

Abstract
Background
Angelman syndrome (AS) is a human neuropsychiatric disorder associated with autism, mental retardation, motor abnormalities, and epilepsy. In most cases, AS is caused by the deletion of the maternal copy of UBE3A gene, which encodes the enzyme ubiquitin ligase E3A, also termed E6-AP. A mouse model of AS has been generated and these mice exhibit many of the observed neurological alterations in humans. Because of clinical and neuroanatomical similarities between AS and schizophrenia, we examined AS model mice for alterations in the neuregulin-ErbB4 pathway, which has been implicated in the pathophysiology of schizophrenia. We focused our studies on the hippocampus, one of the major brain loci impaired in AS mice.
Methods
We determined the expression of NRG1 and ErbB4 receptors in AS mice and wild-type littermates (ages 10-16 weeks), and studied the effects of ErbB inhibition on long-term potentiation (LTP) in hippocampal area CA1 and on hippocampus-dependent contextual fear memory.
Results
We observed enhanced neuregulin-ErbB4 signaling in the hippocampus of AS model mice and found that ErbB inhibitors could reverse deficits in LTP, a cellular substrate for learning and memory. In addition, we found that an ErbB inhibitor enhanced long-term contextual fear memory in AS model mice.
Conclusions
Our findings suggest that neuregulin-ErbB4 signaling is involved in synaptic plasticity and memory impairments in AS model mice, suggesting that ErbB inhibitors have therapeutic potential for the treatment of AS.
Keywords: NRG1, ErbB4, Angelman syndrome, LTP, hippocampus, interneurons
Introduction
Angelman syndrome (AS) is a human neurological disorder associated with symptoms that include autism, mental retardation, motor abnormalities, and epilepsy (1, 2). In most cases, AS is caused by the deletion of small portions on the maternal copy of chromosome 15, which includes the UBE3A gene (3-6). The phenomenon in which only the maternal copy of the UBE3A gene is expressed is called “imprinting”, and is found in multiple brain areas, including excitatory and inhibitory interneurons (7-9). The UBE3A gene encodes ubiquitin ligase E3A (also termed E6-AP) that covalently attaches polyubiquitin chains to specific proteins to signal for their recognition and degradation by the 26S proteasome. A mouse model of AS was generated and exhibits seizures, impaired motor function, and cognitive abnormalities that correlate with neurological alterations observed in humans (10). AS model mice also exhibit impairments in hippocampal long-term potentiation (LTP) and deficits in hippocampus-dependent memory (10, 11), which suggests hippocampal dysfunction in AS.
Neuregulin 1 (NRG1) is one of a family of proteins encoded by four genes (NRG1 through NRG4), each of which contains an epidermal growth factor (EGF)-like domain (12). NRG1 signals via the receptor tyrosine kinase ErbB4, which is related to the EGF family of receptors (13). It has been demonstrated that bath application of NRG1 to hippocampal slices blocks LTP (14). In addition, NRG1 induces depotentiation and constitutively suppresses LTP (15, 16). Moreover, pharmacological and genetic studies showed that the blockade of LTP by NRG1 required stimulation of ErbB4 (16). On the other hand, NRG1 global knockout mice exhibit contextual fear conditioning deficits resembling those in AS model mice, perhaps to compensatory changes in NRG1-ErbB4 signaling during development (17).
The NRG1-ErbB4 pathway has been implicated in the pathophysiology of schizophrenia (18-21). Interestingly, AS and schizophrenia share some common features (Table S1 in the Supplement) and the UBE3A gene has been linked to schizophrenia (22). These findings prompted us to determine whether there were alterations in NRG1-ErbB4 signaling in AS model mice. In addition, we examined whether inhibitors of NRG1-ErbB4 signaling could improve synaptic and cognitive deficits displayed by AS model mice. The results of our studies indicate that altered NRG1-ErbB4 signaling plays a role in abnormal synaptic plasticity and memory impairments in AS model mice.
Methods and Materials
Mice
Angelman syndrome (AS) model mice were generated and genotyped using specific primers as described previously (10). For details regarding mice please see SI Methods.
Tissue preparation, immunoprecipitation, and Western blots
AS mice and wild-type littermates were decapitated, the brain was quickly extracted, and hippocampi from both hemispheres were removed, tissue was prepared, and Western blots were performed using standard protocols. For details concerning homogenization buffers, immunoprecipitation, Western blot procedures and sources and concentrations of antibodies, please see SI Methods.
Extracellular and intracellular electrophysiology
Brains from AS model mice and their wild-type littermates (10-16 weeks of age) were quickly removed and transverse hippocampal slices were prepared using standard techniques. For details concerning the preparation of slices, the various recording conditions and manipulations, please see SI Methods.
Hippocampal Cannulation and Drug infusion
Mice were anesthetized with isoflurane, placed in a stereotaxic frame (Kopf Instruments), and implanted with 22-gauge guide cannulae (Plastics One). PD158780 infusions were given immediately post-training to limit drug effects to the consolidation phase of memory formation. After injection mice were returned to their home cages and transported back to the colony. For details concerning the cannulation procedure and the drug infusion, please see SI Methods.
Contextual Fear Conditioning
Mice (ages 10-16 weeks) were singly housed and handled for 1-2 min on three consecutive days in the same room that fear conditioning was conducted. Training and testing occurred during the light phase. Contextual fear conditioning was performed using a standard technique, as described previously (11). For details concerning the contextual fear conditioning training and test, please see SI Methods.
Results
Altered Expression of NRG1 and ErbB4 in AS Model Mice
Because of several similarities between AS and schizophrenia (Table S1 in the Supplement) and previous studies linking the NRG1-ErbB4 signaling pathway to the pathophysiology of schizophrenia, we examined NRG1 and ErbB4 expression, as well as ErbB4 phosphorylation in the hippocampus of AS model mice. Specifically, we examined NRG1 type I-II protein levels (23) because its mRNA is present in large quantities in all hippocampal subregions (24). We observed a significant increase in NRG1 levels in the hippocampus of AS model mice in comparison to their wild-type littermates (% of wild-type NRG levels: wild-type = 100±7, n=9 and AS = 139±10, n=22, p<0.01) (Figure 1A). We proceeded to examine the phosphorylation state of three ErbB4 tyrosine residues in AS model mice: tyrosine 1056, 1162, and 1188. We observed significant increases in the phosphorylation of ErbB4 on tyrosine 1056 and tyrosine 1162 in the AS model mice (% of wild-type Y1056: wild-type = 100±9, AS = 204±40, p<0.05; % of wild-type Y1162: wild-type = 100±5, n=9 and AS = 138±9, n=11, p<0.01). Although there was a trend, the phosphorylation of ErbB4 on tyrosine 1188 in the AS mice was not significantly enhanced (% of wild-type Y1162: wild-type = 100±9, n=9 and AS = 132±15, n=11, p=0.1) (Figure 1B). We also observed a significant decrease in the total levels of ErbB4 in the AS mice compared to their wild-type littermates (wild-type = 100±6, n=9 and AS=67+6, n=11, p<0.01) (Figure 1C). Significant decreases in ErbB4 expression also were observed in primary hippocampal neuronal cultures from AS mouse embryos (Figure S1A in the Supplement). Taken together with previous studies showing that ErbB4 activation results in its poly-ubiquitination and degradation (25, 26), our current findings suggested that in AS mice, NRG1-ErbB4 signaling is enhanced and that the activated, phosphorylated ErbB4 receptor is most likely degraded, probably via the ubiquitin proteasome system. To examine this possibility, we stimulated primary neuronal cultures from AS mice and their wild-type littermates with the active form of NRG1. We observed that the NRG1 stimulation induced an increase in ErbB4 ubiquitination in both genotypes (Figure S1B in the Supplement).
