid. Graphs represent mean values, and error bars represent SD.
Ott1 maintains HSC quiescence during replicative stress.
To explain the apparent discrepancy between the ability of Ott1-deficient HSCs to maintain life-long function in situ and their failure under engraftment or competitive conditions, we hypothesized that Ott1 function is essential during replicative stress but not steady-state hematopoiesis. Normally quiescent HSCs can be driven into replication after 5-FU treatment.32 Ott1 KO mice and littermate controls were injected with 150 mg/kg 5-FU and followed by serial sampling of peripheral blood (Figure 4A). By 14 days after injection, the control mice had a brisk recovery of peripheral blood neutrophils; however, Ott1 KO mice still had low neutrophil counts, demonstrating an impaired recovery. To determine whether Ott1 KO mice were compromised in their ability to respond to persistent hematopoietic stress, cohorts of Ott1 KO mice and littermate controls were given weekly injections of 150 mg/kg 5-FU and observed for death from BM failure (Figure 4B). Ott1 KO mice died significantly earlier than controls (P < .008).
Figure 4.

BM lacking Ott1 has defective recovery during hematopoietic stress and is accompanied by loss of quiescence in the HSC compartment. (A) Neutrophil counts from peripheral blood samples of Ott1flox/null Mx1-cre (KO) or littermates possessing a WT Ott1 allele (Ctl) 6 weeks after pIpC treated with a single dose of 5-FU (150 mg/kg). Open (Ctl) and closed (KO) boxes represent the mean value, and error bars represent SD (Ctl and KO, n = 4). (B) Kaplan-Meier survival curve of KO and Ctl mice injected with weekly 5-FU (150 mg/kg). P values were calculated using Mantel-Cox test (Ctl, n = 7; KO, n = 15). (C) Cell-cycle analysis of control and KO BM LT-HSC (Lin−Sca-1+c-Kit+ CD34−) and ST-HSC (Lin−Sca-1+c-Kit+CD34+) populations using Hoechst 33342/pyronin Y from panel A at 14 days after 5-FU treatment. Representative flow panels above. (D) Absolute BM cell numbers from mice in panel A of LT-HSCs (Lin−Sca-1+c-Kit+CD34−), ST-HSCs (Lin−Sca-1+c-Kit+CD34+), progenitors (Lin−Sca-1−c-Kit+), and total BM (BM; A) at 14 days after 5-FU treatment. Bars represent mean values, and error bars represent SD (Ctl and KO, n = 4). (E) Cell-cycle analysis of LT- and ST-HSC populations in BM from Ott1flox/null Mx1-cre (KO) or littermates possessing a WT Ott1 allele (control) 6 weeks after pIpC cultured in vitro in IL-3, IL-6, and SCF for 72 hours. Representative flow panels above. Graphs represent mean values, and error bars represent SD (Ctl and KO, n = 4).
HSC populations after a single 5-FU treatment were examined to identify potential etiologies for the poor recovery. No significant changes were seen in apoptosis of LT- or ST-HSCs as measured by annexin V/7-AAD (data not shown). However, cell-cycle analysis of the LT- and ST-HSC populations using Hoechst 33342/pyronin Y revealed a striking decrease in the G0 phase of the Ott1 KO compared with controls: 0.5% plus or minus 0.4% versus 11.6% plus or minus 3.7% (P = .001), respectively (Figure 4C). A corresponding increase in the number of HSCs in G1 was observed in the Ott1 KO but not in the S-G2-M fraction. The absolute numbers of LT-HSCs and ST-HSCs were increased in the Ott1 KO after 5-FU treatment (Figure 4D). Together, these data strongly suggest that proliferative stress shifts Ott1-deleted LT-HSCs out of G0, and they are unable to maintain self-renewal in contrast to steady-state conditions, where G0 is maintained.
