

Abstract
Ebola virus assembles and buds from the inner leaflet of the plasma membrane of mammalian cells, which is primarily attributed to its major matrix protein VP40. Oligomerization of VP40 has been shown to be essential to the life cycle of the virus including formation of virions from infected cells. To date, VP40 oligomerization has mainly been assessed by chemical cross-linking following cell fractionation studies with VP40 transfected cells. This has made it difficult to discern the spatial and temporal dynamics of VP40 oligomerization. To gain a better understanding of the VP40 assembly and oligomerization process in live cells, we have employed real-time imaging of enhanced green fluorescent protein tagged VP40. Here, we use both confocal and total internal reflection microscopy coupled with number and brightness analysis to show that VP40 oligomers are localized on the plasma membrane and are highly enriched at sites of membrane protrusion, consistent with sites of viral budding. These filamentous plasma membrane protrusion sites harbor VP40 hexamers, octamers, and higher order oligomers. Consistent with previous reports, abrogation of VP40 oligomerization through mutagenesis greatly diminished VP40 egress and also abolished membrane protrusion sites enriched with VP40. In sum, real-time single-molecule imaging of fluorescently labeled Ebola VP40 is able to resolve the spatial and temporal dynamics of VP40 oligomerization.
Introduction
Viral hemorrhagic fevers including those stemming from the Ebola virus pose a serious public health threat in central and eastern Africa with fatality rates as high as 90% (1–4). Thus far, there are no FDA approved vaccines or therapeutics to treat or prevent Ebola infections and hence Ebola is classified as a biosafety level 4 (BSL-4) agent (5). In addition to the aforementioned outbreaks in humans there have been incidences of Ebola transmitted in animal facilities in the United States but without human fatalities (6). With the growing threat of bioterrorism, Ebola virus represents a biological agent that could be weaponized by terrorists; hence, the urgent need to identify therapeutic targets (7–9).
The Ebola virus uses a negative sense RNA genome encoding seven proteins to replicate in the host cell (10,11). The nucleoprotein, VP30, VP35, and L protein constitute the nucleocapsid (NC), which is critical for transcription and viral replication (11). The glycoprotein is rooted in the lipid envelope of the virus and is responsible for entry of virions into cells (12). VP40 and VP24 are the matrix proteins of the virus and are important for budding as well as virus structure and stability (7,10,13,14). VP40 (see Fig. 1 A) is the major matrix protein and the most abundant protein of the virus, which plays a central role in the budding of the virus from the plasma membrane. For example, expression of VP40 alone in mammalian cells assembles into virus-like particles (VLPs) that have similar characteristics to the authentic Ebola virus (14–16). Additionally, studies have shown that in the absence of VP40, the nucleocapsid is not effectively transported to the plasma membrane, the site of assembly and budding, where it is incorporated into the virions (17). Therefore, understanding how VP40 regulates assembly of VLPs both in vitro and in live cells is critical for identifying therapeutic targets for inhibiting the replication and spread of the virus (7). The assembly of VLPs by Ebola VP40 also represents an attractive model for studying the assembly of the virus in a BSL-2 setting because the VLPs are noninfectious.
Figure 1.
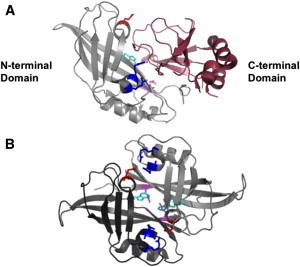
Structure of VP40 and VP40 intradimeric interface. VP40 consists of an N-terminal domain (gray) that has been shown to be involved in oligomerization and a C-terminal domain (raspberry) that has been deemed important for membrane binding. (A) VP40 x-ray structure (PDB: 1ES6) with the N-terminal domain and C-terminal domain colored in gray and raspberry, respectively. Purported residues involved in oligomerization are highlighted: Trp95 (cyan), Arg148 and Arg149 (blue), Glu160 (red), and Glu184 (magenta). (B) Antiparallel monomeric-monomeric interface (intradimeric interface with one monomer in dark gray and the second monomer in light gray) highlighting the Glu160 (red) interaction with Arg148 and Arg149 (blue), and the Trp95 (cyan) interaction with Glu184 (magenta) (PDB: 1H2D).
