

Abstract
Two mechanisms have been postulated to underlie KCNQ3 homomeric current amplitudes, which are small compared with those of KCNQ4 homomers and KCNQ2/Q3 heteromers. The first involves differential channel expression governed by the D-helix within the C-terminus. The second suggests similar channel surface expression but an intrinsically unstable KCNQ3 pore. Here, we find H2O2-enhanced oligomerization of KCNQ4 subunits, as reported by nondenaturing polyacrylamide gel electrophoresis, at C643 at the end of the D-helix, where KCNQ3 possesses a histidine. However, H2O2-mediated enhancement of KCNQ4 currents was identical in the C643A mutant, and KCNQ3 H646C produced homomeric or heteromeric (with KCNQ2) currents similar to those of wild-type KCNQ3, ruling out this divergent residue as underlying the small KCNQ3 amplitudes. In KcsA, F103 in S6 is critical for pore-mediated destabilization of the conductive pathway. We found that mutations at the analogous F344 in KCNQ3 dramatically decreased the KCNQ3 currents. Total internal reflection fluorescence imaging revealed only minor differential surface expression among the wild-type and mutant channels. Homology modeling suggests that the effects of the F344 mutants arise from the disruption of the interaction between F344 and A315 in the pore helix. These data support a secondary role of the C-terminus, compared with pore helix-S6 interactions, in governing KCNQ3 current amplitudes.
Introduction
Voltage-gated KCNQ K+ channels are tetramers of subunits containing six transmembrane domains (S1–S6) that play important roles in the functions of the heart, brain, auditory, vestibular organs, and epithelia (1). KCNQ3 can form functional channels either as homomers or as heteromers with KCNQ2, KCNQ4, or KCNQ5 (2–4). KCNQ2 and KCNQ3 have unitary conductances three to five times greater than those of KCNQ4 or KCNQ1 (5–7), and the maximal open probability of KCNQ3 is 10-fold greater than that of KCNQ4 (8). However, KCNQ4 and KCNQ1 homomers produce a K+ current with an amplitude that is dramatically larger than those of KCNQ2 and KCNQ3 homomers, and KCNQ2/3 heteromers (9–12). Two major mechanisms have been suggested to underlie the divergent expression of KCNQ channels: 1), control over the number of channels at the membrane by C-terminal domains (13,14); and 2), potentiation of the current by pore and pore helix residues (15–17). It has been suggested that an extended region within the distal half of the C-terminus is critical for determining KCNQ current amplitudes (originally called the A domain) (18), in which the first coiled-coiled domain (the C-helix) is required to form functional homomeric and heteromeric channels, and the second coiled-coiled domain (the D-helix) is necessary for tetramerization (19) and efficient transport of channels to the plasma membrane (11). KCNQ3 homomers yield only small currents, whereas the coexpression of KCNQ3 with KCNQ2 yields 10-fold larger heteromeric currents (2,10,11,15,20,21). Several laboratories have suggested a parallel between the number of the channels at the membrane and current levels, after observing such a correlation in heterologous systems (11–13,18). Moreover, Chung and co-workers (22) showed that KCNQ2 and KCNQ3 homomers in nerves display a predominant axoplasmic localization, whereas coexpression of KCNQ3 with KCNQ2 greatly increased axonal surface expression. However, other studies have not found a correlation between surface expression and current amplitude (15–17,23). Indeed, mutation of a single intracellular residue in the pore intracellular vestibule of KCNQ3, A315, to a hydrophilic threonine or serine increased current amplitudes by ∼15-fold without any significant increase in surface expression, as measured by total internal reflection fluorescence (TIRF) microscopy or biotinylation assays (16). Consistent with this, we previously suggested that most KCNQ3 homomers are nonfunctional, or dormant, due to an intrinsically unstable pore.
In a previous work (24), we described augmentation of KCNQ2, KCNQ4, KCNQ5, and KCNQ2/3 currents by hydrogen peroxide (H2O2), accompanied by an increase in the single-channel open probability, which we suggested is neuroprotective during cerebrovascular ischemic stroke. In that work, we localized the H2O2 effect in KCNQ4 to a triplet of cysteines in the S2-S3 linker. However, because KCNQ4 (but not KCNQ3 or KCNQ2) possesses a potentially cross-linkable cysteine at the end of the D-helix, we decided to revisit this issue and to test whether oligomerization by H2O2 at the D-helix may be at least partly responsible for the large KCNQ4 current amplitudes relative to KCNQ3 or KCNQ2 homomers.
Pore instability is thought to underlie C-type inactivation of many K+ channels, involving structural rearrangements of the selectivity filter (SF) (25–27). In KcsA, a recent study highlighted an intrasubunit coupling between S6 (F103) and the pore helix (T74 and T75), as well as I100 in the neighboring subunit, that promotes C-type inactivation by allowing a network of interactions among residues W67, E71, and D80 that destabilizes the SF (27). A number of lines of evidence suggest that a similar mechanism may be involved in KCNQ3 gating. Thus, a parallel network of interactions between the pore helix and the SF has been suggested to underlie the small amplitude of KCNQ3 homomeric currents, manifested by an intrinsically unstable pore that can be wholly stabilized by a single mutation (A315T or A315S) in the intracellular vestibule (16,17). In our companion study (17), we explore the effects of mutation of I312 in KCNQ3, which our modeling suggests is critical to the stabilizing network of interactions between the pore helix and the SF. That work presents data that further support this unstable-pore hypothesis and builds more evidence for the parallel between C-type inactivation of KcsA and putatively dormant KCNQ3 channels.
In this study, we focus on the role of S6-pore helix interactions and a D-helix cysteine in governing KCNQ3 current amplitudes. Using native nondenaturing polyacrylamide gel electrophoresis (PAGE) assays, we found H2O2 oligomerization of KCNQ4 subunits localized to C643 at the end of the D-helix, but such in vitro oligomerization was not associated with determining the current amplitudes of KCNQ3 or KCNQ4. We then focused on a phenylalanine at position 344 in KCNQ3 (analogous to F103 in KcsA) by studying the effects of mutating KCNQ3 at this position. To test for differential membrane protein expression, we performed TIRF assays. The results of homology modeling suggest that pore helix-S6 interactions play a critical role in governing KCNQ3 channel function. We conclude that the D-helix is only weakly implicated in governing KCNQ3 amplitudes compared with the pore-helix-S6 region.