HSC proliferation can be positively regulated through cytokine responses and negatively through niche interaction.33 To determine whether Ott1 KO HSCs have altered response to isolated positive cytokine signaling, proliferative stress was initiated in vitro by incubating Ott1 KO BM and controls for 72 hours in IL-3, IL-6, and SCF, conditions known to promote HSC proliferation (Figure 4E).34 Both Ott1 KO and control HSCs had similar loss of G0 fractions; however, Ott1 KO LT-HSCs had a much higher percentage of cells in S-G2-M than controls: 39.2 ± 3.6 versus 26.3 ± 2.8 (P < .001). A similar increase was seen in Ott1 KO ST-HSCs: 44.5 ± 8.0 versus 28.5 ± 1.0 (P = .012). The discrepancy in cell-cycle regulation between Ott1 KO HSCs at baseline and those in proliferation advocates a distinct role for Ott1 maintaining quiescence during proliferative stress as opposed to steady-state conditions.
Loss of Ott1 leads to aging-associated alterations in HSCs, including elevated ROS
Several characteristics of Ott1 KO hematopoiesis bear a resemblance to changes that occur in aging. These alterations include increased myeloid skewing, decreased lymphopoiesis, increased LSK population, reduced clonagenicity, and decreased stress response ability.5,18,35 Chambers et al used HSCs from mice aged up to 21 months of age to identify genes differentially expressed with age.17 A total of 1667 genes were positively up-regulated in aged HSCs; and of these, 79 were HSC-specific compared with whole BM. To determine whether similar alterations in transcription were occurring in the Ott1 KO, we used GSEA to compare up-regulated-with-age gene sets from Chambers et al with genes up-regulated after Ott1 deletion.
Using sorted LSK cells from Ott1 KO mice and controls, we amplified total RNA and hybridized it on Affymetrix MOE430A microarrays to generate gene expression profiles and identify differentially expressed genes. GSEA comparing Ott1 KO LSK up-regulated genes with the up-regulated-with-age gene set showed significant enrichment with a Normalized Enrichment Score (NES) = 1.167 (P = .012, false discovery rate [FDR] = 0.038) and even greater enrichment with the HSC-specific subset, NES = 2.02 (P < .0001, FDR < 0.001; Figure 5A-B). Significant enrichment was also observed using an up-regulated-with-age gene set generated by Rossi et al comparing HSCs from 2-year-old versus 2-month-old mice (Figure 5C).35 GSEA comparing Ott1 KO down-regulated genes with down-regulated-with-age gene sets were significant for the Rossi et al set (NES = −1.46, P < .001, FDR = 0.054) but not the Chambers et al set (NES = −1.04, P = .34, FDR = 0.33; supplemental Tables 5-6). A caveat of interpreting the GSEA results is that the Ott1 KO profiles are from LSK rather than LT-HSC subpopulations. The full GSEA reports are in supplemental Tables 2 to 6. In summary, Ott1 deletion leads to a transcriptional signature similar to aged HSCs.
Figure 5.

Ott1-deleted HSCs have a gene expression profile similar to aged HSCs with associated physiologic changes. (A-C) GSEA comparing genes up-regulated in sorted LSK cells from Ott1 KO mice compared with WT controls with published gene sets up-regulated in aged HSCs. (A) GSEA using Chambers et al17 1667 genes up-regulated with age in HSCs. (B) GSEA using a 79-gene subset of (A) restricted to genes expressed in HSCs versus whole BM.17 (C) GSEA comparing Ott1 KO LSK with Rossi et al aged HSC profile.35 (D) Localization of p65 NF-κB subunit. Cytospins of sorted control and Ott1 KO LSK cells were stained with rabbit anti-p65 and anti–rabbit-AF488 secondary antibody with DAPI nuclear stain. Cells were imaged by confocal microscopy. A minimum of 30 cells were assessed from each group, and the experiment was performed in duplicate. (E) MFI of DCF-DA–labeled cells from Ott1 KO and control BM. LT-HSCs (Lin−Sca-1+c-Kit+CD34−), ST-HSCs (Lin−Sca-1+c-Kit+CD34+), and progenitors (Lin−Sca-1−c-Kit+). Representative histograms of flow above. (F) Measurement of phospho–γ-H2AX levels in Ott1 and control LT-HSC, ST-HSC, and progenitor populations. Cells stained for intracellular phospho–γ-H2AX using phospho-specific antibody. Representative flow histograms above. Graph bars represent mean of MFI, and error bars represent SD. Control, n = 4; Ott1 KO, n = 3. (G) Phospho-p38Mapk staining of Ott1 KO and control LSK cells. KO or control BM as in panel A was either untreated (baseline) or grown 48 hours in IL-3, IL-6, and SCF (cytokine). Cytospins of sorted LSK cells were stained with anti–phospho-p38Mapk and AF488 secondary antibody and then imaged by confocal microscopy with DAPI nuclear counterstain. Minimum of 40 cells scored. White bar represents 10 μm.
