

Abstract
Cell migration and invasion are largely dependent on the complex organization of the various cytoskeletal components. Whereas the role of actin filaments and microtubules in cell motility is well established, the role of intermediate filaments in this process is incompletely understood. Organization and structure of the keratin cytoskeleton, which consists of heteropolymers of at least one type 1 and one type 2 intermediate filament, are in part regulated by post-translational modifications. In particular, phosphorylation events influence the properties of the keratin network. Sphingosylphosphorylcholine (SPC) is a bioactive lipid with the exceptional ability to change the organization of the keratin cytoskeleton, leading to reorganization of keratin filaments, increased elasticity, and subsequently increased migration of epithelial tumor cells. Here we investigate the signaling pathways that mediate SPC-induced keratin reorganization and the role of keratin phosphorylation in this process. We establish that the MEK–ERK signaling cascade regulates both SPC-induced keratin phosphorylation and reorganization in human pancreatic and gastric cancer cells and identify Ser431 in keratin 8 as the crucial residue whose phosphorylation is required and sufficient to induce keratin reorganization and consequently enhanced migration of human epithelial tumor cells.
Key words: Intermediate filaments, Mitogen-activated protein kinases, Sphingosylphosphorylcholine, Gastric cancer cells, Pancreatic cancer cells
Introduction
Cell migration and invasion are markedly dependent on the complex organization of the cytoskeleton (Ballestrem et al., 2000). The cytoskeleton of epithelial cells is a network of three major classes of filamentous biopolymers: microfilaments, microtubules and intermediate filaments. Intermediate filaments are composed of a large family of cell-specific proteins that organize to form 10 nm filaments sharing sequence homology and structural features. Among the cytoplasmic intermediate filament proteins, keratins are expressed preferentially in epithelial cells (Fuchs and Weber, 1994; Coulombe and Omary, 2002) and constitute nearly 5% of the total protein in these cells (Omary et al., 1998). Keratin filaments are obligate heteropolymers of at least one type I (relatively acidic keratins K9–K28, K31–K40) and one type II keratin (relatively basic keratins K1–K8, K71–K86) (Schweizer et al., 2006). These filaments are usually organized into bundles, the so called tonofibrils, which form cage-like structures around the nucleus and extend from the perinuclear region to the cell periphery (Hatzfeld and Franke, 1985).
K8 and K18 are the major components of intermediate filaments of simple epithelia as found in intestine, liver and exocrine pancreas (Fuchs and Weber, 1994; Coulombe and Omary, 2002). The expression pattern of these proteins is generally persistent in carcinomas arising from tissues that normally express K8 and K18 (Oshima et al., 1996). Keratins play a crucial role in maintaining the structural integrity and the mechanical properties of cells and thereby protect cells from a variety of environmental insults (Yamada et al., 2002). Furthermore, they are major determinants for the mechanical features of the cytoplasm and the nucleus (Maniotis et al., 1997; Fuchs and Cleveland, 1998).
The structure and function of keratins are probably regulated through posttranslational modifications, particularly phosphorylation on serine residues within the so-called ‘head’ (N-terminal) and/or ‘tail’ (C-terminal) non-α-helical end domains (Fuchs and Cleveland, 1998; Omary et al., 2006). Ser52 is the major phosphorylation site of human K18 in vivo. This site has been implicated in increased keratin solubility and altered polymerization (Ku and Omary, 1994; Liao et al., 1995a), keratin reorganization (Ku et al., 1999), apoptosis (Caulín et al., 1997) and cellular stress (Omary et al., 1998). Increased phosphorylation of K18 has also been implicated in the reorganization of keratin filaments in hepatocytes treated with protein phosphatase inhibitors (Toivola et al., 1998).
Ser431 is a major in vivo phosphorylation site in human K8. Ser431 is phosphorylated by mitogen-activated protein kinases (MAPKs) in response to activation of the EGFR (Omary et al., 1998). Phosphorylation at this site has also been described during hyperosmotic stress, whereas hypo-osmotic stress leads to dephosphorylation at Ser431 of K8 (Tao et al., 2006), and also occurs in human and mouse liver upon injury resulting in Mallory–Denk body formation (Stumptner et al., 2000) or during mouse liver and gallbladder injury induced by a high-fat diet (Tao et al., 2003).
Sphingosylphosphorylcholine (SPC) is a naturally occurring bioactive lipid that acts as an intracellular and extracellular signaling molecule in numerous biological processes including proliferation (Seufferlein and Rozengurt, 1995), cell migration (Boguslawski et al., 2000), wound healing (Wakita et al., 1998) and differentiation (Kleger et al., 2007). Similar to other bioactive lipids such as lysophosphatidic acid (LPA) or sphingosine-1-phosphate, many of its actions are mediated by the activation of a subfamily of low- and high-affinity G-protein-coupled receptors (An et al., 1995; Meyer zu Heringdorf et al., 2002).
Previously, we have shown that SPC is one of the few naturally occurring compounds that can induce a perinuclear reorganization of the keratin cytoskeleton in human pancreatic cancer cells. This reorganization is accompanied by keratin phosphorylation, including phosphorylation at K18(S52) and K8(S431), and an increase in cellular elasticity and enhanced migration of cancer cells through size limited pores (Beil et al., 2003). However, the precise downstream signaling mechanisms by which SPC induces keratin reorganization and the role of keratin phosphorylation in this process are as yet unknown.
Here we show that the MEK–ERK signaling cascade regulates both SPC-induced K8 phosphorylation at Ser431 and keratin reorganization in human pancreatic and gastric cancer cells. We identify Ser431 in K8 as the crucial residue whose phosphorylation is required and sufficient to induce keratin reorganization and consequently enhanced migration of human epithelial tumor cells.
Results
Role of the ERK cascade for SPC-induced keratin reorganization
Previously we have demonstrated that SPC reorganizes the keratin cytoskeleton in Panc-1 and AGS human cancer cells from a branched phenotype into a perinuclear, ring-like formation and increases migration of epithelial tumor cells through size-limited pores (Beil et al., 2003). These cell lines express K8 and K18 as their major keratins, as shown using a pan anti-keratin antibody and individual K8 and K18 antibodies (supplementary material Fig. S1A). This effect of SPC is probably mediated by a G-protein-coupled receptor. SPC interacts with S1P receptors 1–5, GPR4 and OGR1 with different affinities (Meyer zu Heringdorf et al., 2002). Both pancreatic and gastric cancer cell lines express S1P1–S1P5, GPR4 and OGR1, as determined by RT-PCR (supplementary material Fig. S1B).
Activation of the ERK signaling cascade has been implicated in cell migration (Huang et al., 2004; Rajalingam et al., 2005; Bove et al., 2008). We have previously shown that SPC potently induces activation of ERKs in fibroblasts (Seufferlein and Rozengurt, 1994). SPC was also able to stimulate ERK activation in human pancreatic and gastric cancer cells, reaching a maximum after 15 and 30 minutes of incubation in Panc-1 and AGS cells, respectively. ERK activation in Panc-1 and AGS cells in response to SPC was prevented by incubation of cells with the selective MEK inhibitors PD98059 or U0126 (Fig. 1). Interestingly, SPC failed to activate other signaling pathways in pancreatic cancer cells including the PI3K–AKT, the PKC–PKD and the PKM2 signaling pathways (supplementary material Fig. S2).
Fig. 1.

SPC-induced activation of p42 and p44 (MAPK1 and MAPK3) phosphorylation. (A) Panc-1 and AGS were incubated with 12.5 μM SPC for the indicated time points. (B) Cells were treated with various concentrations of SPC as indicated for 15 minutes (Panc-1) or 30 minutes (AGS). (C) Panc-1 and AGS were preincubated with PD98059 using the concentrations indicated and subsequently treated with 12.5 μM SPC for 15 minutes (Panc-1) or 30 minutes (AGS). (D) Panc-1 and AGS cells were preincubated with the U0126 as indicated and subsequently treated with 12.5 μM SPC for 15 minutes (Panc-1) or 30 minutes (AGS). Immunoblotting was performed using antibodies against p44 and p42 (p44/42) and phosphorylated p44 and p42 (Ph-p44/42).