Progress has been made in understanding how VP40 forms VLPs. For example, VP40 associates with the plasma membrane (18) where it presumably initiates assembly. Additionally, host factors such as the ESCRT machinery (15,19) as well as COPII proteins (20,21) have been implicated in the budding and transport of VP40, respectively. However, how the virus assembles on the plasma membrane before virion release remains poorly understood. Oligomerization of VP40 is thought to be an important step in this process as studies have shown that VP40 oligomerizes both in the cell (18,22,23) and in solution (24). Moreover, VP40 oligomers have been detected in VLPs and ultraviolet-inactivated virions (18,25). VP40 has primarily been shown to oligomerize into hexamers and octamers (22–25), which share a similar intradimeric (monomer-monomer interface) antiparallel interface (see Fig. 1 B) (26). Although these studies have contributed to our understanding of VP40 behavior, it is difficult to predict the oligomerization dynamics of VP40 in cells from these studies. In addition, the methods used for purification and subsequent identification of these oligomeric structures make it difficult to assess the true size of the oligomers in the real-time assembly and oligomerization of VP40.
Studies have shown that VP40 oligomers are essential for the formation of VLPs (22,27) and have been found to be associated with detergent-resistant membranes (18), suggesting that the plasma membrane may play an important role in the oligomerization of VP40. Oligomerization of the matrix protein on the plasma membrane may serve as a scaffold to recruit host proteins as well as provide the necessary force to bring about membrane deformation and virus particle formation (28,29). Thus, understanding the spatial and temporal dynamics of VP40 oligomerization is critical to unraveling how the virus buds form at the plasma membrane. Furthermore, the origin of these oligomers is still unclear and it is not known whether VP40 oligomers are recruited from the cytoplasm or an intracellular membrane to the site of budding. Therefore, the analysis of formation of VP40 oligomers in live cells would provide keen insight into the spatial and temporal architecture of VP40 assembly.
In this report, we have resolved the oligomerization state of VP40 in live cells by a combination of total internal reflection (TIRF) microscopy and the number and brightness (N&B) analysis (30–32) developed at the Laboratory for Fluorescence Dynamics (www.lfd.uci.edu). This method is noninvasive and is best suited to study assembly at the plasma membrane as molecules that are on the plasma membrane are selectively illuminated (31,33). The studies reveal that the assembly of VP40 is dynamic as monomeric VP40 is recruited from the cytosol to the plasma membrane where VP40 oligomers are observed. In addition, N&B analysis was able to resolve VP40 oligomerization associated with sites of membrane protrusion. This study highlights the value of the N&B approach coupled with TIRF microscopy in defining the oligomeric state of cytosolic and membrane bound viral matrix proteins.
Materials and Methods
Plasmids
The mammalian expression plasmid for VP40 was created by subcloning VP40 into enhanced green fluorescent protein (EGFP)-pcDNA3.1 using EcoRI and XhoI restriction sites with standard cloning methods.
Cell culture
Human embryonic kidney (HEK293T) cells were cultured and maintained at 37°C in a 5% CO2 humidified incubator supplemented with Dulbecco's modified Eagle's medium (low glucose) containing 10% fetal bovine serum and 1% Pen/Strep. After trypsinization, cells were transferred from a T-25 tissue culture flask to a 35 mm glass bottom microwell dish coated with 1–2 μg/ml fibronectin (MatTek, Ashland, MA). Cells were then grown to 50–80% confluency and transfected with 1 μg DNA/dish using lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol.