In addition to similarities in function and gene expression, we investigated whether Ott1 loss resulted in age-associated alterations in cell physiology. Aged HSCs have a proinflammatory profile and exhibit activation of NF-κB.17 A surrogate of NF-κB activation is nuclear localization of the p65 subunit.36 Sorted Ott1 KO LSK cells and controls were cytospun onto slides and stained for anti-p65 with a DAPI nuclear counterstain (Figure 5D). Ott1 KO LSK cells had 78% p65 nuclear localization compared with 26% of controls, indicating increased NF-κB activation in the Ott1 KO similar to that observed in aged cells.
Elevated ROS are thought to be responsible for accumulation of somatic damage to DNA and proteins and contribute to the aging phenotype.3,4,37 Maintaining low ROS levels is also essential to HSC quiescence.4,37 Ott1 KO and controls were examined using the ROS-responsive dye, DCF-DA (Figure 5E). LT-HSCs, ST-HSCs, and myeloid progenitors all displayed markedly increased levels of ROS with 4-, 3-, and 2-fold higher median fluorescence intensities (MFIs), respectively. These data demonstrate that ROS, a known mediator of aging-related processes, is significantly elevated in Ott1 KO HSCs and progenitors and suggests physiologic role for Ott1 in negative regulation of ROS.
A consequence of aging and elevated ROS is increased DNA damage. The extent of DNA damage was assessed using a flow cytometric technique measuring the fluorescence intensity of γH2AX, which binds to damaged DNA sites.21 BM from Ott1 KO and control mice analyzed 6 weeks after pIpC treatment for the amount of γH2AX present during steady-state hematopoiesis (Figure 5F). An increase in the MFI of anti-γH2AX antibody/fluorochrome conjugate complex was observed and significant for LT- (P = .03) and ST-HSC (P = .01) but did not achieve significance for myeloid progenitors (P = .1).
Increases in ROS mediate their physiologic effects in large part because of activation of p38Mapk. Activated p38Mapk has been previously determined to impede HSC function, including self-renewal, and is up-regulated during aging.38 To assess the extent of the phosphorylated, activated form of p38Mapk, sorted LSK cells from steady-state Ott1-deleted BM were used for cytospin preparations and intracellularly stained with anti–phospho-p38Mapk (Figure 5G). Confocal fluorescence microscopy revealed a high percentage of phospho-p38Mapk–containing cells within the Ott1-deleted cohort at baseline compared with controls (67% vs 13%). p38Mapk is phosphorylated in response to cytokine stimulation. In vitro incubation of the cells in cytokine-containing medium resulted in p38Mapk activation of control LSK cells to levels similar to that observed with Ott1 KO LSK cells at baseline.
Elevated ROS are not primarily responsible for the HSC defect
Elevation of ROS disrupts HSC homeostasis as observed in the Atm, Foxo3, and B lymphoma MoMLV insertion region 1 (Bmi1) knockout models, resulting in increased baseline proliferation and/or apoptosis, which can be rescued by reduction of ROS levels with the antioxidant, NAC.19,28 The Ott1 knockout phenotype differs from these models in that ROS-dependent changes to proliferation, apoptosis, and senescence are not observed during steady-state hematopoiesis. If elevated ROS were primarily responsible for the Ott1 KO HSC defect, then NAC treatment should rescue the defect. Unexcised CD45.2 Ott1flox/null Mx1cre and control BM were engrafted into CD45.1 recipients with competitor and then excised with pIpC while being treated with NAC or saline control (Figure 6A). In the presence of NAC, the Ott1 KO engraftment as measured by the CD45.2+/CD45.1− population in Mac1+/Gr1+ peripheral blood cells dropped from a baseline of 59.8% ± 8% to 2.5% ± 2% (P < .01) after 3 weeks (Figure 6B). No significant change was observed for the control NAC-treated population. Comparison between Ott1 KO Mac1+/Gr1+ peripheral blood cells in the NAC and saline groups showed no significant difference, consistent with lack of even a partial rescue (Figure 6C). Specific analysis of the LT- and ST-HSC populations showed minimal remaining Ott1 KO donor chimerism irrespective of the presence or absence of NAC (Figure 6D). The inability of antioxidant treatment to correct the HSC defect in the Ott1 KO suggests that control of ROS is not sufficient to explain the HSC requirement for Ott1.