Materials and Methods
For details about the materials and methods used in this work, see the Supporting Material.
Results
Reactive oxygen species induces oligomerization of KCNQ4 subunits mediated by a cysteine at position 643, but this residue does not govern current amplitudes
In a study by Howard et al. (19), the crystal structure of the KCNQ4 coiled-coiled D-helix highlighted some residues that were suggested to play a critical role in channel oligomerization of KCNQ4 (but not KCNQ3) by promoting salt-bridge formation. In that work, the F622L single mutation, or the D631S, G633E double mutation in the D-helix of KCNQ3, resulted in greater tetramerization of the D-helix when assayed biochemically; however, macroscopic KCNQ3 currents were not enhanced for either mutant when measured by electrophysiology in oocytes. Interestingly, KCNQ4 possesses a cysteine at position 643 at the end of the coiled-coil D-helix, whereas KCNQ3 has a histidine. We wondered whether C643 in KCNQ4 might promote channel oligomerization by forming intersubunit disulfide bonds. As a probe, we used hydrogen peroxide (H2O2), a strong reactive oxygen species (ROS) that is known to induce disulfide bonds between nearby cysteines. We biochemically assayed oligomerization by performing native PAGE on whole-cell lysates from Chinese hamster ovary (CHO) cells transfected with wild-type (WT) or mutant KCNQ4 subunits tagged with the myc epitope. Oligomerization was assayed on lysates from cells incubated in H2O2 alone (500 μM for 15 min), H2O2 followed by the reducing agent dithiothreitol (DTT, 2 mM for 15 min), or only a sham rinse. The gels were then immunoblotted with anti-myc antibodies.
We found that H2O2 markedly augmented oligomerization of KCNQ4 subunits, with dimers and tetramers being the most obvious forms seen (KCNQ4 WT; Fig. 1 A). The extent of oligomerization returned to control levels by DTT treatment, consistent with reversible disulfide bonds. As expected if H2O2 acts to cross-link cysteines, the H2O2-induced augmentation was completely abolished by replacement of all 11 cysteines in KCNQ4 with alanines (KCNQ4 C-less; Fig. 1 A). When only C112, C175, and C643 were left unmutated, H2O2 and DTT had the same effect as on the WT channels, suggesting that one or more of these cysteines are involved in H2O2-induced oligomerization. Individual introduction of cysteines at these three positions in the C-less KCNQ4 background revealed that the enhancement of oligomerization was due to the cysteine at the 643 position, because only that mutant displayed H2O2-induced oligomerization similar to that observed for WT KCNQ4 (KCNQ4 C643; Fig. 1 A).
Figure 1.
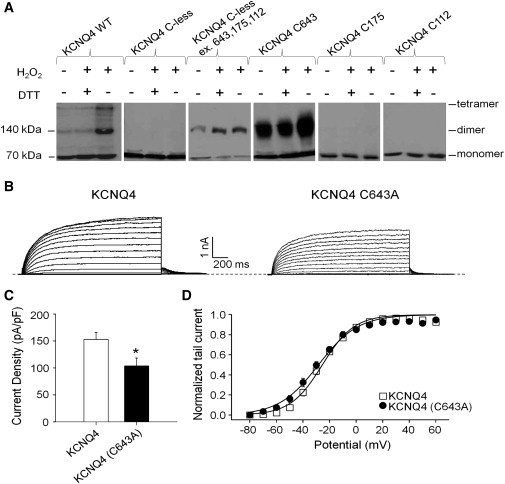
H2O2 induces oligomerization of KCNQ4 channels by intersubunit disulfide bonds between cysteines at the 643 position, but this residue is not a key factor in determining channel currents. (A) Representative immunoblots for cells transfected with the indicated WT or mutant myc-tagged KCNQ4 channels that were subjected to either sham rinse, H2O2 (500 μM, 15 min) alone, or H2O2 followed by DTT (2 mM, 15 min). (B) Representative perforated patch-clamp recordings from KCNQ4 and KCNQ4 (C643A). Cells were held at −80 mV and voltage steps were applied from −80 to 60 mV in 10 mV increments every 3 s. (C) Bars show summarized current densities at 60 mV for the indicated channels under perforated patch-clamp (n = 15–19; ∗p < 0.05). (D) Voltage dependence of activation measured from the normalized amplitudes of the tail currents at −60 mV, plotted as a function of test potential (n = 14–17).
To determine whether C643 plays a critical role in governing KCNQ4 current amplitudes by regulating functional tetramerization, we performed two sets of experiments. In the first set, we tested the effect of replacing this cysteine by an alanine (C643A) on KCNQ4 current amplitudes (Fig. 1, B–D). This mutation modestly but significantly decreased current amplitudes (Fig. 1, B and C). For cells transfected with KCNQ4 or KCNQ4 (C643A), the current densities were 153 ± 13 pA/pF (n = 15) and 104 ± 15 pA/pF (n = 19, p < 0.05), respectively. The voltage dependence of activation was not significantly affected (Fig. 1 D). We previously suggested that a triplet of cysteines (C156, C157, and C158) in the S2-S3 linker is involved in the enhancement of KCNQ4 currents by oxidative modification induced by H2O2 (24). However, given the C643 locus for H2O2-induced oligomerization identified above, we tested the effect of the C643A mutation on H2O2-induced augmentation of KCNQ4 currents under perforated-patch whole-cell clamp. As previously described, bath-application of 500 μM H2O2 increased the KCNQ4 current density by 2.1 ± 0.2-fold (n = 7, p < 0.001), which was completely reversed by 2 mM DTT (Fig. 2, A and C). Importantly, KCNQ4 C643A displayed unaltered H2O2 sensitivity, with the current density augmented by H2O2 by 2.2 ± 0.3-fold (n = 7, p < 0.001; Fig. 2, B and C). These data show that C643 is not involved in H2O2 enhancement of KCNQ4 currents, even though it is the locus of the H2O2-induced oligomerization seen in our in vitro assay.