ERKs also regulate keratin phosphorylation (Ku and Omary, 1997; Huang et al., 2004; Omary et al., 2006). Therefore, we determined whether ERK-mediated phosphorylation of keratins could play a role in SPC-induced keratin reorganization and migration. SPC induced perinuclear reorganization of both endogenous keratin and overexpressed K8 or K18 (Fig. 2A). Keratin perinuclear reorganization was confirmed by quantifying the fluorescence intensity distribution on the y-axis (Fig. 2B, see results below). SPC-induced keratin reorganization was completely prevented upon incubation of Panc-1 or AGS cells with the MEK inhibitors PD90859 or U0126, respectively (Fig. 3). Thus, activation of the MEK–ERK cascade appears to be crucial for perinuclear keratin reorganization in response to SPC.
Fig. 2.

Keratin reorganization in epithelial tumor cells after SPC treatment. (A) Panc-1 and AGS cells were transfected with eCFP-tagged K8, eYFP-tagged K18 or left untransfected and were subsequently incubated with 15 μM SPC for 45 minutes. Endogenous keratins were stained with pan-CK antibody, followed by Alexa Fluor 488 staining. Images were taken using a confocal microscope and keratin was detected in the 488 channel (green emission). Representative images show reorganization of endogenous or ectopic keratin filaments to a perinuclear, ring-like structure upon SPC incubation. Scale bars: 10 μm. (B) Quantification of cytokeratin organization. Cytokeratin organization was quantified in cells treated with or without 12.5 μM SPC for 1 hour, as described in the Materials and Methods. Images represent ortho-max projections of confocal image sections with three linear ROIs in the perinuclear and the cytoplasmic region, respectively. Left graph shows intensity ratio of perinuclear to cytoplasmic ROIs. Cells treated with SPC exhibit an increase in fluorescence intensity in the perinuclear area compared with untreated cells, demonstrating a marked difference in cytokeratin redistribution upon phosphorylation that can be quantified. Right graph shows height of cells (Z-volume) calculated from confocal image stacks. **P<0.01.
Fig. 3.
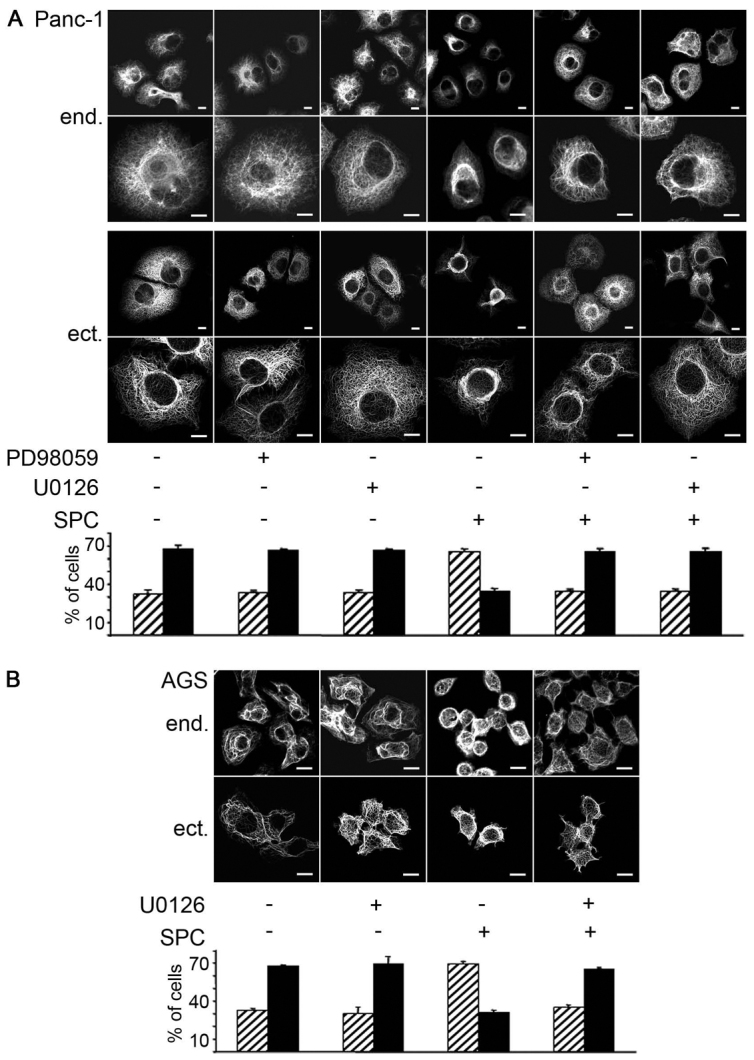
Role of p44 and p42 (MAPK1 and MAPK3) activation in SPC-induced keratin reorganization. (A,B) Cells were plated on coverslips and subsequently transfected with the respective plasmids for 48 hours. Panc-1 (A) and AGS (B) cells were incubated with either 15 μM PD98059 (only Panc-1) or 10 μM U0126 for 1 hour followed by 45 minute incubation with 15 μM SPC. Images of representative cells (two different magnifications in A) show endogenous (end.) stained with a pan-CK antibody, followed by Alexa Fluor 488 staining or transfected eCFP–K8(WT),eYFP–K18(WT) keratin (ectopic = ect.). Images were taken using a confocal microscope and keratin was detected within the 488 channel. Scale bars: 10 μm. In bar graphs in A and B, all cells on the coverslip or all transfected cells, respectively, were counted (between 50 and 200 cells per coverslip) and the ramified versus perinuclear phenotype of keratin was assessed by a person blinded for the specific condition. Data are expressed as the percentage of cells exhibiting a ramified or a perinuclear keratin phenotype and are the means ± s.e.m. of 3–10 independent experiments per condition.
ERKs mediate SPC-induced phosphorylation of K8(Ser431)
Next we wanted to identify the site(s) in keratins that become phosphorylated in response to SPC. SPC stimulated phosphorylation of K8 at Ser431 in both epithelial cancer cell lines – a site that is also phosphorylated by active ERK (Omary et al., 1998; Ku et al., 2005). K8(S431) phosphorylation in response to SPC was prevented when cells were incubated with either MEK inhibitor (Fig. 4A). To determine the effect of ERK-induced K8 phosphorylation on the organization of the keratin cytoskeleton we performed immunocytochemistry using a phosphospecific antibody against phosphorylated K8(S431) [pK8(S431)]. Using this antibody we detected intense pK8(S431) immunoreactivity exclusively upon incubation of cells with SPC, but not in unstimulated, control cells. pK8(S431) immunoreactivity was predominantly detectable in reorganized, perinuclear keratin filaments, indicating that K8 phosphorylation strictly correlates with keratin reorganization (Fig. 4C). In the presence of U0126 or PD98059, the SPC-stimulated increase in Ser431 immunoreactivity was virtually abolished (Fig. 4D). Similarly, the SPC-stimulated increase in K8(S431) immunoreactivity was completely prevented when p42 and p44 (MAPK1 and MAPK3, respectively) were depleted by specific siRNAs (Fig. 4E). Thus, SPC-induced K8(S431) phosphorylation requires ERK activity.
Fig. 4.

