

Abstract
AMPK (AMP-activated protein kinase) is one of the key players in maintaining intracellular homoeostasis. AMPK is well known as an energy sensor and can be activated by increased intracellular AMP levels. Generally, the activation of AMPK turns on catabolic pathways that generate ATP, while inhibiting cell proliferation and biosynthetic processes that consume ATP. In recent years, intensive investigations on the regulation and the function of AMPK indicates that AMPK not only functions as an intracellular energy sensor and regulator, but is also a general stress sensor that is important in maintaining intracellular homoeostasis during many kinds of stress challenges. In the present paper, we will review recent literature showing that AMPK functions far beyond its proposed energy sensor and regulator function. AMPK regulates ROS (reactive oxygen species)/redox balance, autophagy, cell proliferation, cell apoptosis, cellular polarity, mitochondrial function and genotoxic response, either directly or indirectly via numerous downstream pathways under physiological and pathological conditions.
Keywords: AMP-activated protein kinase (AMPK), autophagy, cardiovascular disease, energy sensor, metformin, stress
INTRODUCTION
All living organisms face the challenge of ever-changing environmental stresses, including nutrient stress [1], oxidative stress [2], DNA damage [3] and hypoxia [4]. Appropriate responses and adaptation to these stresses are essential for eliminating cellular injury, and maintaining or re-establishing intracellular homoeostasis and survival. To pursue these goals, cells rely on the co-ordination of multiple stress response pathways that are controlled at the molecular level by a number of highly conserved signalling molecules and transcriptional regulators, such as the TOR (target of rapamycin) pathway, AMPK (AMP-activated protein kinase) pathways and Sirts (sirtuins) and proteins involved in insulin/IGF (insulin-like growth factor) signalling [5].
AMPK is one of the key players in maintaining intracellular homoeostasis. AMPK is well known as an energy sensor and can be activated by increased intracellular AMP levels [6]. Generally, the activation of AMPK turns on catabolic pathways that generate ATP, while inhibiting cell proliferation and biosynthetic processes that consume ATP [6,7]. Recent investigations on the regulation and the function of AMPK indicate that AMPK not only functions as an intracellular energy sensor and regulator, but is also a general stress sensor that is important in maintaining intracellular homoeostasis during many kinds of stress challenges.
AMPK STRUCTURE AND REGULATION
AMPK is an enzyme that is highly conserved in eukaryotic organisms from yeast (snf1 kinase, a homologue of AMPK) to plants (SnRK1, a homologue of AMPK) to humans [8]. AMPK activity is stimulated by intracellular AMP and inhibited by high levels of ATP. AMPK plays an important role in the regulation of whole-body energy homoeostasis by co-ordinating multiple metabolic pathways. Mammalian AMPK is a heterotrimeric complex comprised of a catalytic subunit (α-subunit) and two regulatory subunits (β- and γ-subunits) [7] (Figure 1). Two α-subunit isoforms (α1 and α2), two β-subunit isoforms (β1 and β2) and three γ-subunit isoforms (γ1, γ2, and γ3) are all encoded by distinct genes (PRKAA1, PRKAA2, PRKAB1, PRKAB2, PRKAG1, PRKAG2 and PRKAG3 respectively) that are distributed across five chromosomes (α1, 5p12; α2, 1q31; β1, 12q24.1; β2, 1q21.1; γ1, 12q12–14; γ2, 7q35–36; γ3, 2q35) [9].
Figure 1. Structure of AMPK.

Schematic representation of the components of the AMPK subunits. The catalytic α-subunit can be phosphorylated at Thr172 by upstream kinases including, LKB1, CaMKKβ and TAK1, leading to enzyme activation. The β-subunit contains a glycogen-binding domain (GBD). The γ-subunits contain four nucleotide-binding modules (CBS domains) capable of co-operatively binding to AMP, ADP and ATP.
Subunits of AMPK
The α-subunit contains the catalytic domain of the serine/threonine protein kinase in the N-terminus, as well as the residue Thr172, whose phosphorylation is critical for AMPK activation [10]. The C-terminal domain of the α-subunit is required for interaction with the β-and γ-subunits. An autoinhibitory domain has been mapped to amino acids 312–335 of the α-subunit [11], and the α1 (1–312) truncation is found to be constitutively active even without the β- and γ-subunits [12]. It was originally proposed that AMP promoted AMPK activation by facilitating the phosphorylation of AMPK at Thr172 by upstream kinases, whereas more recent work has suggested that AMP binding also inhibits the dephosphorylation of Thr172 [13]. Interestingly, an inhibitory phosphorylation site, Ser485, was identified in the α-subunit [14], but the detailed mechanism by which this phosphorylation site inhibits AMPK activation and how it is regulated has not been demonstrated.
Even though there is 90 % amino acid identity in the kinase domains of AMPKα1 and α2 isoforms, the AMPK α-subunits have differential tissue-specific expression and activation. AMPKα1 is relatively evenly distributed in the heart, liver, kidney, spleen, lung and brain, whereas AMPKα2 is highly abundant in skeletal muscle and heart, followed by the liver and kidney [15]. AMPKα1 has been reported to be the predominant isoform of AMPK in endothelial cells [16]. Our group has reported that AMPKα1 is the predominant isoform expressed in RBCs (red blood cells) and the mouse vascular wall [17–19]. Salt et al. [20] have reported that the AMPKα2 isoform is more dependent on AMP than is the AMPKα1 isoform, and that AMPKα2 is largely localized to the nucleus.
The β-subunit contributes to the interaction between glycogen and AMPK. The N-terminal region of the β-subunit contains a CBM (carbohydrate-binding module) that is believed to target AMPK to glycogen [21]. In addition, the β-subunit appears to stabilize the interaction between the α- and γ-subunits through a binding domain in its C-terminus (amino acids 186–270) [22,23]. The γ-subunit contains four CBS (cystathionine β-synthase) domains (Figure 1). AMP, ADP and ATP can bind the AMPK γ-subunit to regulate AMPK activation through allosteric structural changes in the catalytic α-subunits, modulating phosphorylation by upstream kinases [24] and regulating the dephosphorylation of Thr172 [13,25].
Regulation of AMPK by AMP/ATP ratios
AMPK plays a key role in regulating cellular energy status. AMPK is activated when the intracellular AMP level increases. The binding of AMP to the AMPK γ-subunit leads to an allosteric structural change in AMPK, resulting in a 2–5-fold increase in AMPK activity [26]. The extent of allosteric activation of AMPK by AMP depends on the specific isoforms of both the α- and γ-subunits present in the enzyme [26]. The greatest activation by AMP occurs in complexes containing the α2 and γ2 isoforms; however, only weak activation occurs in complexes with the γ3 isoforms [26]. The most recent crystal structure for mammalian AMPK indicates that ADP protects AMPK from dephosphorylation after binding to one of the two exchangeable AXP (AMP/ADP/ATP)-binding sites in the γ-subunit [27]. More importantly, the binding of AMP to the γ-subunit of AMPK enhances the phosphorylation of Thr172 by upstream kinases [24] and inhibits the dephosphorylation of Thr172 by PP (protein phosphatase) 2Cα [13] or PP2A [28].
Regulation of AMPK by upstream kinases
In addition to allosteric regulation, AMPK is significantly regulated by upstream kinases (Figure 2). LKB1 (liver kinase B1) and CaMKKβ (Ca2+/calmodulin-dependent protein kinase kinase β) are two well-defined upstream kinases of AMPK. LKB1 was originally discovered as a tumour suppressor in Peutz–Jeghers syndrome patients, who are susceptible to benign intestinal hamartomatous polyps and malignant tumours mainly in the gastrointestinal tract [29]. In vitro, the purified recombinant LKB1 complex directly phosphorylates AMPK on Thr172 [30–32]. LKB1 is not activated by phosphorylation, like most kinases, but is instead regulated by relocation from the nucleus to the cytoplasm upon binding to STRAD (Ste20-related adaptor) and MO25 (mouse protein 25) [33,34]. Research in LKB1-deficient cells revealed CaMKKβ to be another upstream kinase of AMPK [35,36]. Furthermore, CaMKKβ isolated from rat brain or expressed in Escherichia coli phosphorylates and activates AMPK in vitro [37]. The activation of AMPK by CaMKKβ occurs in the absence of a detectable increase in AMP, but depends on an increase in intracellular Ca2+ content [35,36]. More recently, it was reported that TAK1 (transforming growth factor-β-activated kinase 1) can activate AMPK in yeast [38], but the physiological relevance of AMPK activation by TAK1 needs further study.
Figure 2. Regulation of AMPK.

AMPK is a heterotrimeric protein complex which consists of a catalytic α-subunit and regulatory β- and γ-subunits. AMPK is activated by increased intracellular AMP levels, which prevents the catalytic subunit from being dephosphorylated. LKB1 and CaMKK are two major upstream regulators of AMPK that exert their function by phosphorylating the AMPK α-subunit at Thr172. Another reported activator of AMPK is TAK1. Activation of AMPK switches off ATP-consuming processes and promotes ATP-generating processes. TGF, transforming growth factor.
Other pathways in AMPK activity regulation
The phosphorylation of AMPK by upstream kinases is a reversible procedure modulated by protein phosphatases. Both PP2A and PP2C have been reported to be able to dephosphorylate AMPK, thus inhibiting AMPK activation [39,40]. New mechanisms for regulating AMPK activity have been proposed, indicating that AMPK protein can be modulated by ubiquitin-dependent proteasome degradation mediated by CIDEA (cell-death-inducing DNA fragmentation factor-α-like effector A), and a CIDEA-binding site has been identified on the AMPK β-subunit [41]. Oakhill et al. [24] have provided evidence to show that AMPK β-myristoylation plays a key role in the full phosphorylation and activation of AMPK by upstream kinases in response to metabolic stress signals. In a recent paper from Hardie’s group [42], the authors distinguished six pathways that can activate AMPK using isogenic cell lines stably expressing either wild-type AMPK complexes or AMP-insensitive (R531G) γ2 mutants.
AMPK ACTIVATORS AND INHIBITORS
The potential beneficial effects of AMPK activation in disease prevention and treatment have stimulated the interest of researchers in identifying AMPK activators. Thus far, numerous AMPK activators have been reported; some of the direct and indirect activators of AMPK, as well as some AMPK inhibitors, are listed below.
Activation of AMPK in physiological and pathological conditions
AMPK has been implicated in several physiological and pathological conditions, such as hypoxia, CR (caloric restriction) and physiological exercise. In these conditions, activated AMPK modulates numerous downstream targets, allowing the body to adapt to these challenges.
AMPK is activated by hypoxia
Hypoxia is a situation in which the whole body or a region of the body is deprived of adequate oxygen supply. This could happen under physiological conditions, such as during strenuous physical exercise or at high altitudes, and in pathological conditions, such as anaemia, hypoventilation and pulmonary fibrosis. Complete oxygen depletion results in cell death, but cells can survive in reduced oxygen supply conditions by activating broad adaptive processes via HIF (hypoxia-inducible factor)-1, which plays a central role in these adaptations [43]. AMPK is involved in the response to hypoxia in the goldfish, a hypoxia-tolerant species [44], and is activated in hypoxic conditions in rat heart tissue and cultured endothelial cells [45,46]. The mechanisms for the activation of AMPK under hypoxia appear complicated and are dependent on the extent of hypoxia. One of the mechanisms leading to AMPK activation is reduced ATP production due to the inhibition of β-oxidation under hypoxic conditions [47]. On the other hand, ROS (reactive oxygen species) and RNS (reactive nitrogen species) are important in AMPK activation in conditions of non-severe hypoxia. ONOO− (peroxynitrite), generated from NO and O2•− (superoxide anion), has been reported to rapidly activate AMPK in endothelial cells [48,49], whereas mitochondrial RNS are reported to be required for the activation of AMPK by metformin [50]. Under these conditions, intracellular AMP/ATP levels remain unchanged, whereas ROS production by the electron transport chain at complexes I, II and III in mitochondria is significantly increased [51,52].