Figure 2.
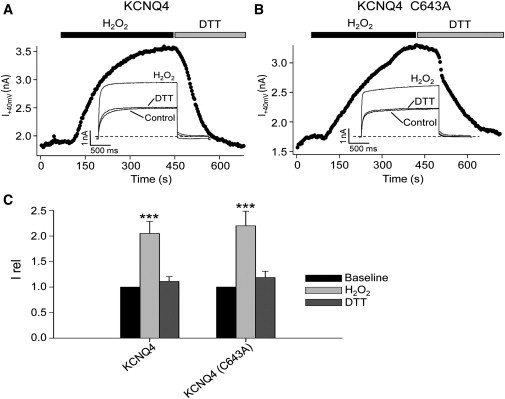
H2O2 induced enhancement of KCNQ4 and KCNQ4 (C643A) currents. (A and B) Plotted are the amplitudes of KCNQ4 and KCNQ4 (C643A) currents, while H2O2 (500 μM), or DTT (2 mM) were bath-applied, as indicated by the bars. Representative traces are shown in the insets. Cells were held at −80 mV and 2-s voltage steps were applied to 40 mV every 3 s. (C) Bars show summarized data. Irel is defined as IH2O2/I0 or IDTT/I0, where IH2O2 and IDTT have the obvious meanings, and I0 is the current before application of H2O2 or DTT. For all, n = 7; ∗∗∗p < 0.001.
Finally, to determine the role of this D-helix cysteine in enabling large macroscopic currents, we replaced the histidine at the analogous 646 position in KCNQ3 with a cysteine and tested the effect of this substitution on homomeric KCNQ3 and heteromeric KCNQ2/3 currents. However, we found that KCNQ3 (H646C) currents displayed a current density (12.5 ± 1.7 pA/pF, n = 8) similar to that of WT KCNQ3 channels (14.4 ± 1.3 pA/pF, n = 11), and KCNQ3 (H646C) subunits expressed with WT KCNQ2 yielded a current density (91 ± 8 pA/pF, n = 12) similar to that of WT KCNQ2+Q3 (92.2 ± 7.0 pA/pF, n = 12; Fig. 3 C). The H646C mutation in KCNQ3 also did not significantly affect the voltage dependence of activation for KCNQ3 homomers or KCNQ2/3 heteromers. For cells transfected with KCNQ3 or KCNQ3 (H646C), the half-activation potentials were −39 ± 3 mV (n = 9) and −42 ± 2 mV (n = 6), respectively. For cells transfected with KCNQ3 WT + KCNQ2 or KCNQ3 (H646C) + KCNQ2, the half-activation potentials were −29.7 ± 2.9 mV (n = 12) and −27.2 ± 2.3 mV (n = 12), respectively (Fig. 3 D). These data rule out the divergent residue at the 643/646 position as underlying the small KCNQ3 versus large KCNQ4 currents. Taken together, our data argue against a role of putative disulfide bonds at the 643 position at the end of the coiled-coil D-helix in governing KCNQ4 current amplitudes or KCNQ4 channel function.
Figure 3.
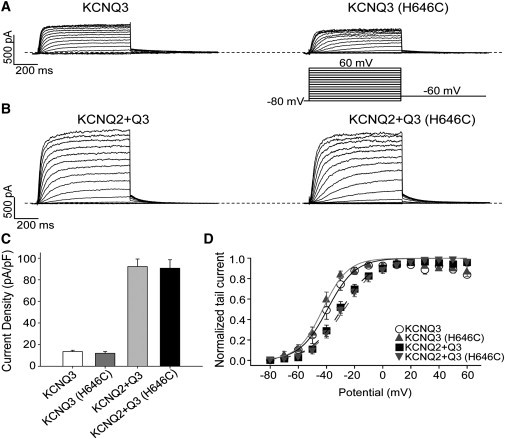
Effects of the H646C mutation on homomeric KCNQ3 and heteromeric KCNQ2 + Q3 currents. (A and B) Representative perforated patch-clamp recordings from cells transfected with WT KCNQ3, KCNQ3 (H646C), KCNQ2 + WT Q3, or KCNQ2 + Q3 (H646C). As shown in the inset, cells were held at −80 mV and voltage steps were applied from −80 to 60 mV in 10 mV increments. (C) Bars show summarized current densities at 60 mV for the indicated channels (n = 8–11). (D) Voltage dependence of activation measured from the normalized amplitudes of the tail currents at −60 mV, plotted as a function of test potential (n = 6–12).
Mutations at the position 344 in the S6 domain have dramatic effects on homomeric KCNQ3 currents
Cordero-Morales et al. (28,29) have described a network of hydrogen bonds among residues W67, E71, and D80 in KcsA that governs the stability of the SF and thus regulates C-type inactivation in those channels. That group recently suggested that a phenylalanine at the 103 position, in close proximity to the C-terminal end of the KcsA pore helix, is necessary to allow the channels to be locked in a nonconductive conformation (27). F103 mutations were found to affect KcsA gating in a side-chain size-dependent way, with substitutions of F103 by small side-chain residues (F103A and F103C) limiting entry into the C-type inactivated state, whereas the larger F103W substitution allowed the channel to inactivate (27). Interestingly, KCNQ3 also possesses a phenylalanine at the analogous position (F344). We thus hypothesized that the F344 mutation might likewise affect KCNQ3 pore stability, as manifested by the amplitude of KCNQ3 currents. To test this hypothesis, we studied the effect of those mutations on KCNQ3 currents by perforated-patch whole-cell clamp (Fig. 4). Indeed, the F344A, F344C, and F344W mutations strongly decreased the current density (Fig. 4 B). For CHO cells transfected with KCNQ3, KCNQ3 (F344A), KCNQ3 (F344C), and KCNQ3 (F344W), the current densities at 60 mV were 13.9 ± 2.1 pA/pF (n = 9), 4.6 ± 0.9 pA/pF (n = 7, p < 0.01), 3.9 ± 0.6 pA/pF (n = 8, p < 0.001), and 4.0 ± 0.3 pA/pF (n = 7, p < 0.001), respectively. Due to the small amplitude of all such homomeric KCNQ3 currents, we were not able to assess changes in channel voltage dependence induced by the F344 mutations. In contrast to KcsA, the effects of the mutations did not correlate with side-chain size. Moreover, they had opposite effects compared with KcsA, i.e., the F344 mutations decreased KCNQ3 currents instead of augmenting them, as in KcsA. These results suggest that the phenylalanine at the 344 position may play a role in stabilizing the SF of KCNQ3, in similarity to the effects of analogous mutations in Kv1.2 (27).