Figure 6.

Ott1-deleted HSCs cannot be rescued with antioxidant treatment and have increased mitochondrial mass. (A) Schema for determining whether NAC is capable of rescuing the maintenance defect in Ott1-deleted HSCs. Unexcised CD45.2 Ott1flox/null Mx1cre and control BM were transplanted into lethally irradiated CD45.1 recipients in a 4:1 ratio with CD45.1/CD45.2 heterozygous WT competitor BM. Four weeks were allowed for stable engraftment; then the 2 cohorts of mice were each split into 2 arms and received either 200 mg/kg NAC or saline placebo intraperitoneally daily for the remainder of the experiment. After 3 days of NAC treatment, the mice were bled to assess baseline engraftment of donor marrow through CD45.1/CD45.2 ratios in the Mac1+/Gr1+ peripheral blood population and then treated with pIpC. At 3 weeks after pIpC, peripheral blood and BM were obtained to analyze chimerism. (B) Precentage of donor chimerism of peripheral blood neutrophils from mice transplanted with BM Ott1flox/null Mx1-cre (KO) or WT Ott1 (control) plus competitor pre-pIpC and after pIpC/NAC for 3 weeks. (C) Peripheral blood neutrophil donor chimerism at 3 weeks after pIpC of Ott1 KO and control donor treated with NAC or saline. (D) Precentage of donor chimerism of LT-HSC (Lin−Sca-1+c-Kit+CD34−) populations from Ott1 KO or control 3 weeks after pIpC and NAC or saline treatment. Donor identified by detection of CD45.2/CD45.2 markers by flow cytometry (n = 4). (E) MFI of Mitotracker deep red of Ott1flox/null Mx1-cre (Ott1 KO) or littermates possessing a WT Ott1 allele (control) 6 weeks after pIpC of LT-HSC (Lin−Sca-1+c-Kit+CD34−), ST-HSC (Lin−Sca-1+c-Kit+CD34+), and progenitor (Lin−Sca-1−c-Kit+) populations. Representative flow histograms at right. Control (gray line), n = 4; Ott1 KO (black line), n = 3. Graph bars represent mean values, and error bars represent SD.
ROS levels may be modulated by increases in overall metabolic activity, ROS synthesis, reduction in scavengers, and variations in extracellular oxygen tension found in the BM niche.37 Increases in metabolic activity are frequently accompanied by corresponding increases in mitochondrial mass.39 Mitochondria can initially increase ROS as a byproduct of oxidative metabolism; however, they also actively degrade excess ROS.39 Quiescent LT-HSCs maintain low mitochondrial mass.40 Given the loss of quiescence observed during stress and the increases in ROS in Ott1 KO HSCs, we hypothesized that Ott1 may participate in mitochondrial regulation. The mitochondrial mass of HSCs from Ott1 KO mice and controls was measured using the Mitotracker dye and found to be significantly increased in LT-HSCs, ST-HSCs, and myeloid progenitors (Figure 6E). We were precluded from simultaneously measuring ROS and mitochondrial mass because of limitations of our flow cytometric assay. The increase in mitochondrial mass in the Ott1 KO implicates Ott1 in the regulation of mitochondrial biogenesis.
Discussion
The inability of Ott1-deleted BM to engraft even within a 2-week period indicates functional defects within both ST- and LT-HSCs. The HSC competitive defect remains despite Ott1 excision after transplantation in a WT recipient, establishing that the HSC requirement for Ott1 is cell autonomous and excluding a homing defect as the etiologic mechanism. Under homeostatic conditions and in the absence of competitor, HSCs lacking Ott1 are able to maintain normal immunophenotypic HSC populations and peripheral blood counts for at least 18 months. No steady-state changes in HSC proliferation, apoptosis, or senescence were observed after Ott1 excision, which might contribute to the defect. However, when 5-FU was used to induce replicative stress in Ott1-deficient mice, the animals had delayed neutrophil recovery and BM failure with repeated doses. The discrepancy between Ott1 dependency in competition or after 5-FU treatment but not during steady-state hematopoiesis argues for a specific requirement in stress hematopoiesis.