Fig. 1.

Altered NRG1-ErbB4 signaling in Angelman syndrome model mice. Western blots of hippocampal homogenates from Angelman syndrome (AS) mice and their wild-type (WT) littermates were probed with various antibodies to examine NRG1-ErbB4 signaling. (A) NRG1 levels are enhanced in AS model mice. (B) Phosphorylated ErbB4 on residues Y1056, Y1162 and Y1188 in AS model mice and their wild-type littermates. The blots were stripped and probed for total ErbB4. (C) Total ErbB4 levels are decreased in AS model mice. (D) E6-AP does not interact with either NRG1 or ErbB4. Hippocampal homogenates were prepared from E6-AP-YFP transgenic mice and immunoprecipitated with antibodies to YFP (left column) Input, supernatants, and immunoprecipitates were examined on Western blots with antibodies for NRG1, ErbB4, YFP, and ERK1/2 as a control. The same homogenates were immunuoprecipated with antibodies to ErbB4 (right column, top) and NRG1 (right column, bottom) and probed for E6-AP-YFP with an antibody to YFP. * denotes statistical significance (p<0.05), ** denotes statistical significance (p<0.01) with a Student’s t-test. For all panels, n=11 for AS mice and n=9 for wild-type mice.
Because NRG1 and ErbB4 levels are altered in AS mice, we determined whether these molecules interact directly with E6-AP. We utilized transgenic mice that express an E6-AP-yellow fluorescent protein (E6-AP-YFP) fusion protein (27) and performed immunoprecipitation experiments with an antibody directed against YFP. We found that neither ErbB4 nor NRG1 co-immunoprecipitated with E6-AP (Figure 1D), suggesting that neither NRG1 nor ErbB4 interact directly with E6-AP.
ErbB Inhibition Reverses LTP Impairments in AS Model Mice
AS model mice display dramatic impairments in hippocampal LTP (10, 11). Because constitutive ErbB4 activity inhibits LTP (15, 16), and our findings suggest that AS mice exhibit enhanced NRG1-ErbB4 signaling (Figure 1), we investigated whether inhibition of ErbB receptors could reverse LTP impairments in AS mice. We stimulated hippocampal slices from AS mice and their wild-type littermates with two trains of high-frequency stimulation (HFS) in the presence of either the ErbB inhibitor PD158780 (10 μM) or vehicle. The LTP impairment displayed by the AS mice was completely reversed by PD158780 (% of baseline fEPSP: wild-type = 163±7, wild-type+PD158780 = 176±19, AS = 93±18, AS+PD158780 = 175±31, at 90 min; n=8 slices and mice for each group, p<0.05 RM-ANOVA) (Figure 2A,B). Paired-pulse facilitation (PPF), before and after HFS in AS hippocampal slices treated with PD158780, was not altered, suggesting that the LTP rescue effect of PD158780 is postsynaptic (Figure 2C). Finally, we conducted additional LTP rescue experiments with slices from AS mice with PD168393, another ErbB inhibitor structurally unrelated to PD158780. Similar to PD158780, we found that PD168393 (10 μM) rescued the LTP impairment in slices from AS mice (Figure S2A in the Supplement). PPF in the PD168393-treated slices before and after HFS was not changed, supporting again a postsynaptic mechanism (Figure S2B in the Supplement). Taken together these findings suggest that inhibiting ErbB signaling rescues LTP impairments in slices from AS model mice.
Fig. 2.
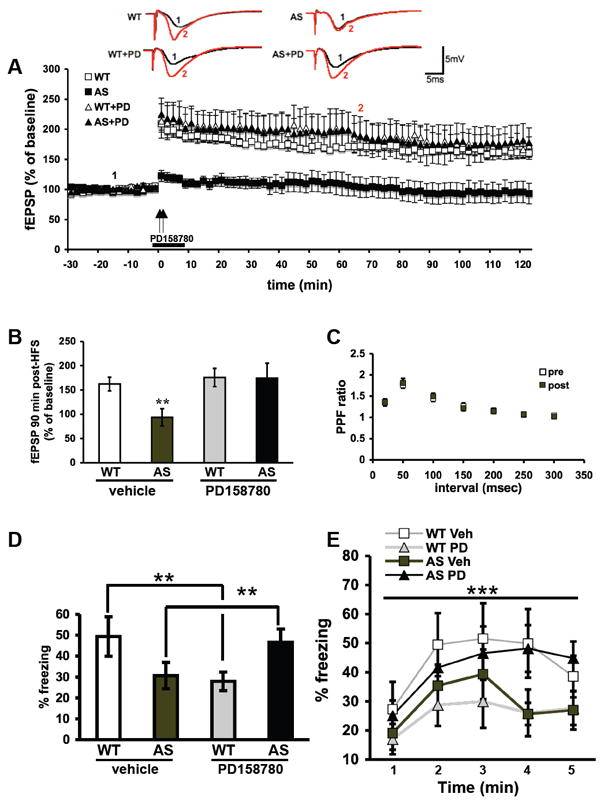
Different hippocampal phenotypes in AS mice, such as LTP in the CA1 and contextual fear conditioning, are rescued by ErbB receptor inhibition. (A) Hippocampal slices from Angelman syndrome (AS) and wild-type (WT) mice were treated with either vehicle (0.1% DMSO) or PD158780 (10 μM) for 10 minutes, beginning 2 minutes prior to two trains of high-frequency stimulation (HFS, indicated by the arrows). For all conditions, n=8 (number of mice and slices). (B) Cumulative data showing the amount of LTP 90 min after the final train of HFS. (C) Paired-pulse facilitation (PPF) curve for AS slices treated with PD158780 10 μM before and after HFS. (D) AS model mice and their WT littermates were treated with either vehicle or PD158780 via bilateral intra-hippocampal infusions. All results are displayed as % freezing. N=8 for all groups. Long-term memory 7 days after training as measured by averaged % freezing during the last three minutes of exposure to context. Posthoc Bonferroni analysis showed that AS mice treated with PD158780 exhibit a significant increase in freezing response compare to AS vehicle treated (p<0.01), and no difference from wild-type littermates treated with vehicle. ** denotes statistical significance of p<0.01 between the AS groups (vehicle and PD158780) and for interaction of group and treatment in 2-Way-ANOVA. (E) The freezing response curve during the entire 5 minutes of exposure 7 days after training shows a significant group effect (group meaning the combination of genotype and treatment) along the exposure time. *** denotes statistical significance p<0.0001 with RM-ANOVA.