Figure 4.
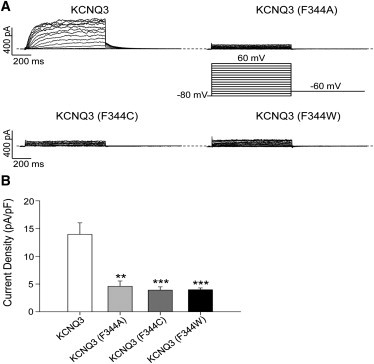
Effects of F344 mutations on KCNQ3 currents. (A) Representative perforated patch-clamp recordings from cells transfected with WT or mutant KCNQ3 channels. The pulse protocol used is as described in Fig. 3. (B) Bars show summarized current densities at 60 mV for the indicated channels (n = 7–9; ∗∗p < 0.01, ∗∗∗p < 0.001).
Small differences in plasma-membrane expression do not explain the effect of pore-region mutations
To determine whether the effects of F344 mutations are due to differences in plasma-membrane expression of KCNQ3 channels, we measured the plasma-membrane expression for WT and mutant KCNQ3 channels by examining individual CHO cells using TIRF microscopy. Under TIRF illumination, we can selectively excite fluorophores located within ∼300 nm of the plasma membrane, whereas any molecules located deeper in the cytoplasm will not be illuminated (30). We expressed YFP-tagged WT and mutant KCNQ3 channels in CHO cells, and estimated the membrane abundance of the channels by measuring the pixel intensity of images taken under TIRF illumination. Fig. 5 A shows representative TIRF images. The emission was quantified as described in our companion article (17). The results are summarized in Fig. 5 B, which shows that the decrease of the current density is not due to a lower expression of mutant KCNQ3 channels in the plasma membrane. We also studied the surface expression of KCNQ3 (H646C) channels. Surprisingly, the YFP emission from KCNQ3 (H646C) subunits was significantly decreased compared with WT KCNQ3, suggesting that the H646C mutation decreased the number of KCNQ3 channels at the plasma membrane without affecting the current density, as observed in Fig. 3 C. These results reinforce the idea that the C-terminus plays only a secondary role in KCNQ3 channel expression.
Figure 5.
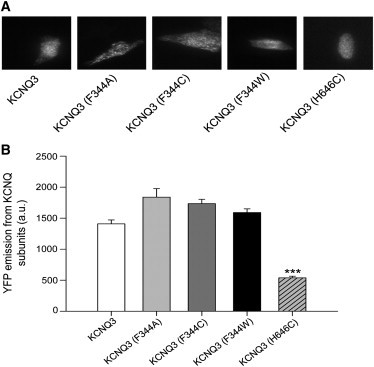
TIRF microscopy indicates that the effects of pore-region mutations result in only small differences in membrane expression of channels. (A) Shown are fluorescent images from TIRF microscopy of CHO cells expressing the indicated YFP-tagged channels. In all cases, CHO cells were transfected with a total of 2 μg cDNA. (B) Bars show summarized data for each channel type (n = 47–86; ∗∗∗p < 0.001).
Mutations at the 344 position in the S6 domain in KCNQ3 also reduce current amplitudes from KCNQ3 (A315T) currents
Because the introduction of a hydrophilic threonine or serine at the 315 position dramatically increases KCNQ3 current amplitudes (15–17), we wondered whether the destabilizing effect of mutating F344 to A, C, or W would also occur in the A315T background. Moreover, unlike KcsA, KCNQ3 channels have an alanine at the 315 position (T74 in KcsA). To test this, we evaluated the effect of F344A, F344C, and F344W mutations made in the KCNQ3 (A315T) background. We found that all F344 mutations strongly decreased the current density from KCNQ3 (A315T) channels (Fig. 6 A). For cells transfected with KCNQ3 (A315T), KCNQ3 (A315T-F344A), KCNQ3 (A315T-F344C), and KCNQ3 (A315T-F344W), the current densities at 60 mV were 174 ± 16 pA/pF (n = 11), 4.3 ± 0.7 pA/pF (n = 7, p < 0.001), 57 ± 6 pA/pF (n = 8, p < 0.001), and 41 ± 5 pA/pF (n = 7, p < 0.001), respectively (Fig. 6 B). All mutants displayed a voltage dependence of activation that was unchanged from KCNQ3 (A315T) (Fig. 6 C). These results indicate that the destabilizing effects of mutating F344 in KCNQ3 dominate over the stabilizing effect of introducing a threonine at the 315 position. We then proceeded to perform further homology modeling to gain structural insight into these results.
Figure 6.
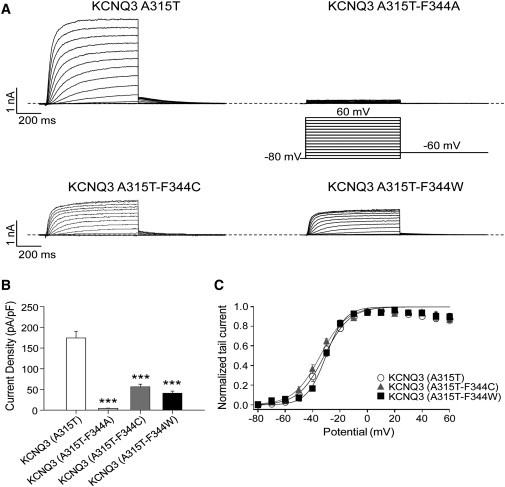
Effects of F344 mutations on KCNQ3 (A315T) currents. (A) Representative perforated patch-clamp recordings from cells transfected with KCNQ3 (A315T), KCNQ3 (A315T-F344A), KCNQ3 (A315T-F344C), or KCNQ3 (A315T-F344W). The pulse protocol used is as described in Fig. 3. (B) Bars show summarized current densities at 60 mV for the indicated channels (n = 7–11; ∗∗∗p < 0.001). (C) Voltage dependence of activation measured from the normalized amplitudes of the tail currents at –60 mV, plotted as a function of test potential (n = 7–11).