ROS/RNS might activate AMPK via different upstream kinases. ONOO− -activated AMPK in endothelial cells is reported to be a c-Src-mediated PI3K (phosphoinositide 3-kinase)-dependent [53]. Further investigations have demonstrated that PKC (protein kinase C)-ζ-mediated phosphorylation of LKB1 is essential in ONOO− -mediated AMPK activation [54]. On other hand, ROS, such as H2O2, can trigger AMPK activation in the apparent absence of increased AMP concentration through ROS-dependent CRAC (Ca2+ release-activated Ca2+ ) channel activation, leading to increases in cytosolic Ca2+ that activate the AMPK upstream kinase CaMKKβ [55]. Interestingly, AMPK has been reported to be required for HIF induction in the response to hypoxia [56], and an AMPK/HIF1 axis has been suggested to be important for cell survival and growth under conditions of hypoxia [52]. However, the detailed mechanism by which AMPK regulates HIF is not completely clear. In addition, AMPK can also regulate cellular survival in conditions of hypoxic stress through autophagic regulation, which is independent of the HIF-1 pathway [57].
AMPK is activated by physical exercise
Regular physical exercise has numerous benefits in the prevention and treatment of diseases [58,59], and AMPK activation is increased during physical exercise [60]. Notably, rodents that are selectively AMPK-deficient in skeletal muscle showed impaired recovery from fatigue after exercise [61]. The exact mechanism of the beneficial effect of AMPK activation by exercise is not clear. It has been reported that activation of AMPK increases fatty acid oxidation and glucose uptake in rat muscles [62]. Similar results were reported in insulin-resistant high-fat-fed rats [63], suggesting a potential beneficial effect of AMPK activation for insulin-resistant patients and those with Type 2 diabetes. Recently, Evans’s group [64] reported that activation of AMPK by AICAR (5-amino-4-imidazolecarboxamide riboside) mimics the effect of training and improves exercise performance in mice. Similar effects are observed in mice treated with GW1516, a PPAR (peroxisome-proliferator-activated receptor)-δ activator. On the basis of these findings, the AMPK/PPAR-δ pathway has been proposed to explain the beneficial effects of AMPK activation in exercise, and it has been suggested that AMPK agonists are exercise mimetics [64]. AMPK has also been implicated in the regulation of muscle fibre type change. A previous study has reported that rats fed with AICAR for 14 days have a significantly increased number of type IIx fibres in their extensor digitorum longus [65]. Similarly, in a transgenic mouse model, Goodyear’s group [66] have demonstrated that AMPK is essential for a fibre type IIb to IIa/x transformation during physical exercise training. Most recently, using skeletal-muscle-specific AMPKβ1/AMPKβ2-knockout mice, O’Neill et al. [68] have demonstrated that AMPK is required in maintaining mitochondrial content and glucose uptake during exercise. AMPK-mediated fibre type change during exercise might result in a significant metabolic switch in muscles, since type IIx fibres are more likely to depend on glycolysis, whereas type IIb fibres rely more on mitochondrial oxidation [69]. Thus increased type IIx fibres during exercise training would promote glucose consumption. Corresponding to this energy-demanding change, there was an increase in glucose uptake capacity supported by an increase in hexokinase expression [66] and increased GLUT4 expression [70] in skeletal muscles of mice after 6 weeks of exercise training. These studies provide molecular mechanisms for the metabolic beneficial effect of AMPK activation during exercise training for obese and diabetic patients.
Natural activators of AMPK
Several physiological hormones and natural plant compounds can activate AMPK. Leptin and adiponectin are hormones secreted by adipocytes. Acute and chronic leptin treatments have been reported to increase AMPK expression and/or phosphorylation in rodent skeletal muscles; this could be an important mechanism by which leptin regulates energy stores [71,72]. Adiponectin has cardioprotective effects against ischaemia/reperfusion injury by a mechanism believed to be due to AMPK activation [73,74]. Resveratrol and berberine are natural compounds found in red grapes and Berberis respectively. Resveratrol exerts an effect similar to CR to extend lifespan, and AMPK is reported to be the key enzyme that mediates this beneficial effect of resveratrol in neurons [75]. Berberine can increase AMPK activity [76] and improve glucose transport in cell lines and animals; this is believed to be the important mechanism by which berberine exerts its beneficial effects in diabetic and insulin-resistant states [77]. Despite existing evidence for the role of AMPK activation by these compounds, the importance of AMPK activation in contributing to the beneficial effects of these chemicals, and the detailed mechanisms by which these compounds activate AMPK, require further investigation.
Chemical activators of AMPK
The most widely used chemical activator of AMPK in research is AICAR. AICAR is a cell-permeant chemical that is converted within the cell into ZMP (monophosphate AICAR), which mimics the effect of AMP in activating AMPK [78]. The glucose analogue 2-DG (2-deoxy-D-glucose) is another strong AMPK activator that is used in experimental research as it can competitively block glucose utilization to mimic the effect of CR [79,80]. Clinically, metformin is the most widely used AMPK activator for the treatment of Type 2 diabetes [81]. TZDs (thiazolidinediones) are PPAR-γactivators used as insulin sensitizers and can rapidly activate AMPK in cell lines and animal tissues [82]. A-769662 is a direct activator of AMPK, identified as the result of screening a chemical library of over 700 000 compounds [83–85]. It is noteworthy that all of these chemicals possess beneficial effects that may be unrelated to AMPK activation. For example, AICAR and metformin have been reported to modulate hepatic glucose metabolism by regulating glucokinase translocation, which is AMPK-independent [86]. Similarly, Viollet’s group [87] have reported that metformin inhibited hepatic glucogenesis in an AMPK-independent manner.
AMPK inhibitors
AMPK is inhibited by high glucose and glycogen
Obesity and diabetes are the fastest growing public health problems in developed and developing countries. Over-nutrition and decreased physical exercise are believed to be the major reasons for these problems. Emerging evidence has suggested that activated AMPK might be beneficial for whole-body energy homoeostasis [88]. On the other hand, AMPK activation is inhibited under high glucose conditions. In animal models, glucose infusion induces acute hyperglycaemia and a reduction in AMPK activation in rat muscle and liver [89]. Similar results are also observed in cultured HepG2 hepatocytes incubated with high glucose [90]. However, the detailed mechanism of AMPK inhibition by high glucose is not completely clear and requires further investigation. High glycogen content represses AMPK activation in rat skeletal muscle without significant changes in adenine nucleotide concentrations [91]. The proposed mechanism suggested McBride and co-workers [92] indicates that glycogen leads to allosteric inhibition of AMPK by binding to the AMPK glycogen-binding domain in the β-subunit.
AMPK is inhibited by lipid overload
High-fat-diet-fed mice have impaired AMPK phosphorylation and protein expression in multiple tissues and organs, including skeletal muscle, heart, liver and hypothalamus [93]. Furthermore, mice fed on a palmitate-enriched diet have a significant inhibition of AMPK activation through the enhancement of PP2A-mediated dephosphorylation of AMPK [28].
AMPK is inhibited by amino acids
A high-protein diet decreases AMPK activity in the hypothalamus, leading to inhibition of NPY (neuropeptide Y) and a reduction in food uptake in mice [94]. Similarly, treatment of C2C12 cells with leucine reduces AMPK phosphorylation with enhanced mTOR (mammalian TOR) activation. Findings further suggest the involvement of an AMPK/mTOR pathway in coordinating amino acid metabolism [95].
Pharmacological AMPK inhibitor
Compound C is a cell-permeant pyrrazolopyrimidine compound that is widely used as an AMPK inhibitor [81], but the non-AMPK-targeted effect need to be considered when using this chemical. Bain et al. [96] have reported that Compound C inhibited AMPK with an IC50 value of 0.1–0.2 μM, whereas it also inhibited other protein kinase, such as ERK8 (extracellular-signal-regulated kinase 1), MNK1 (mitogen-activated protein kinase-interacting kinase 1), PHK (phosphorylase kinase), MELK (maternal embryonic leucine-zipper kinase), DYRK (dual-specificity tyrosine-phosphorylated and -regulated kinase) isoforms, HIPK2 (homeodomain-interacting protein kinase 2), Src, Lck and Yes, FGF-R1 (fibroblast-growth-factor receptor 1) and Eph-A2 (Ephrin A2 receptor) with similar IC50 values, as well as the BMP (bone morphogenetic protein) pathway [97]. Thus some other approaches, such as siRNA (small interfering RNA) or transgenic mice are required to confirm whether these specific effects are AMPK-dependent.
AMPK IS AN ENERGY STRESS SENSOR AND MODULATOR
Limited nutrition, as well as an excessive supply of nutrients for cellular requirements, can both cause cellular stress. Nutrient supply status is sensed at multiple levels by the cell and whole body. AMPK is strongly activated by energy depletion and is inhibited in conditions of over-nutrition, such as in Type 2 diabetic patients. This marks AMPK as one of the master sensors and regulators of nutrient stress in maintaining intracellular energy homoeostasis, which is essential for normal cell function and survival under physiological and pathological conditions (Figure 3).
Figure 3. AMPK is a master regulator of metabolism.
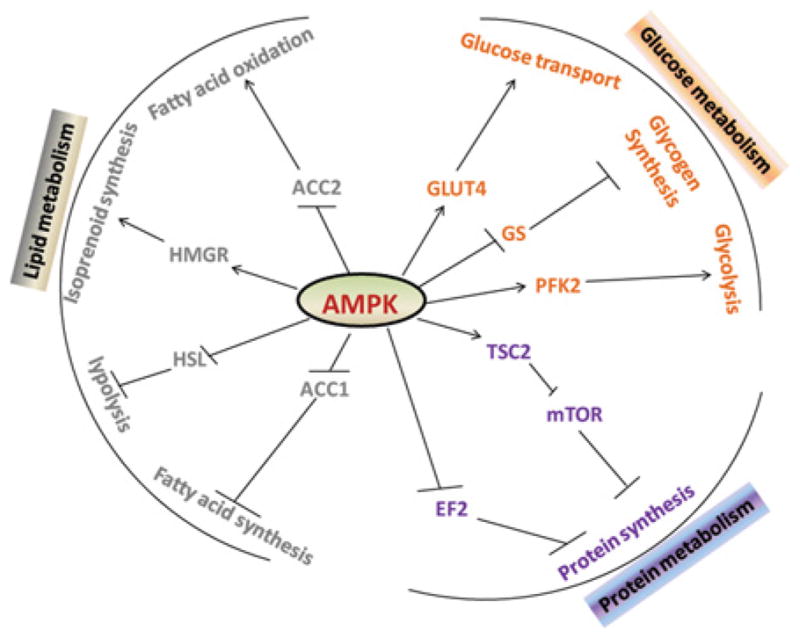
AMPK regulates lipid, protein and glucose metabolism by modulating numerous downstream targets. Activation of AMPK promotes fatty acid oxidation and glucose utilization, but inhibits protein, fatty acid and glycogen synthesis. GS, glycogen synthase; PFK2, phosphofructokinase 2; HMGR, HMG-CoA reductase; HSL, hormone-sensitive lipase.
AMPK activation by nutrient depletion
Nutrient depletion could happen in both physiological and pathological conditions, such as fasting, exercise and ischaemia, and leads to an increase in intracellular AMP levels, which is a direct activator of AMPK. Exercise and CR, which involves a 20–40 % reduction in caloric intake without compromising daily nutritional needs, are believed to benefit human health. CR is the most consistent way to extend the lifespan of yeast, worms, flies and rodents [98] and to lower the incidence of age-related disease [99]. Interestingly, multiple genetic models have confirmed the role of AMPK as a mediator of the beneficial effects of CR on longevity in yeast [100] and Caenorhabditis elegans [101]. In addition, there is an aging-associated reduction in AMPK activity and mitochondrial biogenesis in rodents [102]. More importantly, CR leads to strong activation of AMPK [60]. The mechanisms by which CR activates AMPK and promotes longevity are not completely understood; however, several mechanisms of AMPK activation in CR conditions have been proposed. For example, CR and muscle contraction during exercise are reported to result in an increase in the cellular AMP/ATP ratio. AMPK is therefore activated in mouse skeletal muscle during exercise and increases glucose transport, an effect that is not seen in muscle from LKB1-deficient mice, indicating that LKB1 is important in regulating AMPK activity in response to muscle contraction [103]. Furthermore, it is believed that CR modulates AMPK through ROS-mediated AMPK activation. This hypothesis is supported by results from 2-DG-induced CR conditions in endothelial cells, which trigger autophagy and increased cell survival in response to energy stress. These effects are dependent on AMPK activation [80]. In addition, activated AMPK in CR can modulate mitochondrial function through the regulation of mitochondrial biogenesis via the AMPK/Sirt1/PGC1 (PPAR-γco-activator 1) pathway and mitochondrial elimination by the AMPK/mTOR/ULK1 (UNC-51-like kinase 1) autophagy pathway [104–106]. Consistent with this, a natural compound, resveratrol, found in red wine and red grapes, can mimic the beneficial effects of CR, and this beneficial effect is dependent on the activation of AMPK and Sirt1 [107,108].