Cell-cycle analysis of 5-FU–treated mice revealed a dramatic loss of the G0 HSC population in the absence of Ott1. Several genes have been found to regulate HSC quiescence during replicative stress, including p21Cip and Foxo3; however, their deletion affects steady-state cell-cycle kinetics and HSC numbers as well.19,41 A smaller subset of genes have been identified, which are required for HSC quiescence during stress hematopoiesis, yet their absence does not also reduce quiescence in homeostatic cells. Among these are Hypoxia inducible factor-1α (HIF1α), Thioredoxin interacting protein (Txnip), and Retinoblastoma (Rb).42–44 A common feature of these genes is their involvement in the modulation and response to ROS. Levels of ROS in LT-HSCs are relatively low in part because of the low oxygen tension of the osteoblast niche, overall metabolic state, and ROS scavenger systems.37 Although Ott1-deleted HSCs have high levels of ROS, NAC treatment does not rescue the HSC defect. Furthermore, in contrast to the Atm, FoxO3a, and Bmi1 knockout models, Ott1-deficient HSCs do not show increased steady-state apoptosis, senescence, or abnormal proliferation. Activation of p38Mapk was shown to be the primary mediator of ROS-induced self-renewal defects in HSCs.38 Although p38Mapk is activated in Ott1-deleted HSCs, the physiologic consequences are not apparent, suggesting a requirement for Ott1 in p38Mapk downstream pathways.
Regulation of ROS levels is intimately associated with mitochondrial biogenesis.39 Self-renewing LT-HSC populations have been shown to have lower mitochondrial mass and greater reliance on glycolysis as they inhabit a hypoxic niche.40 Mitochondria may contribute to ROS levels through increased metabolic activity but also provide a mechanism for degradation of excessive ROS levels.45 In the absence of Ott1, LT-HSCs have increased mitochondrial mass during steady-state hematopoiesis, associating them with a more proliferative, non–self-renewing phenotype despite a quiescent cell-cycle profile. Control of ROS levels may be a secondary effect subordinate to Ott1-dependent mitochondrial regulation; however, the inability of NAC to rescue implies control of ROS alone does not define the requirement for Ott1 in HSC function.
Current studies suggest that cellular stress response in aging cells is altered.17,46 p53 has been found to play an important role in HSC stress hematopoiesis and quiescence; however, Ott1-deficent HSCs do not show changes in apoptotic rate or perturbation in p53 target genes Growth factor independence-1 (Gfi-1) and Necdin in the expression array data.47,48 Mutant p53 has been associated with aging-related changes, but in HSCs, the mutant p53-dependent expression profile is separable from the aging-specific profile.17 Telomere dysfunction is associated with increased ROS and manifests in a p53-dependent manner but, in contrast to the Ott1 knockout, shows decreased mitochondrial biogenesis.49
There is a significant phenotypic overlap between Ott1-deficient and aging hematopoiesis, including myeloid skewing, impaired HSC self-renewal, and decreased stress response. At a cellular level, Ott1-deficient HSCs share a gene expression profile with aging HSCs as well as elevated ROS, DNA damage, and activation of p38Mapk and NF-κB.17 Gene expression of Ott1 is stable with age according to expression data generated comparing young and old HSCs, implying that Ott1 does not directly regulate the rate of aging.17,35 Recently, a mouse mutant of SMRT, which binds the conserved SPOC domain of family member Spen, has been shown to possess elevations in cellular ROS and characteristics of aging.50 In contrast to aged HSCs, however, Ott1 deletion results in a much more severe transplantation defect, does not cause an increase in absolute numbers of LT-HSCs and has no apparent effect on overall life span.17,35 We speculate that Ott1 regulates essential cellular pathways, similarly involved in aging, such as quiescence, inflammation, and ROS, which are essential for HSC function and self-renewal.2,3,37
Supplementary Material
Acknowledgments
The authors thank D. Gary Gilliland, Shaoguang Li, and Peter Newberger for critical review of the manuscript; Cong Peng, Yaoyu Chen, and Haojian Zhang for technical assistance; and Paul Furcinitti (University of Massachusetts Bio Imaging Core Facility) for assisting with confocal microscopy.