van der Waals interactions between F344 and the 315 position are required to stabilize KCNQ3 channels in a conductive conformation
Cuello and co-workers (27) showed that F103A and F103C mutations (F344 in KCNQ3) destabilize the nonconductive conformation of KcsA. On the contrary, we showed that the equivalent mutations strongly decreased WT KCNQ3 currents, as well as those from KCNQ3 (A315T) channels. We hypothesized that similar van der Waals interactions exist in KCNQ3 channels, but in this case act to stabilize the SF in a conductive conformation. Among the solved KcsA, KvAP, and Kv1.2 crystal structures, KcsA is the only channel that has a phenylalanine at the analogous position as KCNQ3 and in which such interactions have been described so far; thus, we used that channel as the template for our modeling. We modeled the region of the pore of KCNQ3 using the structure of KcsA (31) as the template. For each channel, the predicted structure of the SF and associated pore-helix, the predicted hydrogen bonds between them, and the predicted van der Waals interactions between S6 at position 344 and the pore helix are shown in Fig. 7. Homology modeling suggests that a phenylalanine at the 344 position is close enough to alanine at the 315 position to allow for van der Waals interactions in WT KCNQ3 (Fig. 7 A). This proximity between A315 and F344 fits with the hypothesis that the interaction between these residues is critical for stabilizing KCNQ3 channels in a conductive conformation. Consistent with this notion, the distance between A315 and the residue at the 344 position increases when the native phenylalanine is mutated to small side-chain residues such as alanine or cysteine, and becomes too large for both residues to maintain close contact (Fig. 7, B and C). In contrast, tryptophan has a side chain as long as that of phenylalanine. Consequently, we could expect the F344W mutation to have little effect. Unexpectedly, though, we observed that this mutation resulted in an effect similar to that observed with mutation to an alanine or a cysteine. However, the modeling predicts that the F103W mutation will also disrupt the interaction between residues at the 315 and 344 positions (Fig. 7 D), probably because tryptophan adopts a different rotamer orientation compared with phenylalanine. In our modeling, we did not bias the orientation of any of the rotamers. Instead, the structure of WT and KCNQ3 (F344W) predicted by the energy minimization algorithm suggests that the aromatic ring of phenylalanine points toward to the nitrogen atom of alanine at the 315 position, whereas that of tryptophan points toward to V341 in another subunit, preventing tryptophan from making contact with A315.
Figure 7.
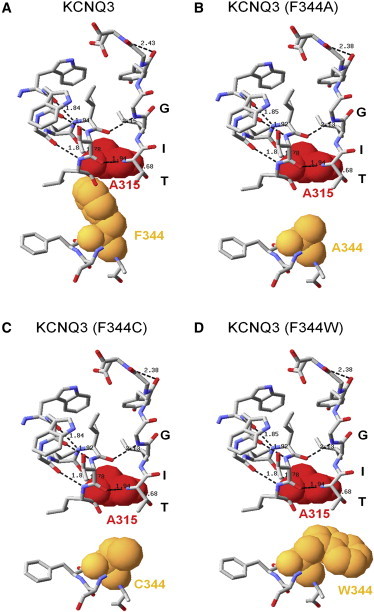
Comparison of the structures of the pore region of WT (A) and mutant KCNQ3 (B–D) predicted by homology modeling. Shown are the predicted structures of the SF and associated pore-helix with the predicted hydrogen bonds between them and the predicted structure of the S6 domain, with key amino acid side chains shown as van der Waals spheres.
We then performed a similar analysis for the F344A, F344C, and F344W mutants in the A315T background as we performed for the F344 mutants of WT KCNQ3. Again, homology modeling predicts that the van der Waals interactions between F344 in S6 and T315 in the pore helix (Fig. 8 A) will be abolished by the F344A, F344C, or F344W mutations (Fig. 8, B–D). These predictions suggest that the decrease of the current density is due to the disruption between S6 and the pore helix. Finally, a similar reorientation of the aromatic ring of tryptophan is suggested to disrupt the interaction between residues at the 315 and 344 positions. We conclude that a coupling between S6 at F344 and A315 in the pore helix, mediated by van der Waals interactions, may be a key factor in governing KCNQ3 channel current amplitudes.
Figure 8.
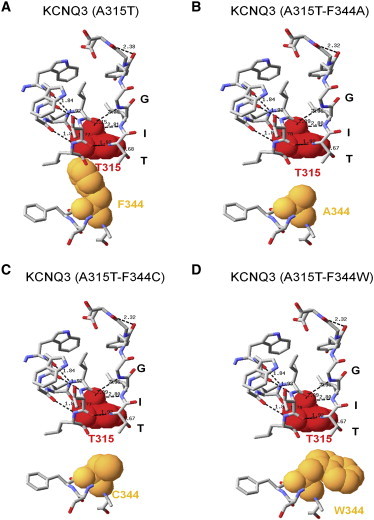
Comparison of the structure of the pore region of KCNQ3 (A315T) (A) and double mutants (A315T-F344) (B–D) predicted by homology modeling. Shown are the predicted structures of the SF and associated pore-helix with the predicted hydrogen bonds between them and the predicted structure of the S6 domain, with key amino acid side chains shown as van der Waals spheres.
Discussion
A number of researchers have investigated the mechanisms underlying the small KCNQ3 current amplitudes. Two major mechanisms have been suggested to be involved in the differential amplitude of their macroscopic currents. The first one is differential surface expression, controlled by the A domain of the distal C-terminus (11). The crystal structure of the D-helix of KCNQ4 indicates that the KCNQ3 coiled-coiled A-domain tail (the D-helix) is divergent at a number of critical positions that hinder tetramerization (19). In particular, the presence of four phenylalanines (one from each subunit) in the coiled-coiled core of the KCNQ3 D-helix was suggested to prevent a stable tetramer, and the F622L mutation to convert the D-helix of KCNQ3 into a form that biochemical analysis indicates is predominantly tetrameric. A similar analysis indicated that the lack of appropriate salt bridges in the KCNQ3 D-helix hinders tetramerization, and that the D631S/G633E double mutation, suggested to restore this salt bridge, promotes tetrameric in vitro properties to the KCNQ3 D-helix. However, neither mutation in the full-length KCNQ3 led to a functional augmentation of KCNQ3 current amplitudes when the channels were expressed in Xenopus oocytes (19), suggesting that the A-domain is critical for channel assembly but is not the key factor in governing KCNQ current amplitudes.