AMPK is a master regulator of energy homoeostasis
AMPK modulates multiple downstream pathways in metabolic regulation
Activation of AMPK has been reported to regulate energy balance at multiple organs by targeting several key substrates. AMPK inactivates ACC (acetyl-CoA carboxylase), leading to the inhibition of fatty acid synthesis and the promotion of mitochondrial β-oxidation. Activated AMPK can inhibit HMG-CoA (3-hydroxy-3-methylglutaryl-CoA) reductase to reduce cholesterol synthesis. AMPK increases cellular NAD+ levels and enhances Sirt1 activity, causing the deacetylation and activation of PGC1α and resulting in an increase in mitochondrial gene expression [109]. In parallel, the activation of AMPK leads to increased food intake [110], and increases GLUT4 (glucose transporter 4) expression and glucose transport in skeletal muscle [111]. Most recently, AMPK has been reported to promote fat absorption in mice [112]. AMPK promotes the initiation of autophagy, a major protective mechanism that allows cells to survive in response to multiple stress conditions, especially nutrient depletion, by inhibiting the mTORC1 (mTOR complex 1) pathway. Under low-nutrient conditions, AMPK is activated and phosphorylates TSC2 (tuberous sclerosis complex 2), leading to the inhibition of mTORC1 [113]. Gwinn et al. [114] have reported that AMPK regulates the mTOR pathway by direct phosphorylation of Raptor (regulatory associated protein of mTOR) at Ser722 and Ser792 residues. Recent research further indicates that AMPK could directly act on initiation of autophagy by the direct phosphorylation of ULK1 at Ser555 and Ser777 [115], or at Ser317 and Ser777 [3], in MEFs (mouse embryonic fibroblasts) under conditions of glucose depletion. A study on C. elegans showed that glucose restriction can significantly extend the lifespan of this organism, and that this requires aak-2, a homologue of AMPK [116]. A recent study indicates further that the effects of AMPK activation on longevity are mediated by CREB (cAMP-response-element-binding protein) and CRTCs (CREB-regulated transcriptional co-activators), especially CRTC-1 [117]. Overall, AMPK will be quickly activated by energy depletion, and activated AMPK can regulate multiple pathways involved in maintaining intracellular energy homoeostasis, which is critical for normal cellular function and survival.
AMPK integrates whole-body energy balance
AMPK acts as a metabolic master sensor and regulator by inducing a cascade of events in multiple organs in response to energy challenge in physiological and pathological conditions. Skeletal muscle is one of the major target organs for insulin in stimulating glucose disposal, and insulin resistance is the major characteristic of the development of Type 2 diabetes. In skeletal muscle, AMPK activity is increased by stimuli such as osmotic stress, hypoxia, ischaemia and exercise [118]. Activation of AMPK in muscle recruits GLUT4 to the plasma membrane and enhances glucose transport, independent of insulin stimulation [119]. AMPK also regulates skeletal muscle energy production by regulating mitochondrial synthesis, which is mediated by several transcription factors, such as PGC-1α, NRF-1 (nuclear respiratory factor-1) and MTFA (mitochondrial transcription factor A) [120,121]. In adipose tissue, AMPK has been reported to be activated by adipocyte hormones, including leptin [122] and adiponectin [123]. In the liver, activation of AMPK leads to the rapid regulation of hepatocyte metabolism by regulating specific enzymes, such as ACC, thereby resulting in increased fatty acid oxidation and inhibited hepatic lipogenesis, cholesterol synthesis and glucose production [124]. Glucose intolerance and hyperglycaemia has been observed in liver-specific AMPKα2−/−mice in a fasted state [125]. Liver AMPK regulates glucose homoeostasis by regulating multiple glucogenesis genes, such as PEPCK (phosphoenolpyruvate carboxykinase), G6Pase (glucose-6-phosphatase) [126,127], and transcription factors, including HNF (hepatocyte nuclear factor)-4α and FoxO1 (forkhead box O1) [128]. In adipose tissue, activated AMPK improves insulin sensitivity by decreasing lipogenesis and increasing fatty acid oxidation, which results in decreased non-esterified fatty acids (free fatty acids) in adipose tissue, and AMPK activation leads to the inhibition of pre-adipocyte differentiation and triacylglycerol (triglyceride) synthesis in adipocytes (for detailed information, see [129]). Low glucose levels activate AMPK [130], whereas high glucose inhibits AMPK activation in β-cells [131]. Pre-activation of AMPK by AICAR blocked the glucose-induced up-regulation of gene expression for insulin secretion in islet β-cells [131]. During food intake regulation, AMPK activation in the hypothalamus is reported to be an important mechanism for anorexigenic and orexigenic signals in integrating whole-body energy homoeostasis [132]. Interestingly, a recent study has highlighted the role of the AMPK/SRC-2 (steroid receptor co-activator-2) axis in fat absorption in the gut to co-ordinate whole-body energy demand [112].
AMPK AND THE OXIDATIVE STRESS RESPONSE
Oxidative stress is defined as an imbalance between the levels of pro-oxidants and antioxidants, resulting in macromolecular damage and disruption of redox signalling and controls [133]. Macromolecular damage usually occurs via oxidative mechanisms linked to free radicals, including O2−, NO and OH− (hydroxyl ions). Free radicals are small diffusible molecules that differ from most biological molecules in that they are highly reactive due to an unpaired electron. Free radicals can participate in chain reactions in which a single free radical initiation event can be propagated to damage multiple molecules, including DNA, damage to membrane ion transport systems, enzymes and lipid peroxidation [134]. Recently, studies including those from our group have shown that AMPK may be an oxidative stress sensor and redox regulator in addition to its traditional role as an energy sensor and regulator.
AMPK is activated by intracellular oxidative species
ROS-induced activation of AMPK is believed to be important for the beneficial effect of many medicinal drugs. For example, metformin has been reported to activate AMPK through mitochondrial-derived RNS [50]. Furthermore, ischaemia/reperfusion experiments with sevoflurane protected rat hearts against ischaemic injury in a manner dependent on the activation of AMPK by intracellular ROS. Application of a ROS scavenger resulted in decreased AMPK activation and diminished the cardioprotective effect of sevoflurane, thereby further substantiating the role of ROS in the activation of AMPK [135]. In mouse skeletal muscle, AMPK is activated by oxidative stress and enhances glucose transport, independent of changes in AMP or the AMP/ATP ratio [136]. Similarly, in cell culture conditions, hypoxia-induced AMPK activation is reported to be dependent on mitochondrial ROS without significant changes in the AMP/ATP ratio [137]. H2O2 has been observed to be able to strongly induce the activation of AMPK [138]. Recent studies have shown that the underlying mechanism of H2O2 activation of AMPK involves the induction of cysteine oxidation [139]. In vascular endothelial cells, our group has reported, in multiple species, that ONOO− activates AMPK by a PKCζ/LKB1-dependent mechanism [140].
AMPK modulates ROS production
As described above, AMPK is functional as a redox sensor that can be quickly activated by increased intracellular ROS/RNS. Activated AMPK appears to be essential in maintaining intracellular redox status by inhibiting oxidant production by NADPH oxidases, mitochondria, etc. or by increasing the expression of antioxidant enzymes such as SOD (superoxide dismutase)-2 and UCP (uncoupling protein)-2.
AMPK regulates NADPH oxidase
NADPH oxidase (Nox) is a membrane-bound complex made of six subunits: a GTPase (usually Rac1 or Rac2) and five phox units: gp91phox, p22phox, p40phox, p47phox and p67phox. NADPH oxidase has been well recognized as an important source of ROS in the vasculature and plays an essential role in normal vascular function and in vascular diseases, such as hypertension, atherosclerosis and diabetes [141]. Overproduction of ROS has been postulated to contribute to a loss of insulin responsiveness. A study that analysed saphenous veins and internal mammary arteries from diabetic patients showed significantly increased NADPH oxidase activity and unregulated expression levels of the p22phox, p67phox and p47phox subunits combined with unpaired endothelial NO synthesis [142] (for reviews, see [143,144]). Activation of AMPK by AICAR has been reported to suppress O2− stimulated by phorbol esters or fMLP (N-formylmethionyl-leucylphenylalanine) by reducing the translocation and phosphorylation of p47phox in human neutrophils [145]. Similarly, rosiglitazone reduced high-glucose-induced NADPH-oxidase-mediated oxidative stress in a manner dependent on AMPK activation in HUVECs (human umbilical vein endothelial cells) [146]. Moreover, metformin suppresses high-glucose-induced oxidative stress in cultured podocytes by an AMPK-mediated reduction of NADPH oxidase activity [147]. In AMPKα2−/− mice, our group has found a significantly increased expression and activation of NADPH oxidase due to enhanced NF-κB (nuclear factor κB) activation [148]. These findings reveal a novel mechanism for the regulation of NADPH oxidase expression by AMPK, and demonstrate that AMPK is a physiological suppressor of NADPH oxidase and ROS production in endothelial cells. Most recently, a similar observation was reported in AMPKα1-deficient mice treated with chronic AngII (angiotensin II), which showed mild endothelial dysfunction associated with increased NADPH oxidase activation and Nox2 up-regulation [149].
AMPK regulates mitochondrial ROS generation
Mitochondria, especially damaged mitochondria, are the major sources of ROS in cells and are related to many chronic human diseases, such as diabetes mellitus, neurodegeneration and cancer [150]. AMPK can regulate mitochondrial ROS generation through several mechanisms.
UCPs are members of the larger family of mitochondrial anion carrier proteins, and UCPs facilitate the transfer of anions across mitochondrial membranes. UCP2 plays an important role in controlling mitochondrial ROS production [151,152] and is believed to be a potential target in the treatment of obesity, diabetes and aging [153]. Studies have shown that one of the mechanisms of AMPK action in redox regulation is to up-regulate the expression of mitochondrial UCP2. For example, activation of AMPK by AICAR, or overexpression of constitutively activated AMPK, inhibited O2− production and reduced tyrosine nitration of prostacyclin synthase under high-glucose treatment in HUVECs [154]. Furthermore, AMPK is involved in the action of ghrelin on NPY/Agouti-related peptide. The latter seems to be due to the up-regulation of UCP2 expression, resulting in the regulation of mitochondrial ROS production in neurons [155] and cultured MIN6 insulinoma cells [156]. Furthermore, in an animal model, it has been found that endurance training activated AMPK, resulting in an increase in UCP2 expression in rat pancreatic islets [157], which is similar to findings in mouse β-cells deficient in AMPKα2 expression [158]. Despite these findings, the detailed mechanisms by which AMPK regulates UCP2 expression and function are still not completely understood.
Emerging evidence suggests that AMPK might modulate autophagy thereby regulating mitochondrial ROS generation. It is well known that damaged proteins and DNA or dysfunctional mitochondria cause enhanced production of ROS from mitochondria [159]. Accordingly, cells have devised specific mechanisms to control these damaged organelles. One such mechanism is autophagy, which is critical in eliminating dysfunctional mitochondria from cells [160]. Autophagy deficiency leads to elevated ROS production; this is supported by numerous experimental results from autophagy-dysfunctional cell lines or animals. For example, skeletal muscle cells from Agt7-deficient mouse exhibit increased defects in mitochondrial respiration and increased ROS levels [161]. Similar observations have been reported in Atg5−/− cells and beclin1+/− iBMK cells, which show elevated ROS production with stress [162]. More importantly, mitochondrial removal is critical for RBC maturation and survival; accordingly, autophagy-defective RBCs from Nix−/− mice have more ROS production and undergo apoptosis [163]. Furthermore, several other autophagy-deficient gene knockout mice (Agt5−/−, Agt7−/− and Ulk1−/−) showed similar anaemic phenotypes due to a shortened RBC lifespan [164]. It has been well established that AMPK regulates autophagy through inactivation of the mTORC1 pathway by phosphorylation and activation of the TSC2 exchange factor [113]. Most recently, a new mechanism involving AMPK in the control of autophagy has been reported by several research groups, involving the direct phosphorylation of Ulk1 by AMPK to regulate autophagy [165], indicating that AMPK can regulate ROS through the regulation of autophagy. Indeed, AMPK inactivation by palmitate leads to defective autophagy and increased generation of mitochondrial ROS in bone-marrow-derived macrophages [166].