This work was supported in part by the National Cancer Institute (grant K08-CA111399).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: N.X. and G.D.R. designed and performed the research, analyzed data, and wrote the manuscript; K.J., K.M., R.O., and D.E.C. performed research and analyzed data; and J.L.J. analyzed expression array data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Glen D. Raffel, Division of Hematology/Oncology, University of Massachusetts Medical School, 364 Plantation St, LRB319, Worcester, MA 01605; e-mail: glen.raffel@umassmed.edu.
References
- 1.Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9(2):115–128. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- 2.Li J. Quiescence regulators for hematopoietic stem cell. Exp Hematol. 2011;39(5):511–520. doi: 10.1016/j.exphem.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 3.Gazit R, Weissman IL, Rossi DJ. Hematopoietic stem cells and the aging hematopoietic system. Semin Hematol. 2008;45(4):218–224. doi: 10.1053/j.seminhematol.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 4.Ergen AV, Goodell MA. Mechanisms of hematopoietic stem cell aging. Exp Gerontol. 2010;45(4):286–290. doi: 10.1016/j.exger.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raffel GD, Mercher T, Shigematsu H, et al. Ott1(Rbm15) has pleiotropic roles in hematopoietic development. Proc Natl Acad Sci U S A. 2007;104(14):6001–6006. doi: 10.1073/pnas.0609041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niu C, Zhang J, Breslin P, Onciu M, Ma Z, Morris SW. c-Myc is a target of RNA-binding motif protein 15 in the regulation of adult hematopoietic stem cell and megakaryocyte development. Blood. 2009;114(10):2087–2096. doi: 10.1182/blood-2009-01-197921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raffel GD, Chu GC, Jesneck JL, et al. Ott1 (Rbm15) is essential for placental vascular branching morphogenesis and embryonic development of the heart and spleen. Mol Cell Biol. 2009;29(2):333–341. doi: 10.1128/MCB.00370-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma Z, Morris SW, Valentine V, et al. Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat Genet. 2001;28(3):220–221. doi: 10.1038/90054. [DOI] [PubMed] [Google Scholar]
- 9.Mercher T, Coniat MB, Monni R, et al. Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22) translocation of acute megakaryocytic leukemia. Proc Natl Acad Sci U S A. 2001;98(10):5776–5779. doi: 10.1073/pnas.101001498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ariyoshi M, Schwabe JW. A conserved structural motif reveals the essential transcriptional repression function of Spen proteins and their role in developmental signaling. Genes Dev. 2003;17(15):1909–1920. doi: 10.1101/gad.266203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindtner S, Zolotukhin AS, Uranishi H, et al. RNA-binding motif protein 15 binds to the RNA transport element RTE and provides a direct link to the NXF1 export pathway. J Biol Chem. 2006;281(48):36915–36928. doi: 10.1074/jbc.M608745200. [DOI] [PubMed] [Google Scholar]
- 12.Majerciak V, Uranishi H, Kruhlak M, et al. Kaposi's sarcoma-associated herpesvirus ORF57 interacts with cellular RNA export cofactors RBM15 and OTT3 to promote expression of viral ORF59. J Virol. 2011;85(4):1528–1540. doi: 10.1128/JVI.01709-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hiriart E, Gruffat H, Buisson M, et al. Interaction of the Epstein-Barr virus mRNA export factor EB2 with human Spen proteins SHARP, OTT1, and a novel member of the family, OTT3, links Spen proteins with splicing regulation and mRNA export. J Biol Chem. 2005;280(44):36935–36945. doi: 10.1074/jbc.M501725200. [DOI] [PubMed] [Google Scholar]
- 14.Ma X, Renda MJ, Wang L, et al. Rbm15 modulates Notch-induced transcriptional activation and affects myeloid differentiation. Mol Cell Biol. 2007;27(8):3056–3064. doi: 10.1128/MCB.01339-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mercher T, Raffel GD, Moore SA, et al. The OTT-MAL fusion oncogene activates RBPJ-mediated transcription and induces acute megakaryoblastic leukemia in a knockin mouse model. J Clin Invest. 2009;119(4):852–864. doi: 10.1172/JCI35901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossi DJ, Bryder D, Weissman IL. Hematopoietic stem cell aging: mechanism and consequence. Exp Gerontol. 2007;42(5):385–390. doi: 10.1016/j.exger.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5(8):e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waterstrat A, Van Zant G. Effects of aging on hematopoietic stem and progenitor cells. Curr Opin Immunol. 2009;21(4):408–413. doi: 10.1016/j.coi.