The Effect of ErbB Inhibitors on Contextual Fear Conditioning in AS Model Mice
Contextual fear conditioning, a hippocampus- and amygdala-dependent form of associative memory, is impaired in AS mice (10). Therefore, we hypothesized that PD158780 would rescue impairments in contextual fear memory displayed by AS mice. We performed experiments using bilateral intra-hippocampal infusions of either vehicle or PD158780 immediately after training. The infusion of the drug occurred immediately after training in order to not affect memory acquisition with the stress of the procedure. Memory acquisition did not differ between groups (WT and AS, with infused with either vehicle or PD158780) (Figure S3 in the Supplement). We observed a differential effect between drug treatment and genotype on long-term memory (LTM). AS mice treated with vehicle exhibited the usual LTM impairment, but AS mice injected with PD158780 exhibited a significant enhancement in LTM, similar to that of wild-type mice treated with vehicle (Figure 2D). In contrast, wild-type mice showed an impairment of memory after PD158780 injection (average % freezing in last three minutes of exposure to context: wild-type+vehicle = 49.4±9.3%, wild-type+PD158780 = 27.9±4.5%, AS+vehicle = 30.6±6.2%, AS+PD158780 = 46.6±6.3%, n=8 mice for all 4 groups, p<0.01, 2-way-ANOVA) (Figure 2D). This differential effect of PD158780 on wild-type and AS mice also was observed during context recognition and the freezing response during the entire 5-minute exposure (p<0.0001 RM-ANOVA) (Figure 2E). Similar results were observed when we examined remote memory 36 days after training as there was a differential effect of PD158780 on the interaction between genotype and treatment (p<0.05 2way ANOVA) (Figure S4A in the Supplement). The increase in freezing response displayed by the AS mice during the 5-minute period due to exposure to the context also was significant (p<0.001 RM-ANOVA). Posthoc Bonferroni analysis indicated that there was no difference between wild-type mice treated with vehicle and AS mice treated with PD158780, and a significant increase between AS mice treated with vehicle and AS mice treated with PD158780 (p<0.05). Wild-type mice treated with vehicle and AS mice treated with vehicle were significantly different as well (p<0.01) (Figure S4B in the Supplement).
Expression and Function of AMPA and NMDA Receptors in AS Model Mice
Because NRG1-ErbB4 signaling has been reported to impair LTP via modulation of AMPA and NMDA receptors (15, 28) and NRG1-ErbB4 signaling is elevated in AS mice (Fig. 1), we examined the expression, phosphorylation, and function of these receptors in the hippocampus of AS mice. We observed no significant differences in the expression of the GluR1 subunit of the AMPA receptor between wild-type and AS mice (% of wild-type GluR1 levels: wild-type = 100±5, AS = 109±3) (Figure 3A). Moreover, there were no differences in the major phosphorylation sites on GluR1 reported to be relevant for LTP (29). Neither serine 831 phosphorylation nor serine 845 phosphorylation were altered in the AS mice compared to their wild-type littermates (% of wild-type S831 levels: wild-type = 100±4, AS = 97±4 for S831; % of wild-type S845 levels: wild-type = 100±3, n=9 and AS = 97±3, n=11 for S845) (Figure 3A). Similar to GluR1, the expression of the NR1, NR2A, and NR2B subunits of the NMDA receptor did not significantly differ between wild-type and AS littermates (% of wild-type NR1: wild-type = 100±5, n=9 and AS = 111±6%, n=11; % of wild-type NR2A: wild-type = 100±6, n=9 and AS = 117±7, n=11; % of wild-type NR2B: wild-type = 100±12, n=9 and AS = 86±10, n=11) (Figure 3B). Furthermore, phosphorylation of NR2B on tyrosine 1472, previously shown to be stimulated by NRG1-ErbB4 signaling via fyn (30), was similar between the wild-type and AS mice (% of wild-type Y1472: wild-type = 100±4, n=9 and AS = 96±3, n=11) (Figure 3B). These findings indicate that the expression and phosphorylation state of AMPA and NMDA receptors are normal in AS mice.
Fig. 3.
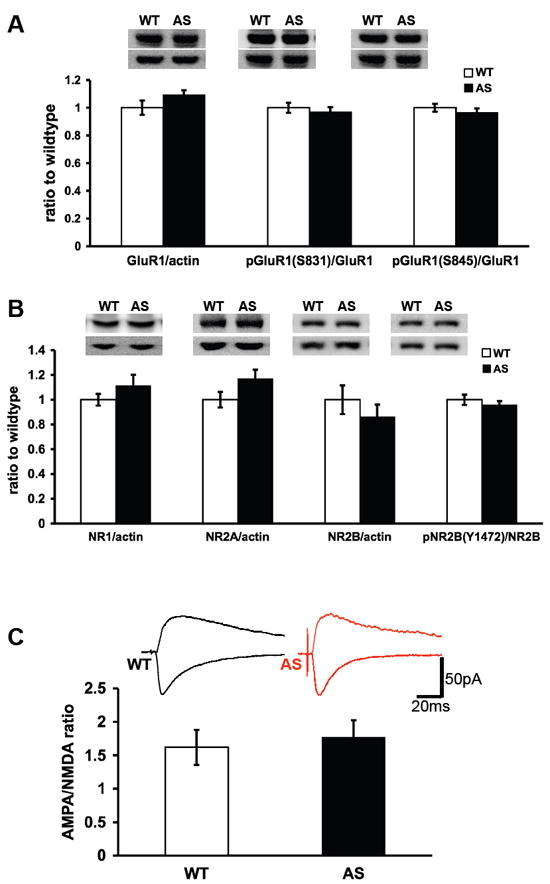
Expression and phosphorylation of AMPA and NMDA receptors in Angelman syndrome model mice. Western blots of hippocampal homogenates from Angelman syndrome (AS) model mice and their wild-type littermates were probed with various antibodies to AMPA and NMDA receptors. (A) Total levels of GluR1, actin, phosphorylated GluR1 on S831 and S845. For total GluR1 expression, GluR1 and actin were probed on the same blot. For phosphorylated GluR1, blots were probed for the phosphorylated form, stripped, and reprobed for total GluR1. (B) Total levels of NR1, NR2A and NR2B. All were compared to actin on the same blots. For phosphorylated NR2B (Y1472), blots were probed for the phosphorylated form, stripped, and reprobed for total NR2B. n=11 for WT mice and n=9 for AS mice in panels A and B. (C) NMDA to AMPA ratio is similar between AS mice and their WT littermates. NMDA/AMPA current ratio was tested in voltage clamp. For AMPA, the peak amplitude was measured at -70 mV (negative current) and for NMDA the amplitude was measured 100 ms after the peak at +40mV (positive current). (WT: n=15 cells, 8 mice; AS: n=15 cells, 8 mice).