AMPK activation increases the antioxidant potential
Oxidative stress is a result of an imbalance of oxidants and antioxidants. Emerging evidence suggests that AMPK also plays an important role in the regulation of cellular antioxidant defence. For instance, activation of AMPK by AICAR is reported to inhibit palmitate-induced ROS levels by increasing the expression of Trx (thioredoxin) in human aortic endothelial cells [167]. Similarly, AMPK activation in endothelial cells increases the expression of Mn-SOD (manganese SOD) by up-regulating the expression of PGC-1α [168]. Consistently, inhibition of AMPKα1 in cultured HUVECs is reported to decrease the expression of some antioxidant genes, including γ-GS (γ-glutamylcysteine synthase), Mn-SOD, catalase and Trx, resulting in the accumulation of ROS and increased apoptosis [169]. Using transgenic mice, our group has reported that AMPKα1−/− mice exhibit an anaemic phenotype because of the imbalance of antioxidants/oxidants in RBCs, leading to a significantly shortened RBC lifespan [19].
ROLE OF AMPK IN CELL PROLIFERATION AND CELL DEATH
AMPK in cell proliferation
Mammalian cell proliferation is controlled by the cell-cycle machinery that is regulated by CDKs (cyclin-dependent kinases), such as Rb (retinoblastoma protein) and CDKIs (CDK inhibitors), including as p21CIP and p27KIP1[170]. AICAR, an AMPK activator, has been reported to induce cell-cycle arrest in multiple types of cells, including carcinoma and non-carcinoma cells [171,172]. AMPK regulates cell proliferation by controlling several key factors in metabolic pathways that are important for cell growth and division (Figure 4). Activated AMPK has been reported to suppress mTOR signalling by growth factors and amino acids to control protein synthesis, which is required for cell growth and division [173]. Furthermore, activation of AMPK by AICAR or rosiglitazone leads to the suppression of prostate cancer cell proliferation by reducing Fas expression [174], which controls fatty acid synthesis. Furthermore, HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis, is a direct substrate of AMPK [175]. Importantly, studies suggest that AMPK has a more direct effect on cell growth control by controlling CDKIs. For instance, Jones et al. [176] have reported that AMPK directly regulates p53 function by modifying the Ser15 phosphorylation site, which controls p21CIP expression in MEFs. Our most recent findings have shown that AMPK, especially AMPKα2, controls mVSMC (mouse vascular smooth muscle cell) proliferation by modulating p27KIP1 expression through p52 NF-κB-2-dependent Skp2 (S-phase kinase-associated protein 2) regulation [17].
Figure 4. Role of AMPK in cellular fate decisions.

Activation of AMPK leads to inhibition of oxidative stress, reduction of DNA damage, promotion of autophagy and restriction of cellular proliferation.
AMPK in cellular survival and apoptosis
The function of AMPK in cellular apoptosis is complicated and sometimes controversial. For example, AICAR has been reported to induce apoptosis in adipocytes through AMPK-dependent eIF2α (eukaryotic initiation factor 2α) regulation [177], whereas another study has reported that AICAR-induced apoptosis is independent of AMPK activation in leukaemia cell lines [178]. Similarly, resveratrol has been reported to induce apoptosis in chemoresistant cancer cells through AMPK modulation [179], but the same group has also reported that resveratrol protects ROS-induced cell death in H9c2 cardiac muscle cells by activating AMPK [180]. Therefore the role of AMPK in cellular fate decision seems to be cell-type- and treatment-specific, and dependent on the duration and tension of the stress. One explanation for these observations is the multiple functions of downstream substrates of AMPK, such as p53, which might play an anti-apoptotic role by inducing cell growth arrest and promoting the expression of genes involved in DNA repair, including GADD45 (growth-arrest and DNA-damage-inducible protein 45) and PCNA (proliferating-cell nuclear antigen), which are important for stem cell survival [181]. At the same time, p53 has a pro-apoptotic effect by up-regulating the expression of several apoptosis genes including Bax [182] or by directly attacking mitochondria [183]. Another example is the function of AMPK in autophagy regulation. Autophagy is generally believed to be a cytoprotective mechanism that enables cells to survive in conditions of unfavourable stress, especially metabolic stress [184]. However, autophagy is also reported to contribute to cell death in some types of damage; for example, damage-regulated modulator-mediated autophagy is reported to play a critical role in p53-modulated apoptosis [185]. In cancer cells, autophagy can increase apoptosis in a caspase-independent manner [186,187]. As discussed above, AMPK can regulate autophagy through the mTOR and ULK1 pathways, and thereby play dual roles in cellular apoptosis. Current evidence supports the theory that AMPK plays important, but complicated, roles in cell fate decisions, which could be cell-type-, stress-tension- and sustention-dependent. It is obvious that the role of AMPK in cell apoptosis or cell fate decisions, particularly the relationship among AMPK, p53 and autophagy, requires further investigation.
ROLE OF AMPK IN CVD (CARDIOVASCULAR DISEASE)
A healthy heart requires a sustained energy supply for its contraction. Many energy substrates, including fatty acids, glucose, ketones and amino acids, can be used by the heart, but fatty acid β-oxidation in mitochondria is its primary source of ATP. AMPK works to restore ATP supply and plays an important role in the heart in both physiological states and stress conditions, such as excessive load and ischaemia/reperfusion. Recent evidence suggests that AMPK might also regulate the oxidative stress response and that dysfunctional AMPK could be the basis of the pathogenesis of several CVDs [188].
Alterations in AMPK cause cardiomyopathy in humans
WPW (Wolff–Parkinson–White) syndrome is a hereditary disease that is characterized by ventricular pre-excitation [189]. WPW syndrome is associated with unexplained LVH [LV (left ventricular) hypertrophy] and is diagnosed as FHC (familial hypertrophic cardiomyopathy) [190]. AMPK γ-subunit mutations have been identified as the molecular basis for the pathogenesis of WPW syndrome and LVH [191,192]. Subsequently, multiple mutations have been reported and distinct clinical features associated with AMPKγ subunit mutations (PRKAG2 cardiac syndrome) [193]. The exact mechanism by which AMPK γ-subunit mutations cause cardiac dysfunction is not clear, but findings suggest that a glycogen storage disorder might be the common reason [194]. Mice overexpressing human PRKAG2 mutant genes, R531G [195], N488I [196] and R302Q [197], closely mimic clinical WPW syndrome, with cardiomyopathy and glycogen accumulation in cardiomyocytes. A glycogen storage problem in PRKAG2 cardiomyopathy patients is also believed to be one of the mechanisms for the cardiac pre-excitation conduction syndrome seen in these patients [198]; notably, other glycogen storage disorders, such as Danon disease, also lead to pre-excitation [199]. Modulating AMPK activation in epithelial cells affects their transmembrane conductance [200,201], potentially indicating a more direct role of AMPK in regulating cardiac electrophysiology; however, this hypothesis needs further investigation.
Protective effect of AMPK activation on cardiomyocytes
In the clinical situation, doxorubicin is a widely used and highly effective medicine in the treatment of cancer, but the significant side effects to non-carcinoma cells, especially cardiomyocytes, limit its utilization [202]. The underlying mechanism of doxorubicin-induced cardiomyocyte apoptosis is not fully known, but findings indicate that AMPK dysfunction might be involved in the development of doxorubicin-induced cardiotoxicity. In isolated perfused hearts, doxorubicin causes a rapid reduction in AMPK activity and protein expression [203]. In line with this observation, activation of AMPK with 2-DG antagonizes doxorubicin-induced cardiomyocyte death in neonatal rat cardiomyocytes, and blocking AMPK activation with Compound C or siRNA attenuates the protective effects of 2-DG, indicating the protective role of AMPK activation against doxorubicin-induced cardiotoxicity [79]. Consistent with this, activation of AMPK with metformin protects adult mouse cardiomyocytes (HL-1 cells) from doxorubicin-induced apoptosis [204]. Furthermore, adiponectin treatment protects mice against doxorubin-induced cardiomyopathy, and this beneficial effect is dependent on AMPK activation [205]. Mice expressing KD-AMPK (kinase-dead AMPK), which has a non-functional mutant kinase domain, do not exhibit dramatic cardiac dysfunction, although their hearts show impaired recovery of LV contractile function with increased caspase 3 activity and TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP nick-end labelling) staining after ischaemia and reperfusion. The results indicate a protective role of AMPK in limiting damage to cardiomyocytes from ischaemic injury [206]. Most recently, our group has reported in OVE26 mice that AMPK activation is reduced along with cardiac dysfunction, and that metformin can preserve cardiac function [207]. More importantly, genetic AMPK inhibition attenuated the protective effect of metformin on cardiomyocytes in a mouse diabetic model [208]. Despite all of these exciting lines of evidence for a beneficial role of AMPK activation in cardiomyocytes under multiple stress conditions, the exact mechanisms by which AMPK protects cardiomyocytes against injury require further investigation.
Anti-inflammatory function of AMPK in CVD
Inflammation has been well known to play an important role in CVD development and progress [209], and inflammation might be a link between the metabolic syndrome and CVD [210]. Extensive evidence suggests that AMPK might be an inflammatory repressor. For example, activation of AMPK by AICAR or metformin have been reported to inhibit inflammation [211,212], whereas inhibition of AMPK leads to increased inflammation in obese humans [213] and in an AMPKα1 genetic knockdown cell culture model [214]. AMPK activation might inhibit inflammation through multiple pathways and recent evidence suggests that inhibition of the NF-κB pathway might play a central role for this inhibitory effect. For example, overexpression of AMPK has been demonstrated to inhibit the NF-κB pathway in endothelial cells [215]. In support of this, NF-κB activity was increased in endothelial cells isolated from AMPKα2-knockout mice [148]. Furthermore, adiponectin application inhibited AngII-induced cardiac hypertrophy in rats by decreasing NF-κB activity and this beneficial effect was blocked by AMPK inhibition [217]. In cultured HUVECs, metformin has been shown to inhibit TNF-α-induced IKK [IκB (inhibitory κB) kinase)α/β phosphorylation, IκBα degradation and IL (interleukin)-6 production in an AMPK-dependent manner. Most recently, Bess et al. [218] have reported that AMPK could contribute to the anti-inflammatory effect of NO in endothelial cells by directly phosphorylating IKK at Ser177 and Ser181. Despite all of these findings, whether there is a direct link between the AMPK/NF-κB pathway and CVD requires further investigation.
Cardioprotective effects of AMPK activators
Metformin lowers the risk of cardiovascular incidents in patients with Type 2 diabetes
Patients with diabetes are at increased risk of CVDs, such as atherosclerotic CHD (coronary heart disease), diabetic cardiomyopathy and stroke (for detailed information, see [219,220]). Metformin is one of the most widely prescribed antidiabetic medicines in the world. Interestingly, a large clinical study showed that patients with Type 2 diabetes treated with metformin have a 36 % lower risk of mortality and 39 % lower risk of myocardial infarction than those exposed to conventional treatment [221]. Similar results were reported later in a clinical study that focused on older patients (over 65 years of age) with diabetes and HF (heart failure) [222]. These results are supported further by a comprehensive study which suggests that metformin is the only antidiabetic medicine that is not associated with deleterious effects, but is instead associated with reduced all-cause mortality in patients with HF and diabetes [223]. The exact mechanisms of the beneficial effects of metformin are not fully known; however, results, including reports from our group, have suggested that AMPK activation is required for the cardiovascular protective effects of metformin [50,81,208]. In vivo administration of metformin increases AMPK phosphorylation in the mouse aorta, resulting in increased NO synthesis and bioavailability [50]. A further study has demonstrated that AMPK activation by metformin increases the association between Hsp90 (heat-shock protein 90) and eNOS (endothelial NO synthase), which reduces eNOS-derived O2− [224]. Although there is still considerable work to be done, current evidence suggests that patients with Type 2 diabetes with CVD will benefit from the activation of AMPK by metformin.