RICS data acquisition
Fluorescence intensity fluctuations within the focal volume of a live cell can be attributed to protein binding to immobile or less mobile fractions, protein conformation changes, and the entry and exit of molecules through the focal volume. Raster image correlation spectroscopy (RICS) is the ideal method for measuring these processes as the scanning movement creates a space-time matrix of pixels within each image. On commercial confocal microscopes the spatial and temporal sampling time of the laser beam (pixel dwell time) is known as is the scan time between scan lines and time between images. Thus, using RICS allows for a generation of spatial-temporal maps of dynamics occurring across the living cell. RICS data were acquired on a commercial laser scanning confocal microscope (Zeiss LSM710 inverted microscope) using a Plan Apochromat 63× 1.4 NA oil objective. The 488 nm line of the Ar ion laser was used for excitation of EGFP. The laser power was maintained at 1% throughout the experiment with the emission collected through a 493–556 nm filter. The data were collected as images of 256 × 256 pixels with a pixel dwell time of 12.6 μs. RICS analysis was done with SimFCS software (Laboratory for Fluorescence Dynamics) using 100 frames of the image series.
TIRF imaging
TIRF imaging was performed using a homebuilt TIRF imaging system (model No. IX81 microscope (Olympus, Melville, NY) as described previously (33). Briefly, images were collected using a Cascade 512B EMCCD camera. Samples were illuminated with the 488 nm line from an Ar ion laser (Melles Griot, Albuquerque, NM) through a 60×, 1.45 NA oil objective (Olympus). To ensure cell integrity, cells were maintained at 37°C using a thermostated stage (Tokai Hit, Shizuoka, Japan). Images were collected at 256 × 256 pixels with a 50 ms exposure time per frame (4000 total frames were collected. Images were saved as 16 bit unsigned and imported into the SimFCS software (Laboratory for Fluorescence Dynamics).
Data analysis
TIRF image series were analyzed using SimFCS (Laboratory for Fluorescence Dynamics). For N&B analysis, 512 frames were analyzed per image series. HEK293T cells expressing monomeric EGFP, a brightness standard, were imaged under the same conditions as EGFP-VP40 and respective mutations. The brightness of the EGFP was used as the brightness of the monomer. The selection window for analysis in the brightness versus intensity plot was based on the average brightness of a monomer (0.104), which allowed for selection of oligomeric size based upon multiple of the monomer. Thus, the selection window for each species is based upon the average brightness, which will yield an average population of each species in the respective area of analysis.
TIRF microscopy and N&B analysis
Determination of the average number and brightness of fluorescent molecules in live cells is a challenging task but an important one as many biological processes often depend upon protein clustering. Recently, determination of fluorescently labeled protein clustering has become much more robust with N&B analysis. N&B analysis is based upon moment analysis and allows for measurement of the average number of molecules as well as brightness in each pixel of a fluorescent microscopy image (31). Here, the average brightness of a particle is determined from the ratio of variance/intensity at each pixel. Fluctuating particles can be determined by dividing the average intensity by the brightness at each pixel. For particles fluctuating in the focal volume, the variance is proportional to the square of the particle brightness; however, the variance of the immobile particles and detector noise is proportional to the intensity of these components. Thus, only fluorescent fluctuations that are dependent upon the mobile particles (square of the brightness) have a ratio of the variance/intensity >1. Brightness maps then allow determination of the aggregation of mobile fluorescent particles with pixel resolution.
For the electron multiplying charge-coupled device camera the following equations were used to compute the number and brightness.
| (1) |
| (2) |
where N and B are the apparent number and the brightness of the molecule, is the average intensity of a pixel throughout a stack of frames (time average), σ2 is the variance, and offset and σ02 are properties that depend on the camera hardware. With these parameters properly calibrated, we obtained the distribution of the brightness of each individual pixel in the image of the cell under investigation.