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Tothova Z, Kollipara R, Huntly BJ, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Challen GA, Boles N, Lin KK, Goodell MA. Mouse hematopoietic stem cell identification and analysis. Cytometry A. 2009;75(1):14–24. doi: 10.1002/cyto.a.20674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci U S A. 2006;103(26):9891–9896. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 23.Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adolfsson J, Mansson R, Buza-Vidas N, et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121(2):295–306. doi: 10.1016/j.cell.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 26.Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 27.Gurumurthy S, Xie SZ, Alagesan B, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468(7324):659–663. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito K, Hirao A, Arai F, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431(7011):997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 29.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92(20):9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell. 2009;36(1):2–14. doi: 10.1016/j.molcel.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 31.Einat M, Resnitzky D, Kimchi A. Close link between reduction of c-myc expression by interferon and, G0/G1 arrest. Nature. 1985;313(6003):597–600. doi: 10.1038/313597a0. [DOI] [PubMed] [Google Scholar]
- 32.Van Zant G. Studies of hematopoietic stem cells spared by 5-fluorouracil. J Exp Med. 1984;159(3):679–690. doi: 10.1084/jem.159.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arai F, Hirao A, Suda T. Regulation of hematopoietic stem cells by the niche. Trends Cardiovasc Med. 2005;15(2):75–79. doi: 10.1016/j.tcm.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 34.Katayama N, Clark SC, Ogawa M. Growth factor requirement for survival in cell-cycle dormancy of primitive murine lymphohematopoietic progenitors. Blood. 1993;81(3):610–616. [PubMed] [Google Scholar]
- 35.Rossi DJ, Bryder D, Zahn JM, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005;102(26):9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 37.Ghaffari S. Oxidative stress in the regulation of normal and neoplastic hematopoiesis. Antioxid Redox Signal. 2008;10(11):1923–1940. doi: 10.1089/ars.2008.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito K, Hirao A, Arai F, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12(4):446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 39.Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84. doi: 10.1042/bse0470069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mantel C, Messina-Graham S, Broxmeyer HE. Upregulation of nascent mitochondrial biogenesis in mouse hematopoietic stem cells parallels upregulation of CD34 and loss of pluripotency: a potential strategy for reducing oxidative risk in stem cells. Cell Cycle. 2010;9(10):2008–2017. doi: 10.4161/cc.9.10.11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng T, Rodrigues N, Shen H, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287(5459):1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 42.Takubo K, Goda N, Yamada W, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 43.Jeong M, Piao ZH, Kim MS, et al. Thioredoxin-interacting protein regulates hematopoietic stem cell quiescence and mobilization under stress conditions. J Immunol. 2009;183(4):2495–2505. doi: 10.4049/jimmunol.0804221. [DOI] [PubMed] [Google Scholar]
- 44.Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129(6):1081–1095. doi: 10.1016/j.cell.2007.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sung HJ, Ma W, Wang PY, et al. Mitochondrial respiration protects against oxygen-associated DNA damage. Nat Commun. 2010;1:5. doi: 10.1038/ncomms1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haigis MC, Yankner BA. The aging stress response. Mol Cell. 2010;40(2):333–344. doi: 10.1016/j.molcel.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2(4):333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milyavsky M, Gan OI, Trottier M, et al. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell. 2010;7(2):186–197. doi: 10.1016/j.stem.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 49.Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470(7334):359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reilly SM, Bhargava P, Liu S, et al. Nuclear receptor corepressor SMRT regulates mitochondrial oxidative metabolism and mediates aging-related metabolic deterioration. Cell Metab. 2010;12(6):643–653. doi: 10.1016/j.cmet.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.