Next we measured AMPA and NMDA currents in hippocampal slices from AS mice and their wild-type littermates. We found that the AMPA/NMDA ratio in the AS mice was comparable to that of wild-type mice. (AMPA/NMDA ratios: wild-type=1.64±0.26, AS=1.77±0.25; n=15 cells from 8 mice) (Figure 3C). Thus, consistent with our biochemical results, the function of AMPA and NMDA receptors in the AS mice appear to be similar to that of their wild-type littermates.
Inhibition of ErbB Receptors Reduces Inhibitory Chloride Currents in AS Model Mice
Because glutamate receptors expression and function was unaltered in AS mice, we postulated that the enhanced NRG1-ErbB4 signaling impairs LTP in AS mice via an increase in inhibitory synaptic transmission. Therefore, we isolated ligand-gated chloride currents in CA1 pyramidal neurons in hippocampal slices from AS and wild-type mice and measured the effect of PD158780 on these currents. PD158780 induced a significantly faster and more robust decrease in chloride inhibitory currents in AS mice compared to their wild-type littermates (p<0.01, RM-ANOVA) (Figure 4A). In AS mice, the PD158780-induced decrease in chloride currents reached its maximum effect within 2 minutes of application. The difference between the AS and wild-type mice was evident from the beginning (after 2 min: 91±3% vs. 75±5% of baseline, wild-type and AS mice respectively, n=15 cells from 8 mice, p<0.01, Student’s t-test) and at the end of the PD158780 application (after 10 min: 80±4 vs. 63±6, wild-type and AS mice, respectively, n=15 cells from 8 mice, p<0.05 Student’s t-test) (Figure 4B). It should be noted that although PD158780 induced a slower and less robust decrease of the chloride currents in the wild-type mice, this inhibition was significant as well (88±3% and 80±4% after 3 and 10 minutes respectively) (Figure 4A). These results suggest that chloride currents are more sensitive to modulation by ErbB receptors in AS model mice compared to their wild-type littermates.
Fig. 4.
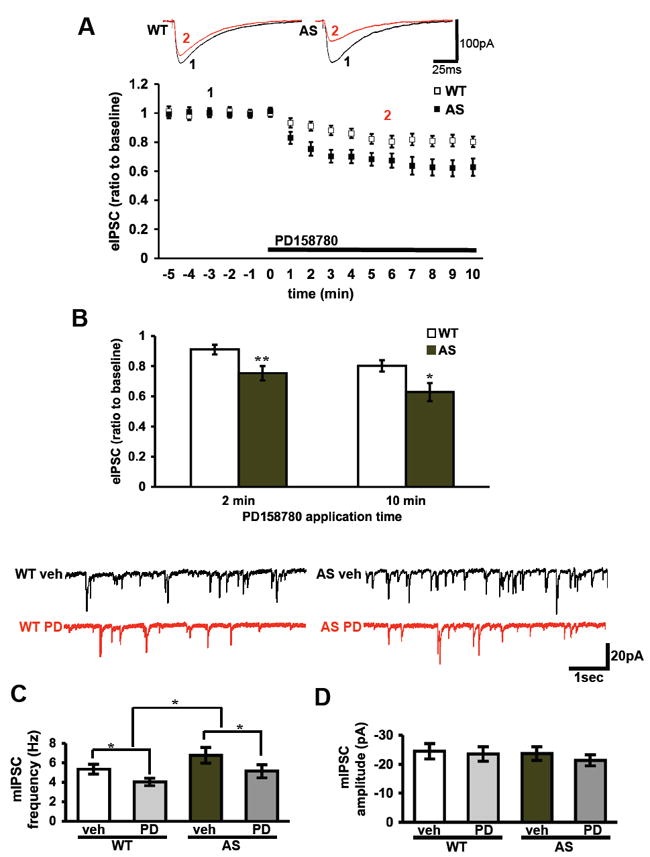
ErbB inhibition induces a larger reduction in evoked inhibitory currents in Angelman syndrome mice compare to wild-type mice. ErbB4 effect on the IPSC reduction is via a presynaptic mechanism. (A) Inhibitory currents were measured as the peak amplitude in voltage clamp at -70 mV with high chloride concentrations in the recording pipette, before and during application of PD158780 (10 μM) application. The bath solution of ACSF contained D-APV 50 μM and DNQX 40 μM. ** denotes p<0.01 RM-ANOVA (B) Cumulative data 2 and 10 min following the application of PD158780 indicates that the reduction of inhibitory currents was larger in the AS mice was compared to their WT littermates. (WT: n=15 cells, 8 mice; AS: n=15 cells, 8 mice). * denotes p<0.05 and ** denotes p<0.01 in student’s t-tests. (C-D) mIPSC recordings from CA1 pyramidal neurons before and after application of PD158780 (10μM) for 4 min resulted in a significant reduction in frequency (C) but not the amplitude (D) of the mIPSCs. The bath solution of ACSF contained D-APV (50 μM), DNQX (40 μM) and tetrodotoxin (1μM). Wild-type (WT) mice: n=13 cells, 4 mice. Angelman syndrome (AS): n=13 cells, 4 mice. * denotes p<0.05 for group effect and for treatment effect in 2-way-ANOVA.