VLP assays
Recently, it has been shown that in addition to VLP formation VP40 is secreted from human cells by an unconventional mechanism (34). This means that VLP assays must be properly controlled by measuring VP40 in VLPs and not a simple analysis of VP40 present in the cellular media as an assessment of VLP formation. To quantify VLP formation, EGFP-VP40, EGFP-VP40-WEA, or EGFP were detected with a GFP ELISA kit (Cell Biolabs, San Diego, CA) according to the manufacturer's protocols. VLPs were isolated from A549 cellular media as previously described (25) 48-h posttransfection. Cells were collected and lysed to measure the cellular concentration of EGFP-VP40, EGFP-VP40-WEA, or EGFP using the BCA protein assay method as a control for total protein content. Relative VLP formation is expressed as the ratio of EGFP in the VLPs/EGFP in the cellular lysate. Experiments were run in triplicate from two separate cellular experiments done on different days.
Results
Dynamic properties of VP40 in the cytosol and at the plasma membrane
To date the dynamics of VP40 in live cells has not been investigated. Thus, we used the RICS method to establish how membrane association regulates the diffusion of VP40. The RICS method (35) is particularly well suited for this approach as it can be used to measure molecular dynamics of fluorescently labeled molecules in live cells. RICS is the ideal method for meeting these objectives as the scanning movement creates a space-time matrix of pixels within each image. From the stack of frames collected for a live cell a spatial correlation map can be obtained to determine a particle's diffusion coefficient. Thus, when the spatial correlation map is obtained at different subcellular locales the diffusion coefficient can be determined at different cellular sites. This provides information on how the membranes, organelles, or other cellular components restrict the movement of fluorescently labeled proteins.
VP40 has been shown to localize to the inner leaflet of the plasma membrane where budding of the Ebola virus occurs. We first sought to confirm the localization of EGFP-VP40 and investigate the dynamics of VP40 when localized to the plasma membrane. For these purposes each cell expressing EGFP-VP40 was scanned with an area of 256 × 256 pixels with a pixel dwell time of 12.6 μs for 100 frames. This allowed collection of a spatial map of the cell with respect to time where expression of EGFP alone was used as a control. To assess the dynamics of VP40 in live cells, EGFP-VP40 diffusion (Fig. S1 in the Supporting Material) was measured in the cytosol and at the plasma membrane 24 h posttransfection and analyzed as described previously (36). EGFP had a diffusion coefficient of 7.7 ± 1 μm2 s−1 and did not show any fluctuations throughout the scan area nor were any changes detected at or near membranes. Shown in Fig. 2, A–F, is the spatial correlation map of VP40 in HEK293T cells, which indeed showed a significant enrichment of VP40 on the plasma membrane. The spatial correlation map was fit to a single species diffusion model to determine the diffusion coefficient of VP40 in the cytoplasm and on the plasma membrane. The diffusion properties of VP40 are summarized in Fig. 2 I. The diffusion of VP40 in the cytosol (2.8 ± 0.4 μm2 s−1) was slower than monomeric EGFP (Fig. 2 I and Fig. S1). This is not unexpected as EGFP-VP40 is 40 kDa larger than EGFP. EGFP-VP40 diffused ∼22 times faster in the cytosol than on the plasma membrane (0.13 ± 0.06 μm2 s−1), which was expected due to restrictions imposed by the membrane. Furthermore, the decreased diffusion may be attributed to membrane binding (37), oligomerization (22,25), or interaction with cellular components (19) that have been previously reported. Taken together, EGFP-VP40 is significantly enriched on the plasma membrane of HEK293T cells where the diffusion is reduced 22-fold.
Figure 2.