p44 and p42 (MAPK1 and MAPK3) mediate SPC-induced keratin phosphorylation. (A–B) Keratins were extracted from Panc-1 and AGS cells after treatment with 15 μM SPC and/or 15 μM PD98059 and 10 μM U0126. Keratin phosphorylation was analyzed by western blotting with antibodies against phosphorylated K8(S431) (Ph-K8-S431; A) or phosphorylated K18(S52) (Ph-K18-S52; B). Representative immunoblots are shown. Graphs display quantifications of luminescence. Error bars indicate the s.e.m. of at least four independent experiments. Significant differences were calculated using Student's t-test (* represents a significant difference between marked columns and untreated control; # represents a significant difference between marked columns and 60 minute SPC treatment). *,#P<0.05; **,##P<0.01; ***,###P<0.001. (C) Panc-1 and AGS cells were treated with 15 μM SPC for 45 minutes and keratin immunocytochemistry was performed using pan-CK and phosphorylated K8(S431) (Ph-K8-S431) antibodies, followed by Alexa Fluor 488 staining. Images show representative cells at two different magnifications. Scale bars: 10 μm. (D) Panc-1 and AGS cells were treated with 15 μM PD98059 or 10 μM U0126 for 1 hour followed by incubation with 15 μM SPC for 45 minutes. Immunocytochemistry was performed using a phosphospecific antibody against K8(S431) (Ph-K8-S431). Graph depicts quantification of pK8(S431)-positive cells compared with all stained cells (* represents significant difference between marked columns and untreated control; # represents significant difference between marked columns and 45 minute SPC treatment). **,##P<0.01; ***,###P<0.001. (E) p44 and p42 were depleted in Panc-1 cells using specific siRNA and confirmed by western blotting (left). p44/42 was depleted in Panc-1 cells followed by SPC treatment (20 minutes; 12.5 μM SPC) and keratin immunocytochemistry was performed using a phosphospecific antibody directed against phosphorylated K8(S431) (p-K8-S431) (right). Intense pK8(S431) immunoreactivity was detected exclusively upon incubation of cells with SPC, but not in unstimulated, control cells or cells in which p44 and p42 were depleted. pK8(S431) immunoreactivity was predominantly detectable in reorganized, perinuclear keratin filaments, indicating that K8 phosphorylation strictly correlates with keratin reorganization. Photographs show representative cells at two different magnifications. Scale bars: 10 μm. Graph depicts quantification of pK8(S431)-positive cells compared with all cells. Error bars indicate s.e.m. of three independent experiments. Significant difference was calculated using the Student's t-test. *P<0.05. M, merged image.
SPC also induces keratin phosphorylation at other sites. Incubation of cells with SPC increased the phosphorylation of K18(S52) in both pancreatic and gastric cancer cells. However, K18(S52) phosphorylation in response to SPC was not prevented by inhibition of the MEK–ERK signaling cascade with PD90859 or U0126, respectively (Fig. 4B). Collectively, these findings indicate that: (1) SPC stimulates ERK activity; (2) SPC-induced keratin reorganization requires ERK activity; (3) SPC-induced phosphorylation of K8(S431) is also dependent on ERK activity; and (4) K8(S431) phosphorylation and keratin reorganization by SPC go hand in hand. These data suggest a relationship between K8(S431) phosphorylation and keratin reorganization in epithelial tumor cells.
Role of phosphorylation at K8(Ser431) and K18(Ser52) in SPC-induced keratin reorganization in human cancer cells
To examine whether keratin phosphorylation at K8(S431) or K18(S52) was required and/or sufficient for SPC-induced keratin reorganization in Panc-1 and AGS cells, we generated eCFP-tagged mutants of K8 and eYFP-tagged mutants of K18 that mimic phosphorylation at K8(S431) and K18(S52), respectively (S→E), or exhibit a non phosphorylatable site (S→A). It has been shown previously that keratin phosphorylation affects its solubility. Indeed, there was more K8(S431E) detectable in the cytosolic/soluble fraction compared with K8(S431A) and K8 WT (supplementary material Fig. S2A).
Upon incubation of cells with SPC, endogenous as well as exogenously expressed K8 and K18 exhibited the typical pattern of ‘reorganized’ keratin, with a predominant perinuclear keratin organization in Panc-1 and AGS cells that was prevented in the presence of U0126 (Fig. 5A; supplementary material Fig. S3A). When cells were transfected with an eCFP–K8(S431A),K18(S52A) double mutant there was no detectable keratin reorganization in response to SPC (Fig. 5B; supplementary material Fig. S3B). In marked contrast, transfection of cells with the K8(S431E),K18(S52E) mutants resulted in a strictly perinuclear redistribution of these keratin mutants that was not further increased in presence of SPC. Incubation of K8(S431E),K18(S52E) transfected cells with U0126 did not prevent the perinuclear organization of the transfected keratins (Fig. 5C; supplementary material Fig. S3C). Thus, phosphorylation of K8 and/or K18 is sufficient to trigger keratin reorganization.
Fig. 5.
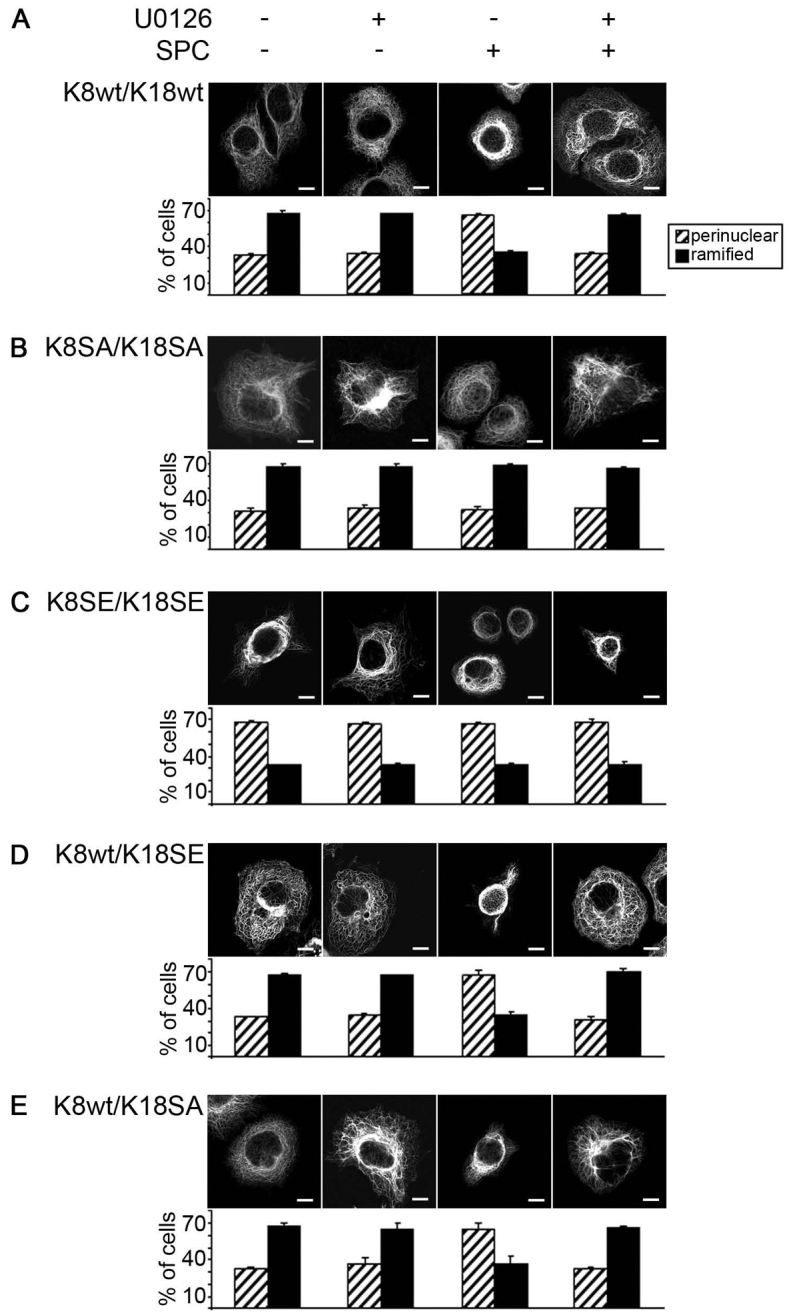
Effect of modifying Ser431 in K8 and Ser52 in K18 on keratin organization in pancreatic cancer cells. (A–E) Panc-1 cells were transfected with K8(WT),K18(WT) (A), K8(SA),K18(SA) (B), K8(SE),K18(SE) (C), K8(WT),K18(SE) (D) or K8(WT),K18(SA) (E) and treated with 15 μM SPC and/or 10 μM U0126 as indicated (eCFP-tagged K8; eYFP-tagged-K18). Images were taken using a confocal microscope and keratin was detected within the 488 channel. Graphs display quantification of cells (means ± s.e.m.) with perinuclear or ramified keratin compared with number of transfected cells.