We then measured miniature inhibitory postsynaptic currents (mIPSCs) before and after four minutes of PD158780 application. We observed that PD158780 induced a significant reduction in the frequency of mIPSCs (wild-type baseline = 5.3±0.5 Hz, wild-type+PD158780 =4.0±0.4 Hz, AS baseline = 6.8±0.8 Hz, AS+PD158780 = 5.1±0.7 Hz; n=13 cells from 4 mice in each group; p<0.05 for treatment and p<0.05 for group in 2-way-ANOVA) (Figure 4C). In contrast, the mIPSC amplitude did not differ between the four groups (wild-type baseline = 24.5±2.7 pA, wild-type+PD158780 = 23.5±2.5 pA, AS baseline = 23.7±2.4 pA, AS+PD158780 = 21.3±1.9 pA; n=13 cells from 4 mice in each group) (Figure 4D). These findings indicate that there is an increase in mIPSC frequency in AS mice (p<0.05 in 2-way-ANOVA), suggesting that there is increased presynaptic inhibition onto CA1 pyramidal neurons. In addition, PD158780 reduces the mIPSC frequency in both AS and wild-type mice, indicating that inhibiting ErbB receptors reduces presynaptic inhibitory input to CA1 pyramidal cells.
Blocking Inhibitory Synaptic Transmission Reverses LTP Impairments in AS Model Mice
Because chloride currents were more sensitive to inhibition of ErbB receptors in AS model mice, we determined whether blocking inhibitory synaptic transmission could reverse LTP impairments in AS mice in a manner similar to ErbB inhibitors. We induced LTP in slices from AS mice and their wild-type littermates in the presence of 20 μM bicuculline, a GABA(A) receptor antagonist. In the absence of bicuculline, LTP was impaired in slices from AS mice (Figure 5A). In contrast, we observed a marked and significant potentiation in bicuculline-treated AS slices that persisted after washout (Figure 5A). In wild-type mice, potentiation was observed in slices treated with and without bicuculline, with no significant differences between them. (% of baseline fEPSP: wild-type = 172±10, wild-type+bicuculline = 204±20, AS = 101±10, AS+bicuculline = 176±16, at 60 min; n=8 slices and mice per group, p<0.05 RM-ANOVA) (Figure 5B). There were no differences in PPF before and after HFS in the bicuculline-treated slices from AS mice (Figure 5C), suggesting that the LTP rescue effect of bicuculline is by a postsynaptic mechanism. These findings indicate that the application of bicuculline, in coincidence with HFS, rescues LTP impairments in the AS model mice.
Fig. 5.
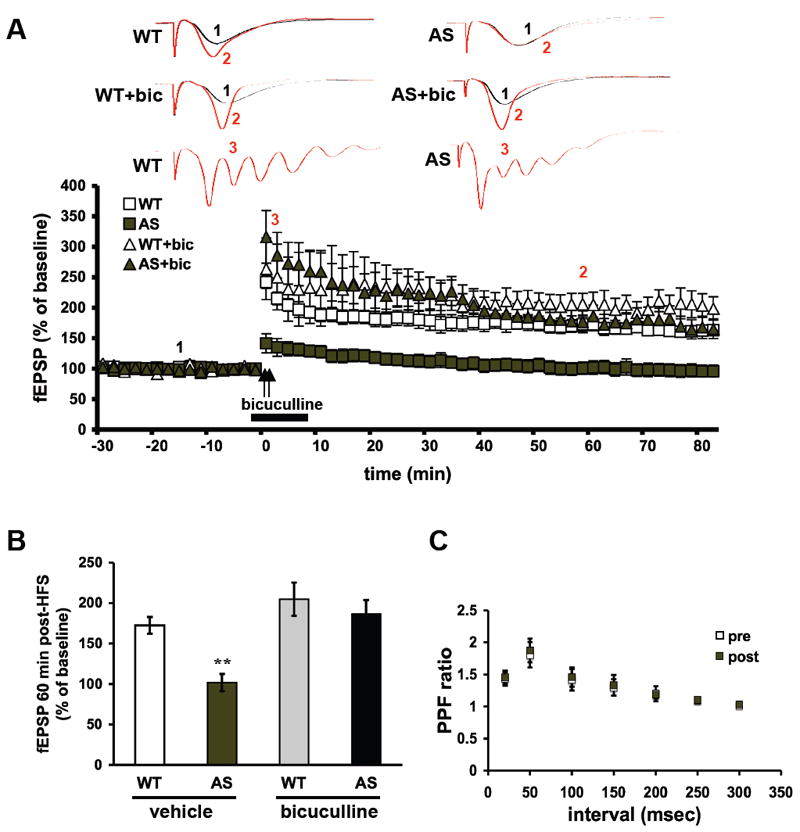
LTP in hippocampal area CA1 in Angelman syndrome model mice is rescued by inhibition of GABA(A) receptors. (A) Hippocampal slices from Angelman syndrome (AS) model mice and their wild-type (WT) littermates were treated with either vehicle (ACSF) or bicuculline (20 μM) for 10 minutes, beginning 2 minutes prior to two trains of high-frequency stimulation (HFS, indicated by the arrows). For all conditions, n=8 (number of mice and slices). (B) Cumulative data showing the amount of LTP 60 min after the final train of HFS. (C) Paired-pulse facilitation (PPF) curves for AS slices treated with bicuculline 20 μM before and after HFS.
Discussion
The involvement of NRG1-ErbB4 signaling pathway in schizophrenia has been well documented (18-21). Due to several shared features between schizophrenia and AS (Table S1 in the Supplement) and a possible genetic linkage of UBE3A gene to schizophrenia (22), we hypothesized that NRG1-ErbB4 signaling is involved in the pathophysiology of AS. We found that NRG1 levels and ErbB4 phosphorylation on two tyrosine residues were significantly increased (Figure 1A,B), whereas total ErbB4 levels were decreased (Figure 1C) in the hippocampus of AS model mice. There is general agreement that enhanced phosphorylation of ErbB4 is correlated with its activation (31). Moreover, activation and phosphorylation of ErbB4 have been shown previously to trigger its degradation via the ubiquitin proteasome system. Thus, our findings suggest that NRG1-ErbB4 signaling is constitutively elevated in the hippocampus of AS mice. Alternatively, a reduction in ErbB4 expression could result in a compensatory feedback upregulation of NRG1 in the AS mice.