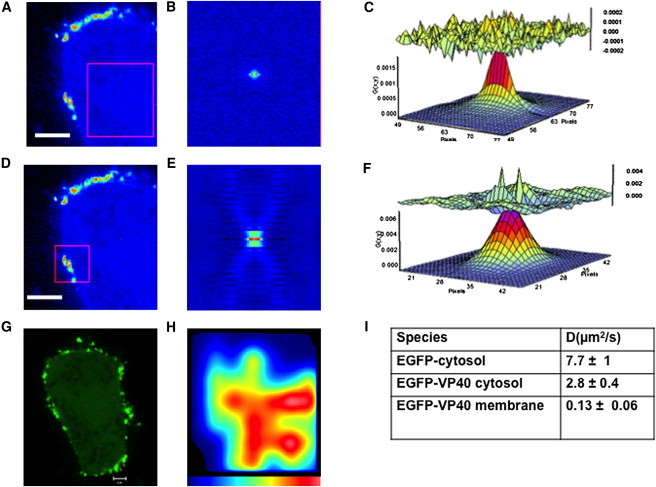
RICS analysis of EGFP-VP40 expressed in HEK293T cells. RICS data were acquired by taking 100 frames (256 × 256 pixels at 0.05 μm/pixel) at a scan speed of 12.60 μs/pixel. (A) A region of interest (White scale bar = 3.2 μm), 4.5 μm × 4.5 μm (pink square box) in the cytosol was analyzed to yield (B) the spatial autocorrelation map of the cytosol. (C) The spatial autocorrelation map is then fit to a single species diffusion model to determine the diffusion coefficient of VP40 in the cytosol (2.8 ± 0.4 μm2 s−1). (D) Region of interest (white scale bar = 3.2 μm) at the plasma membrane from the same cell shown in A is used to calculate (E) the spatial correlation map and (F) fit the spatial correlation map to a single species diffusion model to calculate the diffusion coefficient of VP40 at the plasma membrane (0.13 ± 0.06 μm2 s−1). (G) The same cell shown in A and D (visualized as EGFP localization) is used to create (H) a diffusion map of the VP40 transfected cell, where the color scale represents fast (red or darkest) or slow (blue or lightest) diffusion for the 4.5 μm × 4.5 μm area highlighted in A. Here, the area of interest is fit to a single species diffusion model to investigate the change in diffusion coefficient of EGFP-VP40 with respect to spatial localization. The fastest (or largest diffusion coefficients) moving particles are shown in red (darkest), whereas the slowest (smallest diffusion coefficients) are shown in blue (lightest). (I) The diffusion coefficients determined for EGFP, EGFP-VP40 in the cytosol, and EGFP-VP40 at the plasma membrane. VP40 exhibits 22-fold slower diffusion at the plasma membrane than in the cytosol.
Monomeric VP40 is recruited from the cytosol to the plasma membrane
Infectious Ebola virus as well as VP40 alone assemble and bud from the inner leaflet of the plasma membrane where membrane association has been suggested to trigger oligomerization of VP40 (10). To investigate the assembly or accumulation of EGFP-VP40 in live cells in real time, intensity derivative analysis in the SimFCS program (www.lfd.uci.edu) was used. Intensity derivative analysis allows intensity fluctuations of fluorescently labeled proteins to be monitored at different subcellular locations with respect to time. Thus, to assess the accumulation of VP40 in the cytosol and on the plasma membrane HEK293T cells were scanned for 156 s to obtain plots of fluorescent fluctuations at the plasma membrane and in the cytosol with respect to time. There was a significant accumulation of VP40 on the plasma membrane characterized by the increase in fluorescent intensity as a function of time (Fig. 3, C and D), compared to the cytosol where intensity remained constant over the same time period (Fig. 3, A and B). Taken together, these studies show VP40 is recruited from the cytosol strengthening the notion that VP40 assembly occurs predominately on the plasma membrane. It should also be noted that the transport of VP40 on transport vesicles to the plasma membrane could serve as an alternative interpretation of the recruitment of VP40 to the plasma membrane. Nonetheless, the VP40 bud sites assemble on the plasma membrane as shown in Fig. 3 C.
Figure 3.