To determine whether and which of these phosphorylation sites were required and/or even sufficient to trigger perinuclear keratin reorganization, we expressed K8 and K18 expression plasmids that contained only one keratin modification either in K8 or K18. Organization of K8(WT),K18(S52E) transfected Panc-1 or AGS cells was comparable with that of wild-type keratins. Perinuclear organization of K8(WT),K18(S52E) was only detectable in the presence, but not in the absence of SPC (Fig. 5D; supplementary material Fig. S3D). This SPC-induced perinuclear reorganization of K8(WT),K18(S52E) could be prevented by treatment of cells with U0126. Organization of the K8(WT),K18(S52A) mutant was comparable to that of wild-type K8 or K18 in the presence or absence of SPC (Fig. 3; Fig. 5E; supplementary material Fig. S3E). Thus, K18(S52) phosphorylation was neither sufficient nor required for SPC-induced perinuclear keratin reorganization in epithelial cancer cells.
Transfection of Panc-1 or AGS cells with K8(S431E),K18(WT) resulted in marked perinuclear reorganization of keratin in the absence of SPC. Incubation of cells with SPC did not further enhance perinuclear organization of K8(S431E),K18(WT). Furthermore, incubation of cells with U0126 did not prevent the perinuclear organization of K8(S431E),K18(WT) (Fig. 6A; supplementary material Fig. S4A). In turn, K8(S431A),K18(WT) did not exhibit perinuclear keratin organization either in the absence or in the presence of SPC. The selective MEK inhibitor U0126 had no effect on the subcellular organization of K8(S431A),K18(WT) (Fig. 6B; supplementary material Fig. S4B).
Fig. 6.

K8(S431) phosphorylation is sufficient to trigger perinuclear keratin organization in Panc-1 cells. (A–C) Panc-1 cells were transfected with K8(SE),K18(WT) (A), K8(SA),K18(WT) (B) or K8(SE),K18(SA) (C) and treated with 15 μM SPC and/or 10 μM U0126 as indicated (eCFP-tagged K8; eYFP-tagged K18). Keratin was detected within the 488 channel. Images show representative cells. Graphs display quantification of cells with perinuclear or ramified keratin compared with the total number of transfected cells (means ± s.e.m.). (D) Panc-1 cells were transfected with either eCFP–K8(WT) or eYFP–K8(SE) and stained with pan-CK antibody, followed by labelling with Alexa-Fluor-647 antibody (Pan-CK). Images were taken using a confocal microscope and keratin was detected within two channels. (E) Quantification of cytokeratin organization. Cytokeratin organization was quantified in cells expressing wild-type K8–CFP or K8(SE)–CFP mutant. Images represent ortho-max projections of confocal image sections with three linear ROIs in the perinuclear and the cytoplasmic region, respectively. Left graph shows intensity ratio of perinuclear to cytoplasmic ROIs. Cells overexpressing the phosphomimetic K8 mutant (CFP–K8 se) exhibit an increase in fluorescence intensity in the perinuclear area compared with cells overexpressing wild-type K8, demonstrating a marked difference in cytokeratin redistribution upon phosphorylation that can be quantified. Right graph shows height of cells (Z-volume) calculated from confocal image stacks. **P<0.01; ***P<0.001.
Furthermore, there was no difference in the perinuclear organization of K8(S431E),K18(WT), K8(S431E),K18(S52E) or K8(S431E),K18(S52A), indicating that phosphorylation of K18 at Ser52 is indeed not required for perinuclear keratin organization in human epithelial tumor cells (Fig. 6C; supplementary material Fig. S4C). Interestingly, transfection of K8(S431E) alone was also sufficient to induce perinuclear organization of endogenous K8 and K18 (Fig. 6D). Thus phosphorylation of K8 at Ser431 is required and sufficient for perinuclear keratin reorganization in human pancreatic and gastric cancer cells.
Keratins regulate the motility of epithelial tumor cells
Previously, we have shown that SPC reorganizes the keratin cytoskeleton and facilitates migration of tumor cells through size-limited pores, suggesting a link between keratin organization and tumor cell migration (Beil et al., 2003). To further determine the precise role of keratin in tumor cell migration, we targeted K8 using small interfering (si)RNA oligonucleotides. Depletion of K8 by siRNA was confirmed by qRT-PCR, immunocytochemistry and western blot analysis. K8 mRNA transcripts were reduced by 80% 24 hours after transfection (Fig. 7A; supplementary material Fig. S5). A maximum effect on K8 protein expression was observed after 72 hours of incubation with the specific siRNA (Fig. 7B). Depletion of K8 or K18 resulted in a significantly enhanced random migration of tumor cells (siK8=0.31 μm/minute vs siControl = 0.18 μm/minute; Fig. 7C,D). Thus, under basal conditions, the established keratin cytoskeleton obviously restricts tumor cell motility.
Fig. 7.

Effect of keratin depletion on basal and SPC-induced random migration of pancreatic cancer cells. (A) K8 and/or K18 were depleted in Panc-1 cells using specific siRNAs (siK8, siK18). Relative K8 mRNA (upper panel) and K18 mRNA levels (lower panel) were determined in Panc-1 cells by qRT-PCR using the ICyclerIQ system from Bio-Rad. Graphs depict percentage mRNA expression as compared with control. Figures shows representative data obtained in triplets of at least three independent experiments. (B) Top panel shows Panc-1 cells transfected with specific K8 siRNA (siK8) and/or K18 siRNA (siK18). After 72 hours, keratins were extracted and expression analyzed by western blotting using a pan-CK antibody. Bottom panel is densitometric evaluation of keratin protein levels. Data represent the means ± s.e.m. of three independent experiments. (C) Panc-1 cells were transfected with scrambled siRNA (siCon) or siK8. After 48 hours, cells were incubated with 10 μM SPC (+) or solvent (−) and subjected to a random migration that was analyzed by time-lapse video microscopy. Cells were tracked and the velocity was calculated using the ImageJ program. Data represent the fold increase in migration above control and are the means ± s.e.m. of three independent experiments. (D) Panc-1 cells were transfected with scrambled siRNA (siCon) or siK18. 24 hours after knockdown, cells were incubated with solvent or 10 μM SPC and subjected to random migration. Migration was analyzed as described above. Data represent the fold increase in migration above control and are the means ± s.e.m. of three independent experiments. (E) Panc-1 cells were incubated with 10 μM U0126, 10 μM SPC and 5 ng/ml TGF-β as indicated. Random migration was determined by time-lapse video microscopy as described above. Data represent the fold increase in migration above control and are the means ± s.e.m. of four independent experiments. (F) Panc-1 cells were incubated with 10 μM U0126, 10 μM SPC and 5 ng/ml TGF-β as indicated. TGF-β was used as a positive control (Rahimi and Leof, 2007). Migration through size-limited pores was determined using a Boyden chamber assay as described previously (Beil et al., 2003). Data represent the fold increase in migration above control and are the means ± s.e.m. of three independent experiments. (G) Panc-1 cells were transfected with K8(WT),K18(WT), K8(SE),K18(WT) or K8(SA),K18(WT) and subsequently incubated with 10 μM SPC (+). Random migration was determined as above. Data represent the fold increase in migration above control and are the means ± s.e.m. of four independent experiments. In all experiments described in 7C-G significant differences were tested using the Student's t-test. *P<0.05; **P<0.01; ***P<0.001.
SPC stimulates random motility of Panc-1 cells (Fig. 7C; supplementary material Fig. S6A). This increase in motility corresponds to the changes in keratin organization in response to SPC (supplementary material Fig. S6B) and it was prevented when cells were incubated with the MEK inhibitor U0126 (Fig. 7E). This was not only the case for random motility, but also for tumor cell migration through size-limited pores in Boyden chamber assays (Fig. 7F). Interestingly, SPC stimulated random tumor cell migration to a similar degree as keratin depletion and was not able to further enhance the random motility of K8-depleted cells (Fig. 7C). Thus, SPC-induced cell migration requires ERK activity and SPC affects tumor cell migration by a mechanism that requires MEK-mediated phosphorylation.