Protective role of statins in CVD
Statins are HMG-CoA reductase inhibitors used to lower cholesterol levels in patients. A clinical study has demonstrated that statin treatment reduced total stroke rates, including a substantial reduction in coronary event rates, in patients ranging widely in age [225]. In addition, a nationwide study of older patients with HF indicated that statin therapy significantly lowered mortality and improved outcomes in these patients [226]. The most recent clinical trial results further indicate that statins safely reduce the incidence of major atherosclerotic events in patients with advanced chronic kidney disease. Overall, the beneficial effect of statins in patients with CVD is unquestionable. Mechanistically, the beneficial effect of statins in the cardiovascular system derives from their cholesterol-lowering effect. Recent findings suggest that the direct beneficial effect of statins in the prevention of CVD might be mediated at the molecular level by AMPK. For instance, statins have been shown to increase AMPK activation and NO production in HUVECs and in mouse aortas and myocardium [227]. Similar results have been observed in bovine aortic endothelial cells [228]. Interestingly, the activation of AMPK by statins has been reported to promote the differentiation of EPCs (endothelial progenitor cells) by the activation of eNOS; these effects were blocked by the AMPK antagonist Compound C [229]. Thus activation of AMPK by statins might be an important mechanism involved in the beneficial effects of statins against CVDs. Despite exciting evidence on the role of statins in CVD, more work is required to define the mechanism of action of these compounds and the role of AMPK in its effects.
Other AMPK activators in CVD
As described above, several natural compounds are AMPK activators, including resveratrol and berberine. Studies in humans provide promising evidence of multiple beneficial effects of these two chemicals in both physiological and pathological conditions. For example, resveratrol can modulate cerebral blood flow and cognitive performance in healthy humans [230]. Furthermore, resveratrol supplementation has been reported to improve flow-mediated dilation of the brachial artery in 19 obese volunteers [231]. Consistent with these observations, a study has demonstrated the significant cardiovascular-protective effects of resveratrol in vitro and in vivo [232]. Similarly, berberine supplement has been reported to have beneficial cardiovascular effects in a study including 156 patients with chronic CHF (congestive HF) [233]. Importantly, a clinical study has reported that berberine might be used as a hypolipidaemic drug, as oral administration of berberine for 3 months reduced serum cholesterol by 29 %, triacylglycerols by 35 % and LDL (low-density lipoprotein)-cholesterol by 25 % in 32 hypercholesterolaemic patients [234]. Whether those clinical beneficial effects are AMPK-mediated, and the exact mechanisms by which these compounds activate AMPK, deserve more investigation.
CONCLUSIONS
Taken together, the information described above clearly indicates that AMPK functions far beyond its proposed energy sensor and regulator function. AMPK regulates ROS/redox balance, autophagy, cell proliferation, cell apoptosis, cellular polarity, mitochondria function and genotoxic response, either directly or indirectly via numerous downstream pathways. Further investigations are required to clarify the exact molecular targets of AMPK in physiological and pathological conditions and to develop potential strategies to modulate AMPK activation in diabetes, cancer, and cardiovascular dysfunction.
Abbreviations
- ACC
acetyl-CoA carboxylase
- AICAR
5-amino-4-imidazolecarboxamide riboside
- AMPK
AMP-activated protein kinase
- AngII
angiotension II
- CaMKKβ
Ca2 +/calmodulin-dependent protein kinase kinase β
- CBM
carbohydrate-binding module
- CBS
cystathionine β-synthase
- CDK
cyclin-dependent kinase
- CDKI
CDK inhibitor
- CIDEA
cell-death-inducing DNA fragmentation factor-α-like effector A
- CR
caloric restriction
- CREB
cAMP-response-element-binding protein
- CRTC
CREB-regulated transcriptional co-activator
- CVD
cardiovascular disease
- 2-DG
2-deoxy-D-glucose
- eNOS
endothelial NO synthase
- GLUT4
glucose transporter 4
- HF
heart failure
- HMG-CoA
3-hydroxy-3-methylglutaryl-CoA
- HIF
hypoxia-inducible factor
- HUVEC
human umbilical vein endothelial cell
- IκB
inhibitory κB
- IKK
IκB kinase
- LKB1
liver kinase B1
- LV
left ventricular
- LVH
LV hypertrophy
- MEF
mouse embryonic fibroblast
- NF-κB
nuclear factor κB
- NPY
neuropeptide Y
- O2−
superoxide anion
- ONOO−
peroxynitrite
- PKC
protein kinase C
- PP
protein phosphatase
- PPAR
peroxisome-proliferator-activated receptor
- PGC1
PPAR-γ co-activator 1
- RBC
red blood cell
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- siRNA
small interfering RNA
- Sirt
sirtuin
- SOD
superoxide dismutase
- Mn-SOD
manganese SOD
- TAK1
transforming growth factor-β-activated kinase 1
- TOR
target of rapamycin
- mTOR
mammalian TOR
- mTORC1
mTOR complex 1
- Trx
thioredoxin
- TSC2
tuberous sclerosis complex 2
- TZD
thiazolidinedione
- UCP
uncoupling protein
- ULK1
UNC-51-like kinase 1
- WPW
Wolff–Parkinson–White
References
- 1.Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors and stress. Mol Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calabrese V, Cornelius C, Mancuso C, Lentile R, Stella AM, Butterfield DA. Redox homeostasis and cellular stress response in aging and neurodegeneration. Methods Mol Biol. 2010;610:285–308. doi: 10.1007/978-1-60327-029-8_17. [DOI] [PubMed] [Google Scholar]
- 3.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 6.Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase – development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 8.Hardie DG, Carling D. The AMP-activated protein kinase – fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–273. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- 9.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 10.Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5′-AMP for allosteric stimulation, activation and deactivation of AMP-activated protein kinase. J Biol Chem. 2006;281:32207–32216. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- 11.Pang T, Xiong B, Li JY, Qiu BY, Jin GZ, Shen JK, Li J. Conserved α-helix acts as autoinhibitory sequence in AMP-activated protein kinase α subunits. J Biol Chem. 2007;282:495–506. doi: 10.1074/jbc.M605790200. [DOI] [PubMed] [Google Scholar]
- 12.Crute BE, Seefeld K, Gamble J, Kemp BE, Witters LA. Functional domains of the α1 catalytic subunit of the AMP-activated protein kinase. J Biol Chem. 1998;273:35347–35354. doi: 10.1074/jbc.273.52.35347. [DOI] [PubMed] [Google Scholar]
- 13.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hurley RL, Barre LK, Wood SD, Anderson KA, Kemp BE, Means AR, Witters LA. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J Biol Chem. 2006;281:36662–36672. doi: 10.1074/jbc.M606676200. [DOI] [PubMed] [Google Scholar]
- 15.Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, House CM, Fernandez CS, Cox T, Witters LA, Kemp BE. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996;271:611–614. doi: 10.1074/jbc.271.2.611. [DOI] [PubMed] [Google Scholar]
- 16.Morrow VA, Foufelle F, Connell JM, Petrie JR, Gould GW, Salt IP. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem. 2003;278:31629–31639. doi: 10.1074/jbc.M212831200. [DOI] [PubMed] [Google Scholar]
- 17.Song P, Wang S, He C, Liang B, Viollet B, Zou MH. AMPKα2 deletion exacerbates neointima formation by upregulating Skp2 in vascular smooth muscle cells. Circ Res. 2011;109:1230–1239. doi: 10.1161/CIRCRESAHA.111.250423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, Choi HC, Zou MH. Reduction of AMP-activated protein kinase α2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. 2010;121:792–803. doi: 10.1161/CIRCULATIONAHA.109.900928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Dale GL, Song P, Viollet B, Zou MH. AMPKα1 deletion shortens erythrocyte life span in mice: role of oxidative stress. J Biol Chem. 2010;285:19976–19985. doi: 10.1074/jbc.M110.102467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie DG. AMP-activated protein kinase: greater AMP dependence and preferential nuclear localization, of complexes containing the α2 isoform. Biochem J. 1998;334:177–187. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polekhina G, Gupta A, Michell BJ, van Denderen B, Murthy S, Feil SC, Jennings IG, Campbell DJ, Witters LA, Parker MW, et al. AMPK β subunit targets metabolic stress sensing to glycogen. Curr Biol. 2003;13:867–871. doi: 10.1016/s0960-9822(03)00292-6. [DOI] [PubMed] [Google Scholar]
- 22.Woods A, Cheung PC, Smith FC, Davison MD, Scott J, Beri RK, Carling D. Characterization of AMP-activated protein kinase β and γsubunits. Assembly of the heterotrimeric complex in vitro. J Biol Chem. 1996;271:10282–10290. doi: 10.1074/jbc.271.17.10282. [DOI] [PubMed] [Google Scholar]
- 23.Iseli TJ, Walter M, van Denderen BJ, Katsis F, Witters LA, Kemp BE, Michell BJ, Stapleton D. AMP-activated protein kinase β subunit tethers α and γsubunits via its C-terminal sequence (186–270) J Biol Chem. 2005;280:13395–13400. doi: 10.1074/jbc.M412993200. [DOI] [PubMed] [Google Scholar]
- 24.Oakhill JS, Chen ZP, Scott JW, Steel R, Castelli LA, Ling N, Macaulay SL, Kemp BE. β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK) Proc Natl Acad Sci USA. 2010;107:19237–19241. doi: 10.1073/pnas.1009705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449:496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- 26.Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase γ-subunit isoforms and their role in AMP binding. Biochem J. 2000;346:659–669. [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007;282:9777–9788. doi: 10.1074/jbc.M608310200. [DOI] [PubMed] [Google Scholar]
- 29.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 30.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD α/β and MO25 α/β are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 32.Xie Z, Dong Y, Zhang J, Scholz R, Neumann D, Zou MH. Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Mol Cell Biol. 2009;29:3582–3596. doi: 10.1128/MCB.01417-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, Alessi DR, Clevers HC. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 2003;22:3062–3072. doi: 10.1093/emboj/cdg292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, Prescott AR, Clevers HC, Alessi DR. MO25α/β interact with STRADα/β enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003;22:5102–5114. doi: 10.1093/emboj/cdg490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2 +/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 36.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2 +/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 38.Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- 39.Davies SP, Helps NR, Cohen PT, Hardie DG. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C α and native bovine protein phosphatase-2AC. FEBS Lett. 1995;377:421–425. doi: 10.1016/0014-5793(95)01368-7. [DOI] [PubMed] [Google Scholar]
- 40.Ravnskjaer K, Boergesen M, Dalgaard LT, Mandrup S. Glucose-induced repression of PPARα gene expression in pancreatic β-cells involves PP2A activation and AMPK inactivation. J Mol Endocrinol. 2006;36:289–299. doi: 10.1677/jme.1.01965. [DOI] [PubMed] [Google Scholar]
- 41.Qi J, Gong J, Zhao T, Zhao J, Lam P, Ye J, Li JZ, Wu J, Zhou HM, Li P. Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. EMBO J. 2008;27:1537–1548. doi: 10.1038/emboj.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, Hardie DG. Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11:554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15:660–666. doi: 10.1038/sj.cdd.4402307. [DOI] [PubMed] [Google Scholar]
- 44.Jibb LA, Richards JG. AMP-activated protein kinase activity during metabolic rate depression in the hypoxic goldfish, Carassius auratus. J Exp Biol. 2008;211:3111–3122. doi: 10.1242/jeb.019117. [DOI] [PubMed] [Google Scholar]