Our work here is in full agreement in both aspects. We show that replacement of the native cysteine (C643) with an alanine, at the end of the D-helix in KCNQ4, abolished KCNQ4 oligomerization induced by H2O2 when measured in vitro. However, this substitution led only to a slight decrease of KCNQ4 current amplitudes when the channels were functionally tested. Moreover, the H646C mutation did not modify KCNQ3 homomeric and heteromeric KCNQ2/3 current amplitudes. Furthermore, our TIRF assays showed that the number of KCNQ3 channels at the plasma membrane decreased in the presence of the H646C mutation, but the current amplitude was not altered, arguing against a correlation between surface expression of channels and current amplitudes. Consistent with this, Etxeberria and co-workers (15) did not find such a correlation between current amplitudes and surface expression of various KCNQ2 and KCNQ3 chimeras and point mutants, and suggested that pore regions of the channels are involved in governing KCNQ current amplitudes. Moreover, coexpression of WT KCNQ3 channels with KCNQ3 (A315T), which is known to increase current amplitudes by ∼15-fold, did not modify the ability of channels to form tetramers (23). In accord with those results, our laboratory has suggested that a dramatic increase in current amplitudes arises from a network of interactions between the pore helix (T315 and I312) and the lower part of the SF, which prevents the SF structure from locking closed (16). Thus, we hypothesized that although KCNQ3 homomers are well expressed at the membrane, most of them are in a nonconductive conformation. This mechanism predicts that I312 is critical to this process, and we investigated that issue in our companion study (17).
Much work has suggested that a nonconductive conformation of K+ channels is due to a constriction of the SF induced by a C-type inactivation process (32,33). In KcsA, two networks of interactions have been postulated to underlie C-type inactivation (27,28,34). The first one is located at the top of the SF, where interactions between D80 and W67 in KcsA (D321 and W308 in KCNQ3) are stabilized by the E71 residue (I312 in KCNQ3), converting channels to a structural variant in which C-type inactivation occurs (28). Our homology modeling predicts that a network of interactions between the pore helix (I312) and the top of the SF leads to constriction of the SF (17). The second network involves van der Waals interactions between F103 in S6, the pore helix (T74, T75), and I100 in the adjacent subunit. Disrupting this network of interactions stabilizes the KcsA pore in a conductive conformation (27). KCNQ3 channels also possess a phenylalanine at the equivalent position (F344). Here, we found that mutations at the F344 position dramatically decreased KCNQ3 currents, suggesting that F344 in also intimately involved in pore stabilization in KCNQ3 homomers. TIRF studies revealed only small differences of YFP-tagged channels that cannot explain the differential current amplitudes, suggesting that other mechanisms are dominant. To gain insight into the molecular mechanism involved in this phenomenon, we performed homology modeling of the S6 domain and the pore region of WT and mutant KCNQ3 channels, using KcsA as a template. The modeling suggests that the decrease of current in the F344 mutants is due to the disruption of the van der Waals interactions between F344 in S6 and the pore helix at A315. However, the impact of the pore helix-S6 interactions on the structure of the SF remains to be elucidated in KCNQ3 homomers. In addition, we cannot yet conclude with certainty that a coupling between S6 at F344 and A315 in the pore helix, mediated by van der Waals interactions, is a key factor in governing KCNQ3 channel permeation without comparing the open probability and single-channel current amplitudes of the WT and mutant channels. Finally, our homology modeling does not have the capability to evaluate the structural rearrangements of the SF due to the disruption of the pore helix-S6 interactions. One explanation could be that the previously described network of hydrogen bonds between the pore helix and the SF (16) is destabilized by the disruption of the pore helix-S6 interactions, affecting the stability of the conductive state of the SF. Another hypothesis is that residues between the pore helix and the top of the SF, as described in the companion article (17), are close enough to make interactions, locking channels in the closed, nonconductive state. Consistent with this notion, in KcsA, introducing the mutation F103A in the background of the inactivating mutant E71H abolished the inactivated conformation, suggesting that interactions between the pore helix (D80) and the top of the SF (W67 and E71) are destabilized by mutation of F103. In agreement with this, the crystal structure of the E71H-F103A shows that the SF is in a conductive conformation (27).
In our modeling, we did not bias the orientation of any of the rotamers. At first glance, we were surprised that the substitution of Phe-344 with the similarly sized tryptophan had such a sizable effect on current amplitudes. Instead, the structure of WT and F344W KCNQ3 predicted by the model suggests that the aromatic ring of phenylalanine points toward to the nitrogen atom of alanine at the 315 position, whereas that of tryptophan points toward to V341 in another subunit, preventing tryptophan from making contact with A315. However, this subtle yet important difference in rotamer configuration involved in the F344W substitution precisely parallels the state-dependent rotamer configuration of F103 shown by the open- and closed-state crystal structures of KcsA (27). In those structures, the aromatic ring of F103 changes its rotamer configuration as a consequence of channel opening, with an orientation parallel to the S6 helix in the closed state but a configuration clashing with I100 in the neighboring subunit in the open state, destabilizing the pore. This molecular mechanism was recently accurately modeled with the use of free-energy molecular-dynamics simulations (35).The mechanism by which W344 destabilizes KCNQ3 channels may be identical, involving a steric clash with the V341 of the neighboring subunit, the analogous residue to I100 in KcsA. Apparently, this molecular mechanism is well conserved among a variety of K+ channels, as previously suggested (27).