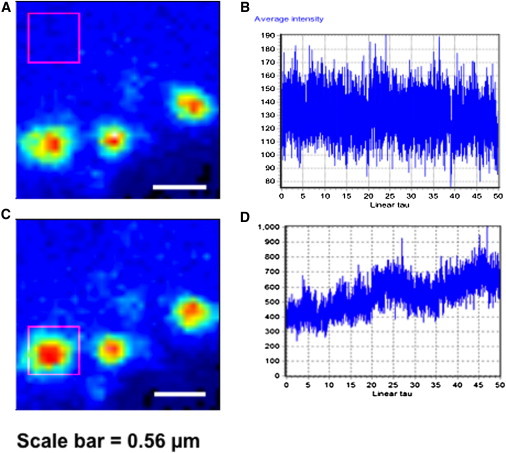
Recruitment of VP40 to the plasma membrane. Analysis of cytosolic and membrane regions of HEK293T cells expressing EGFP-VP40 were done with RICS. (A) Average image of cell showing region of analysis in the cytosol (pink box). (B) Intensity derivative plot obtained by analysis of intensity fluctuations in the cytosol with respect to time. The total scan time was 156 s and thus the x axis represents a total time of 156 s where linear tau represents time with 1 unit representing 3.12 s. (C) Average image showing region of interest at a plasma membrane VP40 bud site where three individual growing bud sites are shown. (D) Intensity plot with respect to time at the membrane. The total scan time was 156 s and thus the x axis represents a total time of 156 s where linear tau represents time with 1 unit representing 3.12 s.
Oligomerization state of VP40 on the plasma membrane
Although many reports have suggested that VP40 oligomerizes in the cell and even specifically on the plasma membrane (18) the origin, true size, and subcellular location of VP40 oligomers in live cells is still unknown. To understand how oligomerization of VP40 varies with cellular localization, a combination of TIRF microscopy and the N&B analysis (31,38) were used to study the oligomerization state of VP40 on the plasma membrane. Additionally, N&B analysis was also done with confocal microscopy to visualize VP40 oligomerization throughout the cell. N&B analysis allows for measurement of the average number of molecules as well as brightness in each pixel of a fluorescent microscopy image. Thus, this method is well suited to resolve the spatial distribution of VP40 oligomers in live cells.
HEK293T cells were transfected with plasmids expressing either monomeric EGFP (N = 5) or Ebola EGFP-VP40 (N = 7). The oligomerization state of VP40 was assessed by analyzing the intrinsic brightness of proteins in the cells. To calibrate for the monomeric state of the proteins, HEK293T cells expressing EGFP were analyzed. The calibration using the brightness of a monomer allows for the selection of oligomers, which are a multiple of the brightness of the monomer. Thus, selection windows in the brightness versus intensity plots can be used to select the average brightness of each species of interest (monomer, dimer, hexamer, etc.) to visualize the cellular localization of each species. As shown in Fig. 4, A–D, we observed that EGFP exists primarily as a monomer and hence did not influence the oligomerization of VP40. N&B analysis of EGFP-VP40 (Fig. 4, E–L), revealed that unlike free EGFP (Fig. 4, A–D), it forms higher order structures as high as 16 mers localized on the plasma membrane (Fig. 4, I–L, and Fig. S2, D and E). Although the frequency of oligomers observed on the plasma membrane was significantly less than the monomeric species (Fig. 4, G and H), oligomers were highly enriched within filamentous structures protruding from the plasma membrane (Fig. 4, J and L). Sites of membrane protrusion were determined by visual inspection of cells using TIRF microscopy (Fig. S3). Expression of EGFP-VP40 but not EGFP alone in HEK293T cells is characterized by a significant number of membrane protrusions extending from the plasma membrane of the cell. Hexamers and octamers of VP40, the predominant oligomeric species of VP40 previously reported (22–25), are found exclusively in these membrane protrusion sites (Fig. 4, I–L). However, the predominant species on the membrane were monomers (Fig. 4, F–H and A), suggesting VP40 either associates with the plasma membrane or is transported to the plasma membrane in a monomeric form where oligomerization is then triggered through membrane association as previously suggested (10). Additionally, other cellular components should be considered such as protein-protein interactions, VP40 cytoskeleton interactions, or VP40 interaction with a specific lipid such as phosphatidylserine (PS) or phosphatidylinositol-3,4,5-trisphosphate, which are enriched on the inner leaflet of the plasma membrane.