Phosphorylation of K8 at Ser431 leads to enhanced tumor cell migration
Because perinuclear keratin reorganization correlates with increased tumor cell motility, we next examined the effect of K8(S431) phosphorylation on random migration of tumor cells. Pancreatic cancer cells expressing wild-type K8 and K18 move with a speed of about 0.19 μm/minute, which corresponds to the speed of untransfected control cells (0.21 μm/minute). K8(S431E),K18(WT)-expressing cells moved twice as fast as wild-type cells (0.39 μm/minute) and as fast as cells expressing wild-type K8 and K18 in the presence of SPC (0.36 μm/minute; Fig. 7G). In addition, the migration speed of K8(S431E),K18(WT)-expressing cells was comparable to that of K8-depleted pancreatic cancer cells using specific siRNA (0.31 μm/minute). Pancreatic cancer cells expressing the K8(S431A),K18(WT) mutant exhibited the same migratory behavior as cells expressing wild-type K8 and K18 (0.21 μm/min). SPC failed to enhance the migration velocity of cells expressing K8(S431A),K18(WT) (0.24 μm/minute; Fig. 7G). This indicates that phosphorylation of K8 at Ser431 plays a crucial role for both SPC-induced keratin organization and SPC-induced release of the keratin-mediated inhibition of human tumor cell migration. It has been reported that K8 and K18 modulate the distribution of focal adhesions in simple epithelial cells (Bordeleau et al., 2010). Our data (supplementary material Fig. S7) show that upon expression of K8(S431E), there was a significant increase in the number but not the size of focal adhesions. The focal adhesions aligned at the margin of the cells. Thus, keratin phosphorylation affects focal adhesion formation in pancreatic cancer cells and could thereby contribute to the pro-migratory effect observed. These data provide a direct link between keratin organization, ERK-mediated keratin phosphorylation and tumor cell migration. They further demonstrate that K8(S431) phosphorylation resembles the effects of knockdown of keratins by releasing the restrictive function of keratins on tumor cell migration.
Discussion
Sphingosylphosphorylcholine (SPC) is a naturally occurring, bioactive lipid that acts as an intracellular and extracellular signaling molecule in numerous biological processes including proliferation (Seufferlein and Rozengurt, 1995), cell migration (Boguslawski et al., 2000), wound healing (Wakita et al., 1998) and differentiation (Kleger et al., 2007). Previously, we established that SPC is one of the few physiological compounds that is able to reorganize the keratin cytoskeleton. SPC induced reorganization of keratin from a widespread, ramified network to a strictly perinuclear, ring-like structure in human epithelial tumor cells (Beil et al., 2003). This reorganization led to an increase in the elasticity of the tumor cells, facilitated migration of cells through size-limited pores and was accompanied by keratin phosphorylation at K8(S431) and K18(S52).
Members of the MAPK family have previously been shown to mediate phosphorylation of keratins (Omary et al., 2006; Omary et al., 2009). Post-translational modifications are major regulators of keratin function (Omary et al., 1998; Ku et al., 1999; Toivola et al., 2004; Magin et al., 2007). In particular, serine phosphorylation of keratins plays a major role in various cellular events including apoptosis and mechanical or non-mechanical stress (Liao et al., 1995b; Liao et al., 1997; Ridge et al., 2005; Jeon et al., 2006; Felder et al., 2008). Serine hyperphosphorylation of keratins, for example by treatment of cells with okadaic acid, leads to disruption of the filament network (Ku and Omary, 1994; Ku et al., 1999; Strnad et al., 2002). Additionally, soluble keratins often exhibit increased serine phosphorylation (Stossel, 1993; Omary et al., 2006). A closer examination of the physiological role of keratin phosphorylation has so far been hampered by the fact that there are only few compounds that physiologically stimulate keratin phosphorylation. Our data point to a central role of K8(S431) in SPC-induced keratin reorganization.
Our data suggested a close relationship between K8(S431) phosphorylation and keratin reorganization in epithelial tumor cells. Phosphorylation of K18 at Ser52 has been described in response to cellular stressors such as heat and viral infection, or during mitosis (Ku and Omary, 1994; Liao et al., 1995a; Ku et al., 1999). Furthermore, K18(S52) phosphorylation seems to protect hepatocytes from toxic injury (Omary et al., 2006) and could therefore act as a protective signal in response to injury or stress (Omary et al., 2009; Toivola et al., 2010). SPC-induced phosphorylation of K18 at Ser52 was independent of MEK–ERK activity in Panc-1 and AGS cells. SPC can induce multiple signaling pathways in different cell lines. However, SPC failed to induce significant activation of other pivotal signaling pathways such as the PI3K–AKT, the PKC–PKD and the PKM2 pathway in our model cancer cell lines. These data underline the importance of ERK-dependent keratin phosphorylation for keratin reorganization in these cell lines. In line with the data described above, experiments using phosphomimetic mutants of K8(S431) and K18(S52) showed that phosphorylation of K18 at Ser52 was neither required nor sufficient for the SPC-induced keratin reorganization. In striking contrast, phosphorylation of K8 at Ser431 was required and sufficient to trigger perinuclear keratin reorganization.
Our previous data suggested that SPC-induced keratin reorganization and cell migration are linked (Beil et al., 2003). In addition, enhanced migration often corresponds to increased activity of the ERK signaling cascade (Huang et al., 2004). Thus, we investigated the role of SPC-induced, ERK-dependent K8 phosphorylation and reorganization in cell migration, both in the absence and presence of SPC. Inhibition of MEK blocked K8(S431) phosphorylation, keratin reorganization and tumor cell migration. Interestingly, depletion of K8 in epithelial tumor cells also resulted in a marked increase in tumor cell motility compared with that in cells transfected with a scrambled siRNA construct. These data are in line with observations in wound healing, where knockdown of K8 with siRNA resulted in accelerated wound closure in vimentin-positive HeLa and Panc-1 cells (Long et al., 2006). Notably, compared with control cells, scratch-wound edges were irregular after K8 depletion and frequently contained cells that were migrating individually in both cell lines (Long et al., 2006). This behavior of K8-knockdown cells is comparable with our single-cell-based experiments, and indicates that the basal organization of the keratin cytoskeleton in simple epithelial tumor cells serves to restrict cellular migration. Interestingly, SPC failed to further increase migration velocity of tumor cells depleted of K8. This shows that keratin organization is crucial for the effect of SPC on tumor cell migration. In addition, our data clearly show that ERK-induced phosphorylation of K8 at Ser431 is sufficient to stimulate tumor cell migration to a similar degree as SPC treatment. Accordingly, phosphorylation at K8(S431) was required for SPC-induced tumor cell migration. The migratory behavior of tumor cells depleted of K8 was similar to that of cells expressing the phosphomimetic K8(S431E) mutant. This shows that ERK-mediated K8(S431) phosphorylation functionally mimics K8 depletion of tumor cells and abolishes the otherwise restrictive function of the keratin cytoskeleton on tumor cell migration. The fact that this effect can be so clearly demonstrated using SPC, but has so far not been described for other activators of the ERK cascade such as EGF–EGFR might lie in the fact that SPC, by acting through a G-protein-coupled receptor (GPCR), induces only few signaling pathways in human epithelial tumor cells, in particular an activation of the ERK cascade (our own unpublished observations). Upon induction of multiple pathways there are likely to be other modifications of keratins that attenuate or modulate the effect of ERK activation with respect to keratin reorganization.
Our data have implications for tumor biology. The ability of tumor cells to migrate is pivotal for tumor progression and metastasis (Chambers et al., 2002). Our data show that ERK-dependent phosphorylation of K8(S431) leads to increased tumor cell migration as a result of reorganization of the keratin cytoskeleton and this is likely to contribute to the metastatic properties of these cells. Thus, we provide a mechanism to explain how ERKs can regulate tumor cell migration and identify keratin phosphorylation as an interesting novel therapeutic target to prevent invasion, and potentially, metastasis.
Materials and Methods
Materials
The MEK inhibitor U0126 was obtained from Promega (Fitchburg, WI). PD98059 and SPC were from Calbiochem (Merck Chemicals, Nottingham, UK), TGF-β from BD Biosciences (San Jose, CA). Antibodies directed against p44 and p42 and phosphorylated p44 and p42 are from Cell Signaling (Danvers, MA), antibodies detecting Pan-Cytokeratin (Pan-CK, clone KL1) are from Immunotech (Praha, Czech Republic).