ErbB receptors have several roles during brain development, including influencing neuronal migration and radial glial development (32). Thus, similar to schizophrenia, a component of the pathological development in the AS brain could be related to an early, aberrant functioning of NRG1-ErbB4 in glial cells (33). Recent studies indicate that the primary ErbB receptor subtype in adult mouse neurons is ErbB4, with diminished expression of ErbB1 in neurons after one week of age and ErbB3 expression restricted to glial cells (34). Moreover, of all the ErbB receptor subtypes, the involvement of ErbB4 in schizophrenia is the most commonly reported, although ErbB1 (35) and ErbB3 (36) also have been implicated in this disorder. Therefore, in the future it will be important to examine the expression and function of other ErbB receptors in AS mice. Previous studies have shown that NRG1-ErbB4 signaling impairs hippocampal LTP (15, 16, 30) and that this inhibition is constitutive (16). The most robust synaptic phenotype described thus far in the hippocampus of AS mice is impaired LTP (10, 11, 37). Because we observed enhanced NRG1-ErbB4 signaling in the hippocampus of AS mice, we posited that inhibiting NRG1-ErbB4 signaling would rescue the impaired hippocampal LTP in AS mice. Indeed, two mechanistically distinct ErbB inhibitors rescued the LTP impairment in AS model mice (Figures 2A and S2 in the Supplement). These findings suggest that constitutively elevated NRG1-ErbB4 signaling specifically contributes to LTP impairments in AS model mice.
Hippocampus-dependent contextual fear memory is impaired in AS mice (10, 11). We found that intra-hippocampal infusion of PD158780 rescued the LTM impairments exhibited by the AS mice, but unlike the LTP experiments, we observed differential effects of the drug on wild-type and AS mice. In AS mice, long-term contextual fear memory was enhanced by PD158780, but in wild-type mice long-term memory was impaired by PD158780 (Figure 2E). These results support the idea that NRG1-ErbB4 signaling is altered in AS mice and impairs hippocampus-dependent function. The impairment in contextual fear memory caused by PD158780 in wild-type mice is consistent with previous findings demonstrating that NRG1 mutant mice (17) and mice with a genetic deletion of ErbB4 in interneurons exhibit similar memory impairments (38). These findings suggest that under normal conditions ErbB receptors are required for hippocampus-dependent memory. Differential effects on hippocampus-dependent memory also have been reported for rapamycin, which inhibits mammalian target of rapamycin complex 1 (mTORC1). Rapamycin was shown to reverse context fear discrimination deficits in a mouse model of tuberous sclerosis complex (TSC), a neurodevelopmental disorder (39), but also has been shown to impair various forms of hippocampus-dependent memory in wild-type mice (40-42). Thus, similar to selective utilization of rapamycin to inhibit upregulation of mTORC1 signaling as a therapy for TSC, selective utilization of ErbB inhibitors might serve as a therapy for AS.
NRG1-ErbB4 signaling has been shown to induce depotentiation of hippocampal LTP via reduction of AMPA receptor currents (15), as well as phosphorylation of NR2B (Y1472) (30), while in the PFC it induces a reduction of NMDA currents (28). However, in the AS model mice, in spite of the NRG1-ErbB4 alterations, there were no changes in either the phosphorylation and expression of total GluR1 or the phosphorylation and expression of NR1, NR2A, and NR2B (Figure 3A,B). Moreover, the NMDA-to-AMPA current ratio did not differ between AS model mice and their wild-type littermates (Figure 3C). These findings suggest that the LTP impairment in AS model mice is not directly related to either AMPA or NMDA receptors.
Several studies have shown that NRG1-ErbB4 signaling regulates inhibitory transmission in the prefrontal cortex (24, 43, 44), including the enhancement of depolarization-induced GABA release inhibitory currents (24). Moreover, in the hippocampus, it was shown that ErbB4 receptors are expressed solely in interneurons and not in pyramidal neurons (45, 46). Therefore, we examined the effect of PD158780 on evoked inhibitory post-synaptic currents (eIPSCs) in CA1 pyramidal neurons and found that it induced a rapid reduction in the eIPSCs in both wild-type and AS model mice. Nevertheless, in the AS mice this reduction was significantly faster and larger (Figure 4A,B), suggesting that inhibitory transmission in AS mice is more sensitive to ErbB inhibition than wild-type mice. Furthermore, we showed that PD158780 significantly reduced mIPSC frequency, but not mIPSC amplitude (Figure 4C,D), indicating that the impact of inhibiting ErbB receptors on inhibitory currents is presynaptic. Thus, inhibiting ErbB4 receptors in the AS mice likely reduces the inhibitory input to CA1 pyramidal neurons, enabling the induction of LTP at the Schaffer collateral excitatory synapses.). We also observed an increase inmIPSC frequency in AS mice compared to their wild-type littermates, which was reversed by inhibiting ErbB receptors (Figure 4C,D).
PD158780 was reported to induce a small increase of fEPSPs in hippocampal area CA1 (15). Furthermore, it was demonstrated that constitutive ErbB4 signaling affects LTP induction (16). These data, when taken together with our results, suggest that in the basal homeostatic condition ErbB4 is constitutively active, affecting basal synaptic transmission by enhancing GABAergic inhibition. Furthermore, these data support the hypothesis that constitutive activity of ErbB4 is enhanced in hippocampal area CA1 of AS mice. Thus, inhibiting ErbB4 in AS mice induces a stronger reduction of inhibition, enabling CA1 pyramidal neurons to reach the threshold for LTP induction. Consistent with this idea, we demonstrated that alleviating inhibition with bicuculline facilitates LTP induction in AS mice (Figure 5A,B). Nevertheless, there is a qualitative difference between the effect of PD158780 and bicuculline on LTP in AS mice. Bicuculline application induced epileptiform activity in hippocampal slices (Figure 5A), whereas PD158780 did not. These observations suggest that compared to bicuculine, PD158780 has a milder effect on inhibitory transmission, though sufficient to enable LTP. Furthermore, these findings suggest that there are additional mechanisms by which ErbB inhibitors contribute to the rescue of LTP in AS mice.
The idea that alleviating enhanced inhibitory synaptic transmission as a potential therapeutic treatment for AS is somewhat counterintuitive. AS mice are prone to epilepsy and in humans the more severe forms of AS are in individuals who have an extended deletion of GABA-A receptor subunit β3 (GABAR-β3) (47-49). However, it is important to note that the epilepsy in AS is not necessarily hippocampal in origin and that other receptor and channel abnormalities could lead to epilepsy (50-52). Furthermore, GABAR-β3 is not the most prevalent GABA-A receptor subunit (53), and in the hippocampus it is mostly an extrasynaptic receptor with slow kinetics that has a minimal impact on fast synaptic transmission (54). Thus, prudent reduction of inhibitory synaptic transmission in the hippocampus might be of therapeutic value for cognitive dysfunction in AS.