- 45.Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10:1247–1255. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 46.Liu C, Liang B, Wang Q, Wu J, Zou MH. Activation of AMP-activated protein kinase α1 alleviates endothelial cell apoptosis by increasing the expression of anti-apoptotic proteins Bcl-2 and survivin. J Biol Chem. 2010;285:15346–15355. doi: 10.1074/jbc.M110.102491. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. BioEssays. 2001;23:1112–1119. doi: 10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- 48.Zou MH, Shi C, Cohen RA. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H2 receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes. 2002;51:198–203. doi: 10.2337/diabetes.51.1.198. [DOI] [PubMed] [Google Scholar]
- 49.Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. 2002;109:817–826. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zou MH, Kirkpatrick SS, Davis BJ, Nelson JS, Wiles WGt, Schlattner U, Neumann D, Brownlee M, Freeman MB, Goldman MH. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem. 2004;279:43940–43951. doi: 10.1074/jbc.M404421200. [DOI] [PubMed] [Google Scholar]
- 51.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr Opin Cell Biol. 2009;21:894–899. doi: 10.1016/j.ceb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–5347. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zou MH, Hou XY, Shi CM, Kirkpatick S, Liu F, Goldman MH, Cohen RA. Activation of 5′-AMP-activated kinase is mediated through c-Src and phosphoinositide 3-kinase activity during hypoxia-reoxygenation of bovine aortic endothelial cells. Role of peroxynitrite. J Biol Chem. 2003;278:34003–34010. doi: 10.1074/jbc.M300215200. [DOI] [PubMed] [Google Scholar]
- 54.Xie Z, Dong Y, Scholz R, Neumann D, Zou MH. Phosphorylation of LKB1 at serine 428 by protein kinase C-ζ is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation. 2008;117:952–962. doi: 10.1161/CIRCULATIONAHA.107.744490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol. 2011;31:3531–3545. doi: 10.1128/MCB.05124-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee M, Hwang JT, Lee HJ, Jung SN, Kang I, Chi SG, Kim SS, Ha J. AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. J Biol Chem. 2003;278:39653–39661. doi: 10.1074/jbc.M306104200. [DOI] [PubMed] [Google Scholar]
- 57.Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3 and BNIP3L. Cell Death Differ. 2008;15:1572–1581. doi: 10.1038/cdd.2008.84. [DOI] [PubMed] [Google Scholar]
- 58.Goodyear LJ. The exercise pill – too good to be true? N Engl J Med. 2008;359:1842–1844. doi: 10.1056/NEJMcibr0806723. [DOI] [PubMed] [Google Scholar]
- 59.Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J. 2009;418:261–275. doi: 10.1042/BJ20082055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol. 1996;270:E299–E304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- 61.Mu J, Barton ER, Birnbaum MJ. Selective suppression of AMP-activated protein kinase in skeletal muscle: update on ‘lazy mice’. Biochem Soc Trans. 2003;31:236–241. doi: 10.1042/bst0310236. [DOI] [PubMed] [Google Scholar]
- 62.Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation and glucose uptake in rat muscle. Am J Physiol. 1997;273:E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- 63.Iglesias MA, Ye JM, Frangioudakis G, Saha AK, Tomas E, Ruderman NB, Cooney GJ, Kraegen EW. AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes. 2002;51:2886–2894. doi: 10.2337/diabetes.51.10.2886. [DOI] [PubMed] [Google Scholar]
- 64.Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, et al. AMPK and PPARδ agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suwa M, Nakano H, Kumagai S. Effects of chronic AICAR treatment on fiber composition, enzyme activity, UCP3 and PGC-1 in rat muscles. J Appl Physiol. 2003;95:960–968. doi: 10.1152/japplphysiol.00349.2003. [DOI] [PubMed] [Google Scholar]
- 66.Rockl KS, Hirshman MF, Brandauer J, Fujii N, Witters LA, Goodyear LJ. Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift. Diabetes. 2007;56:2062–2069. doi: 10.2337/db07-0255. [DOI] [PubMed] [Google Scholar]
- 67.Reference deleted
- 68.O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jorgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, et al. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci USA. 2011;108:16092–16097. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smerdu V, Karsch-Mizrachi I, Campione M, Leinwand L, Schiaffino S. Type IIx myosin heavy chain transcripts are expressed in type IIb fibers of human skeletal muscle. Am J Physiol. 1994;267:C1723–C1728. doi: 10.1152/ajpcell.1994.267.6.C1723. [DOI] [PubMed] [Google Scholar]
- 70.Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5′-AMP-activated protein kinase increases GLUT-4, hexokinase and glycogen in muscle. J Appl Physiol. 1999;87:1990–1995. doi: 10.1152/jappl.1999.87.5.1990. [DOI] [PubMed] [Google Scholar]
- 71.Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 72.Steinberg GR, Rush JW, Dyck DJ. AMPK expression and phosphorylation are increased in rodent muscle after chronic leptin treatment. Am J Physiol Endocrinol Metab. 2003;284:E648–E654. doi: 10.1152/ajpendo.00318.2002. [DOI] [PubMed] [Google Scholar]
- 73.Liao Y, Takashima S, Maeda N, Ouchi N, Komamura K, Shimomura I, Hori M, Matsuzawa Y, Funahashi T, Kitakaze M. Exacerbation of heart failure in adiponectin-deficient mice due to impaired regulation of AMPK and glucose metabolism. Cardiovasc Res. 2005;67:705–713. doi: 10.1016/j.cardiores.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 74.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Ouchi N, Walsh K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–1103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci USA. 2007;104:7217–7222. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Han Y, Wang Q, Song P, Zhu Y, Zou MH. Redox regulation of the AMP-activated protein kinase. PLoS ONE. 2010;5:e15420. doi: 10.1371/journal.pone.0015420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, Ye JM, Lee CH, Oh WK, Kim CT, et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes. 2006;55:2256–2264. doi: 10.2337/db06-0006. [DOI] [PubMed] [Google Scholar]
- 78.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 79.Chen K, Xu X, Kobayashi S, Timm D, Jepperson T, Liang Q. Caloric restriction mimetic 2-deoxyglucose antagonizes doxorubicin-induced cardiomyocyte death by multiple mechanisms. J Biol Chem. 2011;286:21993–22006. doi: 10.1074/jbc.M111.225805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Q, Liang B, Shirwany NA, Zou MH. 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS ONE. 2011;6:e17234. doi: 10.1371/journal.pone.0017234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.LeBrasseur NK, Kelly M, Tsao TS, Farmer SR, Saha AK, Ruderman NB, Tomas E. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am J Physiol Endocrinol Metab. 2006;291:E175–E181. doi: 10.1152/ajpendo.00453.2005. [DOI] [PubMed] [Google Scholar]
- 83.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 84.Anderson SN, Cool BL, Kifle L, Chiou W, Egan DA, Barrett LW, Richardson PL, Frevert EU, Warrior U, Kofron JL, Burns DJ. Microarrayed compound screening (microARCS) to identify activators and inhibitors of AMP-activated protein kinase. J Biomol Screen. 2004;9:112–121. doi: 10.1177/1087057103260592. [DOI] [PubMed] [Google Scholar]
- 85.Pang T, Zhang ZS, Gu M, Qiu BY, Yu LF, Cao PR, Shao W, Su MB, Li JY, Nan FJ, Li J. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J Biol Chem. 2008;283:16051–16060. doi: 10.1074/jbc.M710114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guigas B, Bertrand L, Taleux N, Foretz M, Wiernsperger N, Vertommen D, Andreelli F, Viollet B, Hue L. 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes. 2006;55:865–874. doi: 10.2337/diabetes.55.04.06.db05-1178. [DOI] [PubMed] [Google Scholar]
- 87.Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest. 2006;116:1776–1783. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kraegen EW, Saha AK, Preston E, Wilks D, Hoy AJ, Cooney GJ, Ruderman NB. Increased malonyl-CoA and diacylglycerol content and reduced AMPK activity accompany insulin resistance induced by glucose infusion in muscle and liver of rats. Am J Physiol Endocrinol Metab. 2006;290:E471–E479. doi: 10.1152/ajpendo.00316.2005. [DOI] [PubMed] [Google Scholar]
- 90.Zang M, Zuccollo A, Hou X, Nagata D, Walsh K, Herscovitz H, Brecher P, Ruderman NB, Cohen RA. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem. 2004;279:47898–47905. doi: 10.1074/jbc.M408149200. [DOI] [PubMed] [Google Scholar]
- 91.Wojtaszewski JF, Jorgensen SB, Hellsten Y, Hardie DG, Richter EA. Glycogen-dependent effects of 5-aminoimidazole-4-carboxamide (AICA)-riboside on AMP-activated protein kinase and glycogen synthase activities in rat skeletal muscle. Diabetes. 2002;51:284–292. doi: 10.2337/diabetes.51.2.284. [DOI] [PubMed] [Google Scholar]
- 92.McBride A, Ghilagaber S, Nikolaev A, Hardie DG. The glycogen-binding domain on the AMPK β subunit allows the kinase to act as a glycogen sensor. Cell Metab. 2009;9:23–34. doi: 10.1016/j.cmet.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, Giri S, Andreelli F. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol. 2010;45:276–295. doi: 10.3109/10409238.2010.488215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ropelle ER, Pauli JR, Fernandes MF, Rocco SA, Marin RM, Morari J, Souza KK, Dias MM, Gomes-Marcondes MC, Gontijo JA, et al. A central role for neuronal AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) in high-protein diet-induced weight loss. Diabetes. 2008;57:594–605. doi: 10.2337/db07-0573. [DOI] [PubMed] [Google Scholar]
- 95.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 96.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vogt J, Traynor R, Sapkota GP. The specificities of small molecule inhibitors of the TGFss and BMP pathways. Cell Signalling. 2011;23:1831–1842. doi: 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 98.Fontana L, Partridge L, Longo VD. Extending healthy life span – from yeast to humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ashrafi K, Lin SS, Manchester JK, Gordon JI. Sip2p and its partner snf1p kinase affect aging in S. cerevisiae. Genes Dev. 2000;14:1872–1885. [PMC free article] [PubMed] [Google Scholar]
- 101.Apfeld J, O’Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–156. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A, Alessi DR. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J. 2005;24:1810–1820. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Canto C, Auwerx J. Calorie restriction: is AMPK a key sensor and effector? Physiology. 2011;26:214–224. doi: 10.1152/physiol.00010.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 106.Houtkooper RH, Williams RW, Auwerx J. Metabolic networks of longevity. Cell. 2010;142:9–14. doi: 10.1016/j.cell.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Um JH, Park SJ, Kang H, Yang S, Foretz M, McBurney MW, Kim MK, Viollet B, Chung JH. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes. 2010;59:554–563. doi: 10.2337/db09-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 109.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD + metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lage R, Dieguez C, Vidal-Puig A, Lopez M. AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med. 2008;14:539–549. doi: 10.1016/j.molmed.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 111.McGee SL, van Denderen BJ, Howlett KF, Mollica J, Schertzer JD, Kemp BE, Hargreaves M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes. 2008;57:860–867. doi: 10.2337/db07-0843. [DOI] [PubMed] [Google Scholar]
- 112.Chopra AR, Kommagani R, Saha P, Louet JF, Salazar C, Song J, Jeong J, Finegold M, Viollet B, Demayo F, et al. Cellular energy depletion resets whole-body energy by promoting coactivator-mediated dietary fuel absorption. Cell Metab. 2011;13:35–43. doi: 10.1016/j.cmet.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 114.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Egan DF, Shackelford DB, Mihaylova MM, Gelino SR, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 117.Mair W, Morantte I, Rodrigues AP, Manning G, Montminy M, Shaw RJ, Dillin A. Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature. 2011;470:404–408. doi: 10.1038/nature09706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fujii N, Jessen N, Goodyear LJ. AMP-activated protein kinase and the regulation of glucose transport. Am J Physiol Endocrinol Metab. 2006;291:E867–E877. doi: 10.1152/ajpendo.00207.2006. [DOI] [PubMed] [Google Scholar]