The phosphospecific antibodies detecting K18(S52) (3055) and K8(S431) (5B3) have been described previously (Liao et al., 1995a; Ku et al., 1997). Antibodies directed against Akt, phospho-Akt (Ser 473), phospho-PKD (Ser744/748) and phospho-PKM2 (Tyr105) were obtained from Cell Signaling. The mouse monoclonal antibody against PKM2 was purchased from Abcam (Cambridge, UK). The antibodies detecting PKD2 were purchased from Calbiochem (Merck Chemicals). Phorbol-12-myristate-13-acetate (PMA) was obtained from Calbiochem (Merck Chemicals), IGF1 was from Peprotech (Hamburg, Germany).
Cell culture and reagents
Panc-1 human pancreatic cancer cells were purchased from American Type Culture Collection (Manassas, VA). AGS cells were a gift from Michael Hoecker (Charite, Berlin, Germany). This is a human gastric cancer cell line that also exclusively expresses keratins, but no vimentin. Cells were maintained in DMEM (Gibco, Invitrogen, Carlsbad, CA) supplemented with 10% (v/v) fetal bovine serum (PAA, Pasching, Austria) in a humidified atmosphere and 5% CO2, 95% air at 37°C and passaged every 2–3 days. Cells were incubated in serum-free DMEM (Gibco) for 18 hours according to the experiment.
Cell transfection
For immunofluorescence microscopy and live-time imaging, cells were either transfected with Fugene (Roche, Basel, Switzerland) or Metafectene (Biontex, Martinsried, Germany). For migration assays, transfection was performed with Nucleofector (Amaxa, Cologne, Germany) using Kit R and program X-005, siRNA was transfected with RNAiFect (Qiagen).
Immunofluorescence microscopy
Pan-CK antibody and the phosphospecific antibody detecting K8(S431) (5B3) were used for immunofluorescence microscopy. After the indicated stimulation, cells were fixed with 4% formaldehyde for 10 minutes. Antibodies were added overnight in PBS supplemented with 0.5% Triton X-100 and 0.2% Gelatin (Sigma, St Louis, MO) at 4°C. Cells were then incubated with Alexa Fluor 488, 568 or 647 coupled to secondary antibodies (Invitrogen). Finally, slides were embedded in GelTol Aqueous Mounting Medium (Immunotech). Imaging was performed with confocal laser-scanning microscope LSM510 Meta (Carl Zeiss, Jena, Germany) equipped with a 63× 1.4 NA oil objective using the indicated filters or a Keyence BZ-8000 fluorescence microscope. Images show representative cells from at least three independent experiments.
Cell migration through size-limited pores
Panc-1 cell migration was examined using a modified 48-well Boyden chamber (Nucleopore, Neuro Probe, Gaithersburg, MD) and collagen-coated polycarbonate membranes with a pore diameter of 12 μm (Nucleopore). Panc-1 cells (2×105 cells ml−1) in DMEM were allowed to migrate towards a gradient of the indicated agents for a total of 4 hours in a humidified incubator (37°C; 5% CO2). Adherent cells on the filter membrane were fixed in 99% ethanol for 10 minutes and stained using Giemsa dye. For a quantitative assessment of migrated cells, from three different wells in each case, five high-power fields (15 in total) were counted. The data shown represent the percentage of migrated cells, compared with the unstimulated control.
Migration assays with time-lapse microscopy
Panc-1 cells were seeded on fibronectin (Roche Diagnostics, Penzberg, Germany) covered glass-slides 3 hours before start of migration assay. Glass slides were packed together with serum-free DMEM containing 1% penicillin-streptomycin into a sample sandwich and sealed with wax. The sample sandwich was kept at 37°C and time-lapse was run for 16 hours. Cells expressing K8(WT),K18(WT), K8(SE),K18(WT) and K8(SA),K18(WT) were transfected 24 hours before the start of migration assays. Time-lapse photos were analyzed using ImageJ (NIH).
Western blot analysis and extraction of keratins
For keratin extraction, serum-starved cultures of Panc-1 and AGS cells were treated with factors as indicated and lyzed at 4°C in 20 mM Tris-HCl at pH 7.4, 0.6 M potassium chloride, 1% Triton X-100 and 1 mM PMSF (Triton high-salt buffer). Lysates were incubated for 20 minutes on ice and cleared by centrifugation at 10,000 g for 20 minutes at 4°C. The resulting pellet was subsequently resuspended in the same buffer, incubated for a further 20 minutes on ice and again subjected to centrifugation at 10,000 g for 20 minutes at 4°C. The pellet of insoluble proteins was resuspended in 5 volumes of 8 M urea and the same volume of 5× SDS-PAGE sample buffer was added to the solution. Samples were then separated by SDS-PAGE.
For western blot analysis, serum starved cultures of Panc-1 and AGS cells were treated as indicated and lyzed at 4°C in NP-40 lysis buffer (150 mM NaCl, 20 mM Tris-HCl, 10% glycerol, 1% NP-40, 100 mM Na4O7P2, 100 mM NaVO4, 1M NaF, 10 μg/ml aprotinin, 100 μg/ml leupeptin, 0.7 μg/ml pepstatin). Lysates were incubated for 10 minutes at 4°C and subjected to centrifugation at 14,000 g for 10 minutes at 4°C. Supernatants of the samples were resuspended in 5× SDS-PAGE sample buffer and separated by SDS-PAGE.
Quantification of keratin morphology
Panc-1 and AGS cells were transfected as indicated and keratin morphology was quantified by determining the percentage of cells with a predominant perinuclear or ramified keratin organization using fluorescence microscopy. Keratin organization was classified as predominantly perinuclear when more than 70% of keratin, as assessed by visual judgment, was organized around the nucleus and consequently the cytoplasm contained fewer keratin filaments. The formation of a strict ring structure was frequently observed, but not required for the classification as perinuclear. Keratin organization was classified as predominantly cytoplasmic when more than 70% of keratin was organized in the cytoplasm and consequently the cytoplasm contained more keratin filaments than the perinuclear region. Quantification was performed in a blinded fashion in at least three independent experiments with 50–200 cells per experiment.
Quantification of fluorescence intensity distribution
For quantification of keratin distribution visualized by indirect immunofluorescence or CFP-tagged keratins as described in the figure legends, ortho-max projections of confocal image sections from top to bottom of each cell were analyzed. Cells were imaged using a SP5 confocal microscope (Leica) with similar settings. Three linear ROIs of equal length were placed in perinuclear and cytoplasmic regions of each cell (LAS AF Lite Software, Leica). The normalized intensity profile for all ROI was integrated by calculating area under the curve and the mean intensity ratio of perinuclear to cytoplasmic ROIs for each cell was used an indicator for cytokeratin distribution under different conditions. Height of cells (Z-Volume) was calculated from confocal image stacks. Calculations and statistical analysis was performed using GraphPad Prism version 5.00.
Reverse transcriptase and PCR
mRNA was prepared from either Panc-1 or AGS cells and semi-quantitative RT-PCR analysis was conducted with specific primers for SPC receptors as described previously (Kleger et al., 2007).
Real-time PCR
K8 and K18 mRNA levels were measured using the LightCycler System (Roche Diagnostics, Mannheim, Germany) or the ICyclerIQ system from Bio-Rad. Primers used in real-time PCR were as follows: human keratin 18: QuantiTect Primer Assay Hs_KRT18_2_SG. Human keratin 8: 5′-GCCGTGGTTGTGAAGAA-3′ and 5′-CTGTTCCCAGTGCTACCCT-3′. Human HMBS: 5′-CCCTGGAGAAGAATGAAGTGGA-3′ and 5′-TGGGTGAAAGACAACAGCATC-3′.
siRNA
For K8 knockdown, the siRNA probe: GGGUGACCCAGAAGUCCUA labeled with 3′-fluorescein or 3′-Alexa-Fluor-488 was used. As a control, an AllStar siRNA labeled with Alexa Fluor 546 (Qiagen, Hilden, Germany) was used. To deplete K18, we used a mixture of two different siRNA constructs: Stealth RNAi KRT18-HSS142770 (cat. no. 5194538, Invitrogen) and Hs-KRT18-3 CK (5′-ccgccggatagtggatggcaa-3′, 5′-gccggauaguggauggcaatt-3′) (Qiagen). AllStar siRNA labeled with Alexa Fluor 546 (Qiagen) was used as a control. For knockdown of p44 and p42 MAPKs, ERK1/2 siRNA from Cell Signaling was used. Panc1 cells were transfected using Hyperfect (Qiagen) according to manufacturer's instructions.