In conclusion, we have presented evidence for increased NRG1-ErbB4 signaling in AS mice. Inhibiting this signaling pathway in AS mice reduces the inhibitory input to CA1 pyramidal cells, enabling the induction of LTP and LTM consolidation in AS mice. In support of this idea, we showed that alleviating inhibition in area CA1 in itself is sufficient to rescue the LTP impairment in AS mice. The identification of the involvement of NRG1-ErbB4 signaling, especially its effects on the inhibitory system, in synaptic plasticity impairments in AS mice, might aid in understanding the mechanisms underlying synaptic and behavioral abnormalities observed in AS, as well as other developmental disorders and schizophrenia. Moreover, our findings also support recent animal and clinical studies that indicate the involvement of the GABAergic system in the pathophysiology of Angelman syndrome (55) and other developmental disorders (56-61).
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Arthur L. Beaudet for the generous gift of the founding Angelman syndrome model mice. This work was supported by National Institutes of Health grants NS034007 and NS047384 and the Angelman Syndrome Foundation (E.K.) and NIH Conte Center Grant P50 MH 0645045 (T.A., R. Gur, PI).
Footnotes
Financial Disclosures: The authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet. 2001;38:834–845. doi: 10.1136/jmg.38.12.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140:413–418. doi: 10.1002/ajmg.a.31074. [DOI] [PubMed] [Google Scholar]
- 3.Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 4.Knoll JH, Nicholls RD, Magenis RE, Graham JM, Jr, Lalande M, Latt SA. Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion. Am J Med Genet. 1989;32:285–290. doi: 10.1002/ajmg.1320320235. [DOI] [PubMed] [Google Scholar]
- 5.Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, et al. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- 6.Sutcliffe JS, Jiang YH, Galijaard RJ, Matsuura T, Fang P, Kubota T, et al. The E6-Ap ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region. Genome Res. 1997;7:368–377. doi: 10.1101/gr.7.4.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gustin RM, Bichell TJ, Bubser M, Daily J, Filonova I, Mrelashvili D, et al. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol Dis. 2010;39:283–291. doi: 10.1016/j.nbd.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, et al. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17:75–78. doi: 10.1038/ng0997-75. [DOI] [PubMed] [Google Scholar]
- 9.Jiang Y, Tsai TF, Bressler J, Beaudet AL. Imprinting in Angelman and Prader-Willi syndromes. Curr Opin Genet Dev. 1998;8:334–342. doi: 10.1016/s0959-437x(98)80091-9. [DOI] [PubMed] [Google Scholar]
- 10.Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 11.van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, de Avila Freire R, et al. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci. 2007;10:280–282. doi: 10.1038/nn1845. [DOI] [PubMed] [Google Scholar]
- 12.Holmes WE, Sliwkowski MX, Akita RW, Henzel WJ, Lee J, Park JW, et al. Identification of heregulin, a specific activator of p185erbB2. Science. 1992;256:1205–1210. doi: 10.1126/science.256.5060.1205. [DOI] [PubMed] [Google Scholar]
- 13.Fischbach GD, Rosen KM. ARIA: a neuromuscular junction neuregulin. Annu Rev Neurosci. 1997;20:429–458. doi: 10.1146/annurev.neuro.20.1.429. [DOI] [PubMed] [Google Scholar]
- 14.Huang YZ, Won S, Ali DW, Wang Q, Tanowitz M, Du QS, et al. Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron. 2000;26:443–455. doi: 10.1016/s0896-6273(00)81176-9. [DOI] [PubMed] [Google Scholar]
- 15.Kwon OB, Longart M, Vullhorst D, Hoffman DA, Buonanno A. Neuregulin-1 reverses long-term potentiation at CA1 hippocampal synapses. J Neurosci. 2005;25:9378–9383. doi: 10.1523/JNEUROSCI.2100-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pitcher GM, Beggs S, Woo RS, Mei L, Salter MW. ErbB4 is a suppressor of long-term potentiation in the adult hippocampus. Neuroreport. 2008;19:139–143. doi: 10.1097/WNR.0b013e3282f3da10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ehrlichman RS, Luminais SN, White SL, Rudnick ND, Ma N, Dow HC, et al. Neuregulin 1 transgenic mice display reduced mismatch negativity, contextual fear conditioning and social interactions. Brain Res. 2009;1294:116–127. doi: 10.1016/j.brainres.2009.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH, et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med. 2006;12:824–828. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- 19.Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9:437–452. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corfas G, Roy K, Buxbaum JD. Neuregulin 1-erbB signaling and the molecular/cellular basis of schizophrenia. Nat Neurosci. 2004;7:575–580. doi: 10.1038/nn1258. [DOI] [PubMed] [Google Scholar]
- 21.Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iossifov I, Zheng T, Baron M, Gilliam TC, Rzhetsky A. Genetic-linkage mapping of complex hereditary disorders to a whole-genome molecular-interaction network. Genome Res. 2008;18:1150–1162. doi: 10.1101/gr.075622.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003;284:14–30. doi: 10.1016/s0014-4827(02)00102-7. [DOI] [PubMed] [Google Scholar]
- 24.Woo RS, Li XM, Tao Y, Carpenter-Hyland E, Huang YZ, Weber J, et al. Neuregulin-1 enhances depolarization-induced GABA release. Neuron. 2007;54:599–610. doi: 10.1016/j.neuron.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 25.Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009;315:683–696. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 26.Sundvall M, Korhonen A, Paatero I, Gaudio E, Melino G, Croce CM, et al. Isoform-specific monoubiquitination, endocytosis, and degradation of alternatively spliced ErbB4 isoforms. Proc Natl Acad Sci U S A. 2008;105:4162–4167. doi: 10.1073/pnas.0708333105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2007;17:111–118. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- 28.Gu Z, Jiang Q, Fu AK, Ip NY, Yan Z. Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J Neurosci. 2005;25:4974–4984. doi: 10.1523/JNEUROSCI.1086-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 30.Bjarnadottir M, Misner DL, Haverfield-Gross S, Bruun S, Helgason VG, Stefansson H, et al. Neuregulin1 (NRG1) signaling through Fyn modulates NMDA receptor phosphorylation: differential synaptic function in NRG1+/- knock-outs compared with wild-type mice. J Neurosci. 2007;27:4519–4529. doi: 10.1523/JNEUROSCI.4314-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaushansky A, Gordus A, Budnik BA, Lane WS, Rush J, MacBeath G. System-wide investigation of ErbB4 reveals 19 sites of Tyr phosphorylation that are unusually selective in their recruitment properties. Chem Biol. 2008;15:808–817. doi: 10.1016/j.chembiol.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anton ES, Marchionni MA, Lee KF, Rakic P. Role of GGF/neuregulin signaling in interactions between migrating neurons and radial glia in the developing cerebral cortex. Development. 1997;124:3501–3510. doi: 10.1242/dev.124.18.3501. [DOI] [PubMed] [Google Scholar]