Interestingly, the phenylalanine at the 344 position in KCNQ3 is highly conserved in KCNQ1-5 α-subunits. Mutations of the analogous F340 in KCNQ1 lead to channel inactivation (36,37). Based on the closed channel structure of KcsA, homology modeling of the pore of KCNQ1 suggests that this phenylalanine is capable of interacting with V310, which is located in the pore helix, and with L273 in S5, and may destabilize the open state of the pore (36). V310 of KCNQ1 is located next to the residue homologous to A315 of KCNQ3. Moreover, Gibor and colleagues (38) showed that the L273F mutation causes a decrease in the flexibility of the SF at the level of the S1 permeant ion-binding site, mediating the inactivation process. Taken together, these studies suggest that disruption of pore helix-S6 interactions likely affects SF flexibility. It is quite possible that the disruption of pore helix-S6 interactions in KCNQ3 likewise leads to a decrease of SF flexibility, causing destabilization of KCNQ3 channels. Finally, the network of residues that form this interaction has also been suggested in Kv1.2 and Shaker (27) and may also be applicable to hERG, in which the analogous F656 in S6 has been suggested to be involved in inactivation (39).
In this study and our companion article (17), we highlight two networks of interactions that affect the stability of the SF, as summarized in Fig. 9. The first involves hydrogen bonds between the pore helix and the SF. In WT KCNQ3, I312 is free to rotate, which destabilizes the hydrogen bonds it can make with the lower part of the SF. Thus, most WT KCNQ3 homomers are dormant, and currents are only modest. A hydrophilic residue at position 312 (I312E, I312K, and I312R) leads to the creation of hydrogen bonds between this residue in the pore helix and the top of the SF, locking channels in a nonconductive conformation (17). In contrast, a hydrophilic substitution at position 315 (A315T,S) stabilizes the position of I312 via a hydrogen bond, and consequently stabilizes the interaction that I312 makes with the lower part of the SF, which adopts a reliably conductive conformation. However, this conformation is disrupted in the presence of a hydrophilic residue at position 312, indicating that the destabilizing effect of interactions between the pore helix at the 312 position and the top of the SF dominates over the stabilizing effect of interactions between the residues in the inner vestibule at positions 312 and 315. The second network involves van der Waals interactions between the S6 domain at position 344 and the pore helix. In WT channels or the A315T mutant, this interaction is strong, allowing a weakly stable or strongly stable SF, respectively. However, disruption of this van der Waals interaction (F344A, F344C, and F344W) between the S6 domain at position 344 and the pore helix at position 315 is inconsistent with the stability of the KCNQ3 SF, even in the presence of an isoleucine at 312 and a stabilizing threonine or serine at position 315. Thus, the destabilizing effect of removing this van der Waals interaction also dominates over the stabilizing interactions between T315 and I312. We conclude that a stable KCNQ3 conductive pathway is mediated by S6-pore helix-SF interactions, the presence or absence of which is largely responsible for the widely variable amplitude of KCNQ3 currents. The intrinsically weakly unstable pore of KCNQ3 homomers probably serves to make transcriptional control of KCNQ2 subunit expression the master switch in determining M-current amplitudes in the nervous system, consistent with the large number of inherited KCNQ2 mutations underlying syndromes of human epilepsy (40).
Figure 9.
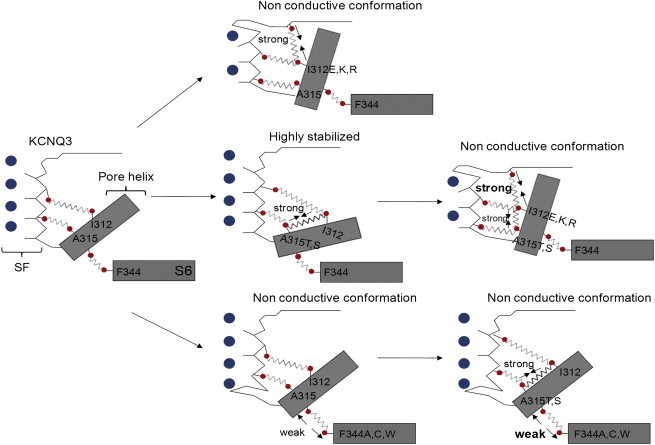
Structural rearrangements of the pore region involved in KCNQ3 gating. Shown is a schematic representation of the network of interactions suggested to be involved in KCNQ3 gating, taken from this work and the companion study (17). For details, see the last paragraph of the Discussion section.
Acknowledgments
We thank Ciria C. Hernandez for assistance with the modeling, and Pamela Reed for expert technical assistance. All authors declare that there is no conflict of interest.
This work was supported by National Institutes of Health grants R01 NS043394 and R01 NS065138.