Site-directed mutagenesis
To obtain amino acid exchange of K8(Ser431) and K18(Ser52), site-directed mutagenesis with pK8–eCFP and pK18–eYFP (Rudolf Leube, Universitätsklinikum Aachen, Germany) (Wöll et al., 2005) as matrices were performed using QuikChange XL-sdm-Kit (Stratagene, La Jolla, CA) according to the instruction manual. To replace K8(S431) and K8(S52) with Alanine (A) or Glutamic acid (E), primers as follows were used: K8(S431A): TATGGGGGCCTCCAGCCCCCGGCCTCA; K8(S431E): CTATGGGGGCCTCACAGAACCCGGGCTCAGCTACAG; K18(S52A): ATCTCCGTGTCCCGGTCGACCGCCTTCAGGGGC; K-18(S52E): GTGTCCCGCTCCACCGAAAGGGGCGGCATG.
Random migration
Serum-starved Panc-1 cells were subjected to a random migration on fibronectin and imaged by the time-lapse video microscopy. Glass bottom culture dishes (MakTek Corporation) were coated with 50 μg/ml of fibronectin (Roche). Cells were allowed to spread for 3 hours on the fibronectin-coated dishes in DMEM with 1% FCS. Imaging was performed using a BZ-8000 Keyence microscope. During imaging cells were kept at 37°C in an atmosphere containing 5% CO2. A motion picture (AVI format) was created from time-lapse images using the BZ-Analyzer software (Keyence Corporation). Cell movement was analyzed using tracking routines implemented in ImageJ software. Three independent experiments were done for each condition.
Isolation and analysis of keratin fractions
Keratins were isolated from three cellular fractions. The cytosolic fraction was obtained after disrupting cells by centrifugation at 100,000 g for 90 minutes in Buffer A [PBS with 10 mM EDTA and protease and phosphatase inhibitor cocktail (Roche)]. The pellet was then solubilized using 1% NP-40 in buffer A (30 minutes at 4°C) followed by centrifugation (16,000 g, 15 minutes, 4°C) and collecting the NP-40 fraction. The remaining cytoskeletal fraction was solubilized in 50 mM Tris-HCl, pH 7.4, 2 mM EDTA with 9.5 M urea.
Acknowledgements
We thank Rudolf Leube (Aachen) for providing the keratin vectors pK8–eCFP and pK18–eYFP, and Claudia Ruhland and Ulrike Mayr-Beyrle for expert technical assistance.
Footnotes
Funding
This work was supported by the Deutsche Krebshilfe [grant number 107344 to T.S.]; and National Institutes of Health [grant number DK47918 to M.B.O.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.080127/-/DC1
References
- An S., Tsai C., Goetzl E. J. (1995). Cloning, sequencing and tissue distribution of two related G protein-coupled receptor candidates expressed prominently in human lung tissue. FEBS Lett. 375, 121-124 [DOI] [PubMed] [Google Scholar]
- Ballestrem C., Wehrle-Haller B., Hinz B., Imhof B. A. (2000). Actin-dependent lamellipodia formation and microtubule-dependent tail retraction control-directed cell migration. Mol. Biol. Cell 11, 2999-3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beil M., Micoulet A., von Wichert G., Paschke S., Walther P., Omary M. B., Van Veldhoven P. P., Gern U., Wolff-Hieber E., Eggermann J., et al. (2003). Sphingosylphosphorylcholine regulates keratin network architecture and visco-elastic properties of human cancer cells. Nat. Cell Biol. 5, 803-811 [DOI] [PubMed] [Google Scholar]
- Boguslawski G., Lyons D., Harvey K. A., Kovala A. T., English D. (2000). Sphingosylphosphorylcholine induces endothelial cell migration and morphogenesis. Biochem. Biophys. Res. Commun. 272, 603-609 [DOI] [PubMed] [Google Scholar]
- Bordeleau F., Galarneau L., Gilbert S., Loranger A., Marceau N. (2010). Keratin 8/18 modulation of protein kinase C-mediated integrin-dependent adhesion and migration of liver epithelial cells. Mol. Biol. Cell 21, 1698-1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove P. F., Hristova M., Wesley U. V., Olson N., Lounsbury K. M., van der Vliet A. (2008). Inflammatory levels of nitric oxide inhibit airway epithelial cell migration by inhibition of the kinase ERK1/2 and activation of hypoxia-inducible factor-1 alpha. J. Biol. Chem. 283, 17919-17928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulín C., Salvesen G. S., Oshima R. G. (1997). Caspase cleavage of keratin 18 and reorganization of intermediate filaments during epithelial cell apoptosis. J. Cell Biol. 138, 1379-1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers A. F., Groom A. C., MacDonald I. C. (2002). Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2, 563-572 [DOI] [PubMed] [Google Scholar]
- Chou C. F., Riopel C. L., Omary M. B. (1994). Identification of a keratin associated protein that localizes to a membrane compartment. Biochem. J. 298, 457-463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe P. A., Omary M. B. (2002). ‘Hard’ and ‘soft’ principles defining the structure, function and regulation of keratin intermediate filaments. Curr. Opin. Cell Biol. 14, 110-122 [DOI] [PubMed] [Google Scholar]
- Felder E., Siebenbrunner M., Busch T., Fois G., Miklavc P., Walther P., Dietl P. (2008). Mechanical strain of alveolar type II cells in culture: changes in the transcellular cytokeratin network and adaptations. Am. J. Physiol. Lung Cell. Mol. Physiol. 295, L849-L857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E., Weber K. (1994). Intermediate filaments: structure, dynamics, function, and disease. Annu. Rev. Biochem. 63, 345-382 [DOI] [PubMed] [Google Scholar]
- Fuchs E., Cleveland D. W. (1998). A structural scaffolding of intermediate filaments in health and disease. Science 279, 514-519 [DOI] [PubMed] [Google Scholar]
- Hatzfeld M., Franke W. W. (1985). Pair formation and promiscuity of cytokeratins: formation in vitro of heterotypic complexes and intermediate-sized filaments by homologous and heterologous recombinations of purified polypeptides. J. Cell Biol. 101, 1826-1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Jacobson K., Schaller M. D. (2004). MAP kinases and cell migration. J. Cell Sci. 117, 4619-4628 [DOI] [PubMed] [Google Scholar]
- Jeon E. S., Song H. Y., Kim M. R., Moon H. J., Bae Y. C., Jung J. S., Kim J. H. (2006). Sphingosylphosphorylcholine induces proliferation of human adipose tissue-derived mesenchymal stem cells via activation of JNK. J. Lipid Res. 47, 653-664 [DOI] [PubMed] [Google Scholar]
- Kleger A., Busch T., Liebau S., Prelle K., Paschke S., Beil M., Rolletschek A., Wobus A., Wolf E., Adler G., et al. (2007). The bioactive lipid sphingosylphosphorylcholine induces differentiation of mouse embryonic stem cells and human promyelocytic leukaemia cells. Cell. Signal. 19, 367-377 [DOI] [PubMed] [Google Scholar]
- Ku N. O., Omary M. B. (1994). Identification of the major physiologic phosphorylation site of human keratin 18: potential kinases and a role in filament reorganization. J. Cell Biol. 127, 161-171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N. O., Omary M. B. (1997). Phosphorylation of human keratin 8 in vivo at conserved head domain serine 23 and at epidermal growth factor-stimulated tail domain serine 431. J. Biol. Chem. 272, 7556-7564 [DOI] [PubMed] [Google Scholar]