- 119.Fujii N, Aschenbach WG, Musi N, Hirshman MF, Goodyear LJ. Regulation of glucose transport by the AMP-activated protein kinase. Proc Nutr Soc. 2004;63:205–210. doi: 10.1079/PNS2004340. [DOI] [PubMed] [Google Scholar]
- 120.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci USA. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Garcia-Roves PM, Osler ME, Holmstrom MH, Zierath JR. Gain-of-function R225Q mutation in AMP-activated protein kinase γ3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J Biol Chem. 2008;283:35724–35734. doi: 10.1074/jbc.M805078200. [DOI] [PubMed] [Google Scholar]
- 122.Orci L, Cook WS, Ravazzola M, Wang MY, Park BH, Montesano R, Unger RH. Rapid transformation of white adipocytes into fat-oxidizing machines. Proc Natl Acad Sci USA. 2004;101:2058–2063. doi: 10.1073/pnas.0308258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wu X, Motoshima H, Mahadev K, Stalker TJ, Scalia R, Goldstein BJ. Involvement of AMP-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes. 2003;52:1355–1363. doi: 10.2337/diabetes.52.6.1355. [DOI] [PubMed] [Google Scholar]
- 124.Viollet B, Guigas B, Leclerc J, Hebrard S, Lantier L, Mounier R, Andreelli F, Foretz M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol. 2009;196:81–98. doi: 10.1111/j.1748-1716.2009.01970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Andreelli F, Foretz M, Knauf C, Cani PD, Perrin C, Iglesias MA, Pillot B, Bado A, Tronche F, Mithieux G, et al. Liver adenosine monophosphate-activated kinase-α2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology. 2006;147:2432–2441. doi: 10.1210/en.2005-0898. [DOI] [PubMed] [Google Scholar]
- 126.Lochhead PA, Salt IP, Walker KS, Hardie DG, Sutherland C. 5-Aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes. 2000;49:896–903. doi: 10.2337/diabetes.49.6.896. [DOI] [PubMed] [Google Scholar]
- 127.Horike N, Sakoda H, Kushiyama A, Ono H, Fujishiro M, Kamata H, Nishiyama K, Uchijima Y, Kurihara Y, Kurihara H, Asano T. AMP-activated protein kinase activation increases phosphorylation of glycogen synthase kinase 3β and thereby reduces cAMP-responsive element transcriptional activity and phosphoenolpyruvate carboxykinase C gene expression in the liver. J Biol Chem. 2008;283:33902–33910. doi: 10.1074/jbc.M802537200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Viollet B, Foretz M, Guigas B, Horman S, Dentin R, Bertrand L, Hue L, Andreelli F. Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J Physiol. 2006;574:41–53. doi: 10.1113/jphysiol.2006.108506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Daval M, Foufelle F, Ferre P. Functions of AMP-activated protein kinase in adipose tissue. J Physiol. 2006;574:55–62. doi: 10.1113/jphysiol.2006.111484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Salt IP, Johnson G, Ashcroft SJ, Hardie DG. AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic β cells and may regulate insulin release. Biochem J. 1998;335:533–539. doi: 10.1042/bj3350533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.da Silva Xavier G, Leclerc I, Salt IP, Doiron B, Hardie DG, Kahn A, Rutter GA. Role of AMP-activated protein kinase in the regulation by glucose of islet β cell gene expression. Proc Natl Acad Sci USA. 2000;97:4023–4028. doi: 10.1073/pnas.97.8.4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 133.Sies H. Biological redox systems and oxidative stress. Cell Mol Life Sci. 2007;64:2181–2188. doi: 10.1007/s00018-007-7230-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lamberts RR, Onderwater G, Hamdani N, Vreden MJ, Steenhuisen J, Eringa EC, Loer SA, Stienen GJ, Bouwman RA. Reactive oxygen species-induced stimulation of 5′AMP-activated protein kinase mediates sevoflurane-induced cardioprotection. Circulation. 2009;120:S10–S15. doi: 10.1161/CIRCULATIONAHA.108.828426. [DOI] [PubMed] [Google Scholar]
- 136.Toyoda T, Hayashi T, Miyamoto L, Yonemitsu S, Nakano M, Tanaka S, Ebihara K, Masuzaki H, Hosoda K, Inoue G, et al. Possible involvement of the α1 isoform of 5′AMP-activated protein kinase in oxidative stress-stimulated glucose transport in skeletal muscle. Am J Physiol Endocrinol Metab. 2004;287:E166–E173. doi: 10.1152/ajpendo.00487.2003. [DOI] [PubMed] [Google Scholar]
- 137.Emerling BM, Weinberg F, Snyder C, Burgess Z, Mutlu GM, Viollet B, Budinger GR, Chandel NS. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radical Biol Med. 2009;46:1386–1391. doi: 10.1016/j.freeradbiomed.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Choi SL, Kim SJ, Lee KT, Kim J, Mu J, Birnbaum MJ, Soo Kim S, Ha J. The regulation of AMP-activated protein kinase by H2O2. Biochem Biophys Res Commun. 2001;287:92–97. doi: 10.1006/bbrc.2001.5544. [DOI] [PubMed] [Google Scholar]
- 139.Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem. 2010;285:33154–33164. doi: 10.1074/jbc.M110.143685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xie Z, Dong Y, Zhang M, Cui MZ, Cohen RA, Riek U, Neumann D, Schlattner U, Zou MH. Activation of protein kinase Cζ by peroxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J Biol Chem. 2006;281:6366–6375. doi: 10.1074/jbc.M511178200. [DOI] [PubMed] [Google Scholar]
- 141.Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–1662. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- 143.Gao L, Mann GE. Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc Res. 2009;82:9–20. doi: 10.1093/cvr/cvp031. [DOI] [PubMed] [Google Scholar]
- 144.Brandes RP, Weissmann N, Schroder K. NADPH oxidases in cardiovascular disease. Free Radical Biol Med. 2010;49:687–706. doi: 10.1016/j.freeradbiomed.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 145.Alba G, El Bekay R, Alvarez-Maqueda M, Chacon P, Vega A, Monteseirin J, Santa Maria C, Pintado E, Bedoya FJ, Bartrons R, Sobrino F. Stimulators of AMP-activated protein kinase inhibit the respiratory burst in human neutrophils. FEBS Lett. 2004;573:219–225. doi: 10.1016/j.febslet.2004.07.077. [DOI] [PubMed] [Google Scholar]
- 146.Hwang J, Kleinhenz DJ, Rupnow HL, Campbell AG, Thule PM, Sutliff RL, Hart CM. The PPARγ ligand, rosiglitazone, reduces vascular oxidative stress and NADPH oxidase expression in diabetic mice. Vasc Pharmacol. 2007;46:456–462. doi: 10.1016/j.vph.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 147.Piwkowska A, Rogacka D, Jankowski M, Dominiczak MH, Stepinski JK, Angielski S. Metformin induces suppression of NAD(P)H oxidase activity in podocytes. Biochem Biophys Res Commun. 2010;393:268–273. doi: 10.1016/j.bbrc.2010.01.119. [DOI] [PubMed] [Google Scholar]
- 148.Wang S, Zhang M, Liang B, Xu J, Xie Z, Liu C, Viollet B, Yan D, Zou MH. AMPKα2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes. Circ Res. 2010;106:1117–1128. doi: 10.1161/CIRCRESAHA.109.212530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schuhmacher S, Foretz M, Knorr M, Jansen T, Hortmann M, Wenzel P, Oelze M, Kleschyov AL, Daiber A, Keaney JF, Jr, et al. α1AMP-activated protein kinase preserves endothelial function during chronic angiotensin II treatment by limiting Nox2 upregulation. Arterioscler Thromb Vasc Biol. 2011;31:560–566. doi: 10.1161/ATVBAHA.110.219543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011;208:417–420. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- 152.Leloup C, Casteilla L, Carriere A, Galinier A, Benani A, Carneiro L, Penicaud L. Balancing mitochondrial redox signaling: a key point in metabolic regulation. Antioxid Redox Signaling. 2011;14:519–530. doi: 10.1089/ars.2010.3424. [DOI] [PubMed] [Google Scholar]
- 153.Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005;2:85–93. doi: 10.1016/j.cmet.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 154.Xie Z, Zhang J, Wu J, Viollet B, Zou MH. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes. 2008;57:3222–3230. doi: 10.2337/db08-0610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 155.Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschop MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, Horvath TL, Diano S. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454:846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wang Y, Nishi M, Doi A, Shono T, Furukawa Y, Shimada T, Furuta H, Sasaki H, Nanjo K. Ghrelin inhibits insulin secretion through the AMPK-UCP2 pathway in β cells. FEBS Lett. 2010;584:1503–1508. doi: 10.1016/j.febslet.2010.02.069. [DOI] [PubMed] [Google Scholar]
- 157.Calegari VC, Zoppi CC, Rezende LF, Silveira LR, Carneiro EM, Boschero AC. Endurance training activates AMP-activated protein kinase, increases expression of uncoupling protein 2 and reduces insulin secretion from rat pancreatic islets. J Endocrinol. 2011;208:257–264. doi: 10.1530/JOE-10-0450. [DOI] [PubMed] [Google Scholar]
- 158.Beall C, Piipari K, Al-Qassab H, Smith MA, Parker N, Carling D, Viollet B, Withers DJ, Ashford ML. Loss of AMP-activated protein kinase α2 subunit in mouse β-cells impairs glucose-stimulated insulin secretion and inhibits their sensitivity to hypoglycaemia. Biochem J. 2010;429:323–333. doi: 10.1042/BJ20100231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, Rovira II, Gutkind S, Daniels MP, Komatsu M, Finkel T. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging. 2009;1:425–437. doi: 10.18632/aging.100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mortensen M, Ferguson DJ, Simon AK. Mitochondrial clearance by autophagy in developing erythrocytes: clearly important, but just how much so? Cell Cycle. 2010;9:1901–1906. doi: 10.4161/cc.9.10.11603. [DOI] [PubMed] [Google Scholar]
- 165.Roach PJ. AMPK→ULK1→autophagy. Mol Cell Biol. 2011;31:3082–3084. doi: 10.1128/MCB.05565-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Li XN, Song J, Zhang L, LeMaire SA, Hou X, Zhang C, Coselli JS, Chen L, Wang XL, Zhang Y, Shen YH. Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes. 2009;58:2246–2257. doi: 10.2337/db08-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, Yano M, Motoshima H, Taguchi T, Matsumura T, Araki E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55:120–127. [PubMed] [Google Scholar]
- 169.Colombo SL, Moncada S. AMPKα1 regulates the antioxidant status of vascular endothelial cells. Biochem J. 2009;421:163–169. doi: 10.1042/BJ20090613. [DOI] [PubMed] [Google Scholar]
- 170.Grana X, Reddy EP. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs) Oncogene. 1995;11:211–219. [PubMed] [Google Scholar]
- 171.Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001;287:562–567. doi: 10.1006/bbrc.2001.5627. [DOI] [PubMed] [Google Scholar]
- 172.Igata M, Motoshima H, Tsuruzoe K, Kojima K, Matsumura T, Kondo T, Taguchi T, Nakamaru K, Yano M, Kukidome D, et al. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ Res. 2005;97:837–844. doi: 10.1161/01.RES.0000185823.73556.06. [DOI] [PubMed] [Google Scholar]
- 173.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277:23977–23980. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 174.Xiang X, Saha AK, Wen R, Ruderman NB, Luo Z. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem Biophys Res Commun. 2004;321:161–167. doi: 10.1016/j.bbrc.2004.06.133. [DOI] [PubMed] [Google Scholar]
- 175.Henin N, Vincent MF, Gruber HE, Van den Berghe G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J. 1995;9:541–546. doi: 10.1096/fasebj.9.7.7737463. [DOI] [PubMed] [Google Scholar]
- 176.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 177.Dagon Y, Avraham Y, Berry EM. AMPK activation regulates apoptosis, adipogenesis and lipolysis by eIF2α in adipocytes. Biochem Biophys Res Commun. 2006;340:43–47. doi: 10.1016/j.bbrc.2005.11.159. [DOI] [PubMed] [Google Scholar]
- 178.Santidrian AF, Gonzalez-Girones DM, Iglesias-Serret D, Coll-Mulet L, Cosialls AM, de Frias M, Campas C, Gonzalez-Barca E, Alonso E, Labi V, et al. AICAR induces apoptosis independently of AMPK and p53 through up-regulation of the BH3-only proteins BIM and NOXA in chronic lymphocytic leukemia cells. Blood. 2010;116:3023–3032. doi: 10.1182/blood-2010-05-283960. [DOI] [PubMed] [Google Scholar]