Supporting Material
References
- 1.Jentsch T.J. Neuronal KCNQ potassium channels: physiology and role in disease. Nat. Rev. Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- 2.Wang H.S., Pan Z., McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 3.Schroeder B.C., Hechenberger M., Jentsch T.J. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- 4.Bal M., Zhang J., Shapiro M.S. Homomeric and heteromeric assembly of KCNQ (Kv7) K+ channels assayed by total internal reflection fluorescence/fluorescence resonance energy transfer and patch clamp analysis. J. Biol. Chem. 2008;283:30668–30676. doi: 10.1074/jbc.M805216200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y., Gamper N., Shapiro M.S. Single-channel analysis of KCNQ K+ channels reveals the mechanism of augmentation by a cysteine-modifying reagent. J. Neurosci. 2004;24:5079–5090. doi: 10.1523/JNEUROSCI.0882-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selyanko A.A., Hadley J.K., Brown D.A. Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J. Physiol. 2001;534:15–24. doi: 10.1111/j.1469-7793.2001.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pusch M. Increase of the single-channel conductance of KvLQT1 potassium channels induced by the association with minK. Pflugers Arch. 1998;437:172–174. doi: 10.1007/s004240050765. [DOI] [PubMed] [Google Scholar]
- 8.Li Y., Gamper N., Shapiro M.S. Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 2005;25:9825–9835. doi: 10.1523/JNEUROSCI.2597-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kubisch C., Schroeder B.C., Jentsch T.J. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437–446. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- 10.Gamper N., Stockand J.D., Shapiro M.S. Subunit-specific modulation of KCNQ potassium channels by Src tyrosine kinase. J. Neurosci. 2003;23:84–95. doi: 10.1523/JNEUROSCI.23-01-00084.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwake M., Athanasiadu D., Friedrich T. Structural determinants of M-type KCNQ (Kv7) K+ channel assembly. J. Neurosci. 2006;26:3757–3766. doi: 10.1523/JNEUROSCI.5017-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schenzer A., Friedrich T., Schwake M. Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J. Neurosci. 2005;25:5051–5060. doi: 10.1523/JNEUROSCI.0128-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwake M., Pusch M., Jentsch T.J. Surface expression and single channel properties of KCNQ2/KCNQ3, M-type K+ channels involved in epilepsy. J. Biol. Chem. 2000;275:13343–13348. doi: 10.1074/jbc.275.18.13343. [DOI] [PubMed] [Google Scholar]
- 14.Maljevic S., Lerche C., Lerche H. C-terminal interaction of KCNQ2 and KCNQ3 K+ channels. J. Physiol. 2003;548:353–360. doi: 10.1113/jphysiol.2003.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Etxeberria A., Santana-Castro I., Villarroel A. Three mechanisms underlie KCNQ2/3 heteromeric potassium M-channel potentiation. J. Neurosci. 2004;24:9146–9152. doi: 10.1523/JNEUROSCI.3194-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zaika O., Hernandez C.C., Shapiro M.S. Determinants within the turret and pore-loop domains of KCNQ3 K+ channels governing functional activity. Biophys. J. 2008;95:5121–5137. doi: 10.1529/biophysj.108.137604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choveau F.S., Hernandez C.C., Shapiro M.S. Pore determinants of KCNQ3 K+ current expression. Biophys. J. 2012;102:2489–2498. doi: 10.1016/j.bpj.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwake M., Jentsch T.J., Friedrich T. A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep. 2003;4:76–81. doi: 10.1038/sj.embor.embor715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howard R.J., Clark K.A., Minor D.L., Jr. Structural insight into KCNQ (Kv7) channel assembly and channelopathy. Neuron. 2007;53:663–675. doi: 10.1016/j.neuron.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schroeder B.C., Kubisch C., Jentsch T.J. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature. 1998;396:687–690. doi: 10.1038/25367. [DOI] [PubMed] [Google Scholar]
- 21.Yang W.P., Levesque P.C., Blanar M.A. Functional expression of two KvLQT1-related potassium channels responsible for an inherited idiopathic epilepsy. J. Biol. Chem. 1998;273:19419–19423. doi: 10.1074/jbc.273.31.19419. [DOI] [PubMed] [Google Scholar]
- 22.Chung H.J., Jan Y.N., Jan L.Y. Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in the KCNQ2 and KCNQ3 C-terminal domains. Proc. Natl. Acad. Sci. USA. 2006;103:8870–8875. doi: 10.1073/pnas.0603376103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gómez-Posada J.C., Etxeberría A., Villarroel A. A pore residue of the KCNQ3 potassium M-channel subunit controls surface expression. J. Neurosci. 2010;30:9316–9323. doi: 10.1523/JNEUROSCI.0851-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gamper N., Zaika O., Shapiro M.S. Oxidative modification of M-type K(+) channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 2006;25:4996–5004. doi: 10.1038/sj.emboj.7601374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoshi T., Zagotta W.N., Aldrich R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 26.Kiss L., LoTurco J., Korn S.J. Contribution of the selectivity filter to inactivation in potassium channels. Biophys. J. 1999;76:253–263. doi: 10.1016/S0006-3495(99)77194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuello L.G., Jogini V., Perozo E. Structural basis for the coupling between activation and inactivation gates in K+ channels. Nature. 2010;466:272–275. doi: 10.1038/nature09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cordero-Morales J.F., Cuello L.G., Perozo E. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 2006;13:311–318. doi: 10.1038/nsmb1069. [DOI] [PubMed] [Google Scholar]
- 29.Cordero-Morales J.F., Jogini V., Perozo E. Molecular driving forces determining potassium channel slow inactivation. Nat. Struct. Mol. Biol. 2007;14:1062–1069. doi: 10.1038/nsmb1309. [DOI] [PubMed] [Google Scholar]
- 30.Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Methods Enzymol. 2003;361:1–33. doi: 10.1016/s0076-6879(03)61003-7. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Y., Morais-Cabral J.H., MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 32.Yellen G., Sodickson D., Jurman M.E. An engineered cysteine in the external mouth of a K+ channel allows inactivation to be modulated by metal binding. Biophys. J. 1994;66:1068–1075. doi: 10.1016/S0006-3495(94)80888-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y., Jurman M.E., Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- 34.Cordero-Morales J.F., Jogini V., Perozo E. A multipoint hydrogen-bond network underlying KcsA C-type inactivation. Biophys. J. 2011;100:2387–2393. doi: 10.1016/j.bpj.2011.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan A.C., Cuello L.G., Roux B. Thermodynamic coupling between activation and inactivation gating in potassium channels revealed by free energy molecular dynamics simulations. J. Gen. Physiol. 2011;138:571–580. doi: 10.1085/jgp.201110670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seebohm G., Westenskow P., Sanguinetti M.C. Mutation of colocalized residues of the pore helix and transmembrane segments S5 and S6 disrupt deactivation and modify inactivation of KCNQ1 K+ channels. J. Physiol. 2005;563:359–368. doi: 10.1113/jphysiol.2004.080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Panaghie G., Purtell K., Abbott G.W. Voltage-dependent C-type inactivation in a constitutively open K+ channel. Biophys. J. 2008;95:2759–2778. doi: 10.1529/biophysj.108.133678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibor G., Yakubovich D., Attali B. An inactivation gate in the selectivity filter of KCNQ1 potassium channels. Biophys. J. 2007;93:4159–4172. doi: 10.1529/biophysj.107.107987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J., Seebohm G., Sanguinetti M.C. Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc. Natl. Acad. Sci. USA. 2002;99:12461–12466. doi: 10.1073/pnas.192367299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maljevic S., Wuttke T.V., Lerche H. Nervous system KV7 disorders: breakdown of a subthreshold brake. J. Physiol. 2008;586:1791–1801. doi: 10.1113/jphysiol.2008.150656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.