- Ku N. O., Liao J., Omary M. B. (1997). Apoptosis generates stable fragments of human type I keratins. J. Biol. Chem. 272, 33197-33203 [DOI] [PubMed] [Google Scholar]
- Ku N. O., Zhou X., Toivola D. M., Omary M. B. (1999). The cytoskeleton of digestive epithelia in health and disease. Am. J. Physiol. 277, G1108-G1137 [DOI] [PubMed] [Google Scholar]
- Ku N. O., Lim J. K., Krams S. M., Esquivel C. O., Keeffe E. B., Wright T. L., Parry D. A., Omary M. B. (2005). Keratins as susceptibility genes for end-stage liver disease. Gastroenterology 129, 885-893 [DOI] [PubMed] [Google Scholar]
- Liao J., Lowthert L. A., Omary M. B. (1995a). Heat stress or rotavirus infection of human epithelial cells generates a distinct hyperphosphorylated form of keratin 8. Exp. Cell Res. 219, 348-357 [DOI] [PubMed] [Google Scholar]
- Liao J., Lowthert L. A., Ku N. O., Fernandez R., Omary M. B. (1995b). Dynamics of human keratin 18 phosphorylation: polarized distribution of phosphory-lated keratins in simple epithelial tissues. J. Cell Biol. 131, 1291-1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J., Ku N. O., Omary M. B. (1997). Stress, apoptosis, and mitosis induce phosphorylation of human keratin 8 at Ser-73 in tissues and cultured cells. J. Biol. Chem. 272, 17565-17573 [DOI] [PubMed] [Google Scholar]
- Long H. A., Boczonadi V., McInroy L., Goldberg M., Määttä A. (2006). Periplakin-dependent re-organisation of keratin cytoskeleton and loss of collective migration in keratin-8-downregulated epithelial sheets. J. Cell Sci. 119, 5147-5159 [DOI] [PubMed] [Google Scholar]
- Magin T. M., Vijayaraj P., Leube R. E. (2007). Structural and regulatory functions of keratins. Exp. Cell Res. 313, 2021-2032 [DOI] [PubMed] [Google Scholar]
- Maniotis A. J., Chen C. S., Ingber D. E. (1997). Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc. Natl. Acad. Sci. USA 94, 849-854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer zu Heringdorf D., Himmel H. M., Jakobs K. H. (2002). Sphingosylphosphorylcholine-biological functions and mechanisms of action. Biochim. Biophys. Acta 1582, 178-189 [DOI] [PubMed] [Google Scholar]
- Omary M. B., Ku N. O., Liao J., Price D. (1998). Keratin modifications and solubility properties in epithelial cells and in vitro. Subcell. Biochem. 31, 105-140 [PubMed] [Google Scholar]
- Omary M. B., Ku N. O., Tao G. Z., Toivola D. M., Liao J. (2006). “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem. Sci. 31, 383-394 [DOI] [PubMed] [Google Scholar]
- Omary M. B., Ku N. O., Strnad P., Hanada S. (2009). Toward unraveling the complexity of simple epithelial keratins in human disease. J. Clin. Invest. 119, 1794-1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima R. G., Baribault H., Caulín C. (1996). Oncogenic regulation and function of keratins 8 and 18. Cancer Metastasis Rev. 15, 445-471 [DOI] [PubMed] [Google Scholar]
- Rahimi R. A., Leof E. B. (2007). TGF-beta signaling: a tale of two responses. J. Cell. Biochem. 102, 593-608 [DOI] [PubMed] [Google Scholar]
- Rajalingam K., Wunder C., Brinkmann V., Churin Y., Hekman M., Sievers C., Rapp U. R., Rudel T. (2005). Prohibitin is required for Ras-induced Raf-MEK-ERK activation and epithelial cell migration. Nat. Cell Biol. 7, 837-843 [DOI] [PubMed] [Google Scholar]
- Ridge K. M., Linz L., Flitney F. W., Kuczmarski E. R., Chou Y. H., Omary M. B., Sznajder J. I., Goldman R. D. (2005). Keratin 8 phosphorylation by protein kinase C delta regulates shear stress-mediated disassembly of keratin intermediate filaments in alveolar epithelial cells. J. Biol. Chem. 280, 30400-30405 [DOI] [PubMed] [Google Scholar]
- Schweizer J., Bowden P. E., Coulombe P. A., Langbein L., Lane E. B., Magin T. M., Maltais L., Omary M. B., Parry D. A., Rogers M. A., et al. (2006). New consensus nomenclature for mammalian keratins. J. Cell Biol. 174, 169-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seufferlein T., Rozengurt E. (1994). Sphingosine induces p125FAK and paxillin tyrosine phosphorylation, actin stress fiber formation, and focal contact assembly in Swiss 3T3 cells. J. Biol. Chem. 269, 27610-27617 [PubMed] [Google Scholar]
- Seufferlein T., Rozengurt E. (1995). Sphingosylphosphorylcholine activation of mitogen-activated protein kinase in Swiss 3T3 cells requires protein kinase C and a pertussis toxin-sensitive G protein. J. Biol. Chem. 270, 24334-24342 [DOI] [PubMed] [Google Scholar]
- Stossel T. P. (1993). On the crawling of animal cells. Science 260, 1086-1094 [DOI] [PubMed] [Google Scholar]
- Strnad P., Windoffer R., Leube R. E. (2002). Induction of rapid and reversible cytokeratin filament network remodeling by inhibition of tyrosine phosphatases. J. Cell Sci. 115, 4133-4148 [DOI] [PubMed] [Google Scholar]
- Stumptner C., Omary M. B., Fickert P., Denk H., Zatloukal K. (2000). Hepatocyte cytokeratins are hyperphosphorylated at multiple sites in human alcoholic hepatitis and in a mallory body mouse model. Am. J. Pathol. 156, 77-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao G. Z., Toivola D. M., Zhong B., Michie S. A., Resurreccion E. Z., Tamai Y., Taketo M. M., Omary M. B. (2003). Keratin-8 null mice have different gallbladder and liver susceptibility to lithogenic diet-induced injury. J. Cell Sci. 116, 4629-4638 [DOI] [PubMed] [Google Scholar]
- Tao G. Z., Toivola D. M., Zhou Q., Strnad P., Xu B., Michie S. A., Omary M. B. (2006). Protein phosphatase-2A associates with and dephosphorylates keratin 8 after hyposmotic stress in a site- and cell-specific manner. J. Cell Sci. 119, 1425-1432 [DOI] [PubMed] [Google Scholar]
- Toivola D. M., Omary M. B., Ku N. O., Peltola O., Baribault H., Eriksson J. E. (1998). Protein phosphatase inhibition in normal and keratin 8/18 assembly-incompetent mouse strains supports a functional role of keratin intermediate filaments in preserving hepatocyte integrity. Hepatology 28, 116-128 [DOI] [PubMed] [Google Scholar]
- Toivola D. M., Ku N. O., Resurreccion E. Z., Nelson D. R., Wright T. L., Omary M. B. (2004). Keratin 8 and 18 hyperphosphorylation is a marker of progression of human liver disease. Hepatology 40, 459-466 [DOI] [PubMed] [Google Scholar]
- Toivola D. M., Strnad P., Habtezion A., Omary M. B. (2010). Intermediate filaments take the heat as stress proteins. Trends Cell Biol. 20, 79-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakita H., Matsushita K., Nishimura K., Tokura Y., Furukawa F., Takigawa M. (1998). Sphingosylphosphorylcholine stimulates proliferation and upregulates cell surface-associated plasminogen activator activity in cultured human keratinocytes. J. Invest. Dermatol. 110, 253-258 [DOI] [PubMed] [Google Scholar]
- Wöll S., Windoffer R., Leube R. E. (2005). Dissection of keratin dynamics: different contributions of the actin and microtubule systems. Eur. J. Cell. Biol. 84, 311-328 [DOI] [PubMed] [Google Scholar]
- Yamada S., Wirtz D., Coulombe P. A. (2002). Pairwise assembly determines the intrinsic potential for self-organization and mechanical properties of keratin filaments. Mol. Biol. Cell 13, 382-391 [DOI] [PMC free article] [PubMed] [Google Scholar]