- 179.Hwang JT, Kwak DW, Lin SK, Kim HM, Kim YM, Park OJ. Resveratrol induces apoptosis in chemoresistant cancer cells via modulation of AMPK signaling pathway. Ann NY Acad Sci. 2007;1095:441–448. doi: 10.1196/annals.1397.047. [DOI] [PubMed] [Google Scholar]
- 180.Hwang JT, Kwon DY, Park OJ, Kim MS. Resveratrol protects ROS-induced cell death by activating AMPK in H9c2 cardiac muscle cells. Genes Nutr. 2008;2:323–326. doi: 10.1007/s12263-007-0069-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Asai T, Liu Y, Bae N, Nimer SD. The p53 tumor suppressor protein regulates hematopoietic stem cell fate. J Cell Physiol. 2011;226:2215–2221. doi: 10.1002/jcp.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 183.Moll UM, Zaika A. Nuclear and mitochondrial apoptotic pathways of p53. FEBS Lett. 2001;493:65–69. doi: 10.1016/s0014-5793(01)02284-0. [DOI] [PubMed] [Google Scholar]
- 184.Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Sem Cell Dev Biol. 2010;21:683–690. doi: 10.1016/j.semcdb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 185.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 186.Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–1635. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Shirwany NA, Zou MH. AMPK in cardiovascular health and disease. Acta Pharmacol Sin. 2010;31:1075–1084. doi: 10.1038/aps.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gollob MH, Green MS, Tang AS, Roberts R. PRKAG2 cardiac syndrome: familial ventricular preexcitation, conduction system disease and cardiac hypertrophy. Curr Opin Cardiol. 2002;17:229–234. doi: 10.1097/00001573-200205000-00004. [DOI] [PubMed] [Google Scholar]
- 190.MacRae CA, Ghaisas N, Kass S, Donnelly S, Basson CT, Watkins HC, Anan R, Thierfelder LH, McGarry K, Rowland E, et al. Familial Hypertrophic cardiomyopathy with Wolff–Parkinson–White syndrome maps to a locus on chromosome 7q3. J Clin Invest. 1995;96:1216–1220. doi: 10.1172/JCI118154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, Salmon A, Ostman-Smith I, Watkins H. Mutations in the γ2 subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. 2001;10:1215–1220. doi: 10.1093/hmg/10.11.1215. [DOI] [PubMed] [Google Scholar]
- 192.Gollob MH, Green MS, Tang AS, Gollob T, Karibe A, Ali Hassan AS, Ahmad F, Lozado R, Shah G, Fananapazir L, et al. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med. 2001;344:1823–1831. doi: 10.1056/NEJM200106143442403. [DOI] [PubMed] [Google Scholar]
- 193.Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circ Res. 2007;100:474–488. doi: 10.1161/01.RES.0000258446.23525.37. [DOI] [PubMed] [Google Scholar]
- 194.Arad M, Benson DW, Perez-Atayde AR, McKenna WJ, Sparks EA, Kanter RJ, McGarry K, Seidman JG, Seidman CE. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest. 2002;109:357–362. doi: 10.1172/JCI14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Davies JK, Wells DJ, Liu K, Whitrow HR, Daniel TD, Grignani R, Lygate CA, Schneider JE, Noel G, Watkins H, Carling D. Characterization of the role of γ2 R531G mutation in AMP-activated protein kinase in cardiac hypertrophy and Wolff–Parkinson–White syndrome. Am J Physiol Heart Circ Physiol. 2006;290:H1942–H1951. doi: 10.1152/ajpheart.01020.2005. [DOI] [PubMed] [Google Scholar]
- 196.Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez-Atayde AR, Sawyer DB, Walter M, Li GH, Burgon PG, Maguire CT, et al. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff–Parkinson–White syndrome in glycogen storage cardiomyopathy. Circulation. 2003;107:2850–2856. doi: 10.1161/01.CIR.0000075270.13497.2B. [DOI] [PubMed] [Google Scholar]
- 197.Sidhu JS, Rajawat YS, Rami TG, Gollob MH, Wang Z, Yuan R, Marian AJ, DeMayo FJ, Weilbacher D, Taffet GE, et al. Transgenic mouse model of ventricular preexcitation and atrioventricular reentrant tachycardia induced by an AMP-activated protein kinase loss-of-function mutation responsible for Wolff–Parkinson–White syndrome. Circulation. 2005;111:21–29. doi: 10.1161/01.CIR.0000151291.32974.D5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Patel VV, Arad M, Moskowitz IP, Maguire CT, Branco D, Seidman JG, Seidman CE, Berul CI. Electrophysiologic characterization and postnatal development of ventricular pre-excitation in a mouse model of cardiac hypertrophy and Wolff–Parkinson–White syndrome. J Am Coll Cardiol. 2003;42:942–951. doi: 10.1016/s0735-1097(03)00850-7. [DOI] [PubMed] [Google Scholar]
- 199.Arad M, Maron BJ, Gorham JM, Johnson WH, Jr, Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, Seidman JG. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med. 2005;352:362–372. doi: 10.1056/NEJMoa033349. [DOI] [PubMed] [Google Scholar]
- 200.Carattino MD, Edinger RS, Grieser HJ, Wise R, Neumann D, Schlattner U, Johnson JP, Kleyman TR, Hallows KR. Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J Biol Chem. 2005;280:17608–17616. doi: 10.1074/jbc.M501770200. [DOI] [PubMed] [Google Scholar]
- 201.Hallows KR, Raghuram V, Kemp BE, Witters LA, Foskett JK. Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J Clin Invest. 2000;105:1711–1721. doi: 10.1172/JCI9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kalyanaraman B, Joseph J, Kalivendi S, Wang S, Konorev E, Kotamraju S. Doxorubicin-induced apoptosis: implications in cardiotoxicity. Mol Cell Biochem. 2002;234–235:119–124. [PubMed] [Google Scholar]
- 203.Tokarska-Schlattner M, Zaugg M, da Silva R, Lucchinetti E, Schaub MC, Wallimann T, Schlattner U. Acute toxicity of doxorubicin on isolated perfused heart: response of kinases regulating energy supply. Am J Physiol Heart Circ Physiol. 2005;289:H37–H47. doi: 10.1152/ajpheart.01057.2004. [DOI] [PubMed] [Google Scholar]
- 204.Asensio-Lopez MC, Lax A, Pascual-Figal DA, Valdes M, Sanchez-Mas J. Metformin protects against doxorubicin-induced cardiotoxicity: Involvement of the adiponectin cardiac system. Free Radical Biol Med. 2011;51:1861–1871. doi: 10.1016/j.freeradbiomed.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 205.Konishi M, Haraguchi G, Ohigashi H, Ishihara T, Saito K, Nakano Y, Isobe M. Adiponectin protects against doxorubicin-induced cardiomyopathy by anti-apoptotic effects through AMPK up-regulation. Cardiovasc Res. 2011;89:309–319. doi: 10.1093/cvr/cvq335. [DOI] [PubMed] [Google Scholar]
- 206.Russell RR, III, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Xie Z, He C, Zou MH. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy. 2011;7:1254–1255. doi: 10.4161/auto.7.10.16740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hansson GK. Inflammation, atherosclerosis and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 210.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 211.Zhao X, Zmijewski JW, Lorne E, Liu G, Park YJ, Tsuruta Y, Abraham E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L497–L504. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Akbar DH. Effect of metformin and sulfonylurea on C-reactive protein level in well-controlled type 2 diabetics with metabolic syndrome. Endocrine. 2003;20:215–218. doi: 10.1385/ENDO:20:3:215. [DOI] [PubMed] [Google Scholar]
- 213.Gauthier MS, O’Brien EL, Bigornia S, Mott M, Cacicedo JM, Xu XJ, Gokce N, Apovian C, Ruderman N. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun. 2011;404:382–387. doi: 10.1016/j.bbrc.2010.11.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–8641. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Katerelos M, Mudge SJ, Stapleton D, Auwardt RB, Fraser SA, Chen CG, Kemp BE, Power DA. 5-Aminoimidazole-4-carboxamide ribonucleoside and AMP-activated protein kinase inhibit signalling through NF-κB. Immunol. Cell Biol. 2010;88:754–760. doi: 10.1038/icb.2010.44. [DOI] [PubMed] [Google Scholar]
- 216.Reference deleted
- 217.Wang C, Li L, Zhang ZG, Fan D, Zhu Y, Wu LL. Globular adiponectin inhibits angiotensin II-induced nuclear factor κB activation through AMP-activated protein kinase in cardiac hypertrophy. J Cell Physiol. 2010;222:149–155. doi: 10.1002/jcp.21931. [DOI] [PubMed] [Google Scholar]
- 218.Bess E, Fisslthaler B, Fromel T, Fleming I. Nitric oxide-induced activation of the AMP-activated protein kinase α2 subunit attenuates IκB kinase activity and inflammatory responses in endothelial cells. PLoS ONE. 2011;6:e20848. doi: 10.1371/journal.pone.0020848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, Mitch W, Smith SC, Jr, Sowers JR. Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation. 1999;100:1134–1146. doi: 10.1161/01.cir.100.10.1134. [DOI] [PubMed] [Google Scholar]
- 220.Asghar O, Al-Sunni A, Khavandi K, Khavandi A, Withers S, Greenstein A, Heagerty AM, Malik RA. Diabetic cardiomyopathy. Clin Sci. 2009;116:741–760. doi: 10.1042/CS20080500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- 222.Masoudi FA, Inzucchi SE, Wang Y, Havranek EP, Foody JM, Krumholz HM. Thiazolidinediones, metformin and outcomes in older patients with diabetes and heart failure: an observational study. Circulation. 2005;111:583–590. doi: 10.1161/01.CIR.0000154542.13412.B1. [DOI] [PubMed] [Google Scholar]
- 223.Eurich DT, McAlister FA, Blackburn DF, Majumdar SR, Tsuyuki RT, Varney J, Johnson JA. Benefits and harms of antidiabetic agents in patients with diabetes and heart failure: systematic review. BMJ. 2007;335:497. doi: 10.1136/bmj.39314.620174.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Davis BJ, Xie Z, Viollet B, Zou MH. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55:496–505. doi: 10.2337/diabetes.55.02.06.db05-1064. [DOI] [PubMed] [Google Scholar]
- 225.Lewington S, Whitlock G, Clarke R, Sherliker P, Emberson J, Halsey J, Qizilbash N, Peto R, Collins R. Blood cholesterol and vascular mortality by age, sex and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007;370:1829–1839. doi: 10.1016/S0140-6736(07)61778-4. [DOI] [PubMed] [Google Scholar]
- 226.Foody JM, Shah R, Galusha D, Masoudi FA, Havranek EP, Krumholz HM. Statins and mortality among elderly patients hospitalized with heart failure. Circulation. 2006;113:1086–1092. doi: 10.1161/CIRCULATIONAHA.105.591446. [DOI] [PubMed] [Google Scholar]
- 227.Sun W, Lee TS, Zhu M, Gu C, Wang Y, Zhu Y, Shyy JY. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation. 2006;114:2655–2662. doi: 10.1161/CIRCULATIONAHA.106.630194. [DOI] [PubMed] [Google Scholar]
- 228.Choi HC, Song P, Xie Z, Wu Y, Xu J, Zhang M, Dong Y, Wang S, Lau K, Zou MH. Reactive nitrogen species is required for the activation of the AMP-activated protein kinase by statin in vivo. J Biol Chem. 2008;283:20186–20197. doi: 10.1074/jbc.M803020200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 229.Li X, Han Y, Pang W, Li C, Xie X, Shyy JY, Zhu Y. AMP-activated protein kinase promotes the differentiation of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:1789–1795. doi: 10.1161/ATVBAHA.108.172452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Kennedy DO, Wightman EL, Reay JL, Lietz G, Okello EJ, Wilde A, Haskell CF. Effects of resveratrol on cerebral blood flow variables and cognitive performance in humans: a double-blind, placebo-controlled, crossover investigation. Am J Clin Nutr. 2010;91:1590–1597. doi: 10.3945/ajcn.2009.28641. [DOI] [PubMed] [Google Scholar]
- 231.Wong RH, Howe PR, Buckley JD, Coates AM, Kunz I, Berry NM. Acute resveratrol supplementation improves flow-mediated dilatation in overweight/obese individuals with mildly elevated blood pressure. Nutr Metab Cardiovasc Dis. 2011;21:851–856. doi: 10.1016/j.numecd.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 232.Bradamante S, Barenghi L, Villa A. Cardiovascular protective effects of resveratrol. Cardiovasc Drug Rev. 2004;22:169–188. doi: 10.1111/j.1527-3466.2004.tb00139.x. [DOI] [PubMed] [Google Scholar]
- 233.Zeng XH, Zeng XJ, Li YY. Efficacy and safety of berberine for congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 2003;92:173–176. doi: 10.1016/s0002-9149(03)00533-2. [DOI] [PubMed] [Google Scholar]
- 234.Kong W, Wei J, Abidi P, Lin M, Inaba S, Li C, Wang Y, Wang Z, Si S, Pan H, et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med. 2004;10:1344–1351. doi: 10.1038/nm1135. [DOI] [PubMed] [Google Scholar]