- 33.Roy K, Murtie JC, El-Khodor BF, Edgar N, Sardi SP, Hooks BM, et al. Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc Natl Acad Sci U S A. 2007;104:8131–8136. doi: 10.1073/pnas.0702157104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerecke KM, Wyss JM, Karavanova I, Buonanno A, Carroll SL. ErbB transmembrane tyrosine kinase receptors are differentially expressed throughout the adult rat central nervous system. J Comp Neurol. 2001;433:86–100. doi: 10.1002/cne.1127. [DOI] [PubMed] [Google Scholar]
- 35.Mizuno M, Iwakura Y, Shibuya M, Zheng Y, Eda T, Kato T, et al. Antipsychotic potential of quinazoline ErbB1 inhibitors in a schizophrenia model established with neonatal hippocampal lesioning. J Pharmacol Sci. 2010;114:320–331. doi: 10.1254/jphs.10099fp. [DOI] [PubMed] [Google Scholar]
- 36.Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A. 2001;98:4746–4751. doi: 10.1073/pnas.081071198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weeber EJ, Jiang YH, Elgersma Y, Varga AW, Carrasquillo Y, Brown SE, et al. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J Neurosci. 2003;23:2634–2644. doi: 10.1523/JNEUROSCI.23-07-02634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen YJ, Zhang M, Yin DM, Wen L, Ting A, Wang P, et al. ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc Natl Acad Sci U S A. 2010;107:21818–21823. doi: 10.1073/pnas.1010669107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, et al. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blundell J, Kouser M, Powell CM. Systemic inhibition of mammalian target of rapamycin inhibits fear memory reconsolidation. Neurobiol Learn Mem. 2008;90:28–35. doi: 10.1016/j.nlm.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gafford GM, Parsons RG, Helmstetter FJ. Consolidation and reconsolidation of contextual fear memory requires mammalian target of rapamycin-dependent translation in the dorsal hippocampus. Neuroscience. 2011;182:98–104. doi: 10.1016/j.neuroscience.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krivosheya D, Tapia L, Levinson JN, Huang K, Kang Y, Hines R, et al. ErbB4-neuregulin signaling modulates synapse development and dendritic arborization through distinct mechanisms. J Biol Chem. 2008;283:32944–32956. doi: 10.1074/jbc.M800073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wen L, Lu YS, Zhu XH, Li XM, Woo RS, Chen YJ, et al. Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc Natl Acad Sci U S A. 2010;107:1211–1216. doi: 10.1073/pnas.0910302107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neddens J, Buonanno A. Selective populations of hippocampal interneurons express ErbB4 and their number and distribution is altered in ErbB4 knockout mice. Hippocampus. 2009 doi: 10.1002/hipo.20675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vullhorst D, Neddens J, Karavanova I, Tricoire L, Petralia RS, McBain CJ, et al. Selective expression of ErbB4 in interneurons, but not pyramidal cells, of the rodent hippocampus. J Neurosci. 2009;29:12255–12264. doi: 10.1523/JNEUROSCI.2454-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laan LA, Vein AA. Angelman syndrome: is there a characteristic EEG? Brain Dev. 2005;27:80–87. doi: 10.1016/j.braindev.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 48.Fiumara A, Pittala A, Cocuzza M, Sorge G. Epilepsy in patients with Angelman syndrome. Ital J Pediatr. 2010;36:31. doi: 10.1186/1824-7288-36-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clayton-Smith J, Laan L. Angelman syndrome: a review of the clinical and genetic aspects. J Med Genet. 2003;40:87–95. doi: 10.1136/jmg.40.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noebels JL. The biology of epilepsy genes. Annu Rev Neurosci. 2003;26:599–625. doi: 10.1146/annurev.neuro.26.010302.081210. [DOI] [PubMed] [Google Scholar]
- 51.Steinlein OK. Channelopathies can cause epilepsy in man. Eur J Pain. 2002;6(Suppl A):27–34. doi: 10.1053/eujp.2001.0319. [DOI] [PubMed] [Google Scholar]
- 52.Steinlein OK, Bertrand D. Nicotinic receptor channelopathies and epilepsy. Pflugers Arch. 2010;460:495–503. doi: 10.1007/s00424-009-0766-8. [DOI] [PubMed] [Google Scholar]
- 53.Sotiriou E, Papatheodoropoulos C, Angelatou F. Differential expression of gamma-aminobutyric acid--a receptor subunits in rat dorsal and ventral hippocampus. J Neurosci Res. 2005;82:690–700. doi: 10.1002/jnr.20670. [DOI] [PubMed] [Google Scholar]
- 54.Hentschke H, Benkwitz C, Banks MI, Perkins MG, Homanics GE, Pearce RA. Altered GABAA, slow inhibition and network oscillations in mice lacking the GABAA receptor beta3 subunit. J Neurophysiol. 2009;102:3643–3655. doi: 10.1152/jn.00651.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dan B. Angelman syndrome: current understanding and research prospects. Epilepsia. 2009;50:2331–2339. doi: 10.1111/j.1528-1167.2009.02311.x. [DOI] [PubMed] [Google Scholar]
- 56.Fernandez F, Morishita W, Zuniga E, Nguyen J, Blank M, Malenka RC, et al. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci. 2007;10:411–413. doi: 10.1038/nn1860. [DOI] [PubMed] [Google Scholar]
- 57.Bhattacharyya A, McMillan E, Chen SI, Wallace K, Svendsen CN. A critical period in cortical interneuron neurogenesis in down syndrome revealed by human neural progenitor cells. Dev Neurosci. 2009;31:497–510. doi: 10.1159/000236899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perez-Cremades D, Hernandez S, Blasco-Ibanez JM, Crespo C, Nacher J, Varea E. Alteration of inhibitory circuits in the somatosensory cortex of Ts65Dn mice, a model for Down’s syndrome. J Neural Transm. 2010;117:445–455. doi: 10.1007/s00702-010-0376-9. [DOI] [PubMed] [Google Scholar]
- 59.Rachidi M, Lopes C. Mental retardation and associated neurological dysfunctions in Down syndrome: a consequence of dysregulation in critical chromosome 21 genes and associated molecular pathways. Eur J Paediatr Neurol. 2008;12:168–182. doi: 10.1016/j.ejpn.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 60.Olmos-Serrano JL, Paluszkiewicz SM, Martin BS, Kaufmann WE, Corbin JG, Huntsman MM. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J Neurosci. 2010;30:9929–9938. doi: 10.1523/JNEUROSCI.1714-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Selby L, Zhang C, Sun QQ. Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci Lett. 2007;412:227–232. doi: 10.1016/j.neulet.2006.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.