Figure 4.
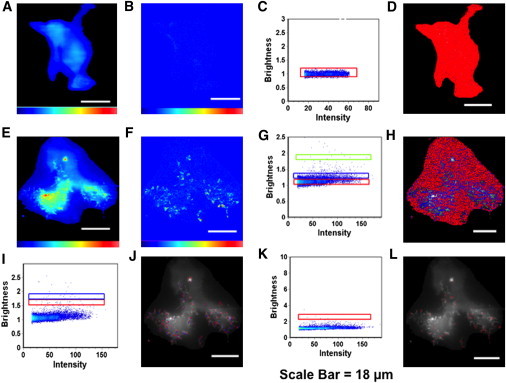
Brightness analysis of VP40 and EGFP in HEK293T cells. (A) TIRF intensity image of a HEK293T cell transfected with plasmid expressing EGFP. (B) Brightness image of the same cell shows the lack of EGFP clustering. (C) Brightness versus intensity map for the EGFP expressing cell with monomers highlighted by the red box. (D) Selected pixels from C displaying the localization of monomeric EGFP in red. (E) TIRF intensity image of a HEK293T cell expressing EGFP-VP40. (F) Brightness distribution of the VP40 expressing cell in E demonstrates enrichment of EGFP-VP40 clustering on the plasma membrane. Specifically, EGFP-VP40 is clustered at sites of membrane protrusion that can also be viewed in Fig. S3. (G) Brightness versus intensity map of VP40 showing monomers (red box or lower box), trimers (blue box or middle box), and octamers (green box or upper box). (H) Selected pixels from G showing monomers (red), trimers (blue), and octamers (green). The majority of VP40 localized to the plasma membrane is present in the monomeric form. (I) Brightness versus intensity plot displaying hexamers (red box or lower box) and octamers (blue box or upper box). (J) Brightness distribution of VP40 with selected pixels displaying hexamers (red) and octamers (blue), which are significantly enriched at sites of membrane protrusions. (K) Brightness versus intensity plot displaying 16 mers (red box). (L) Image of the cell showing pixels with brightness of 16 mers (red) localized at sites of membrane protrusion. White scale bar = 18 μm on all panels.
N&B analysis was also performed using the confocal image stacks acquired from RICS analysis of HEK293T cells expressing either EGFP (Fig. S1) or EGFP-VP40 (Fig. S2). In consonance with the TIRF imaging data, VP40 oligomers were highly enriched on the plasma membrane (Fig. S2, D and E), when compared to other cellular sites. This underscores the unique localization of VP40 oligomers to sites of membrane protrusion from the plasma membrane.
To control for nonspecific oligomerization of VP40 we employed a double mutant control of the VP40 intradimeric interface (W95A/E160A or VP40-WEA), which has been shown to abolish VP40 oligomerization and diminish egress (22). EGFP-VP40-WEA displayed a similar cytosolic diffusion coefficient as wild-type EGFP-VP40 but did not display detectable oligomers (Fig. S4, E and F). Additionally, sites of membrane protrusion were no longer visible (Fig. S4, A, B, D, and F) in EGFP-VP40-WEA transfected cells consistent with a recent study that showed VP40-WEA significantly reduced viral egress (22). A frequency versus apparent brightness plot for EGFP (Fig. 5 A), EGFP-VP40 (Fig. 5 B), or EGFP-VP40-WEA (Fig. 5 C) expressing cells further demonstrates the specific and significant oligomerization of EGFP-VP40 compared to EGFP-VP40-WEA or EGFP controls. As shown in Fig. 5 D VP40-WEA significantly reduced VLP formation compared to wild-type VP40, which is consistent with the loss of filamentous sites of viral egress (Fig. S4, A, D, and F) as well as previous studies demonstrating the role of oligomerization in VP40-mediated exit (22,25).
Figure 5.
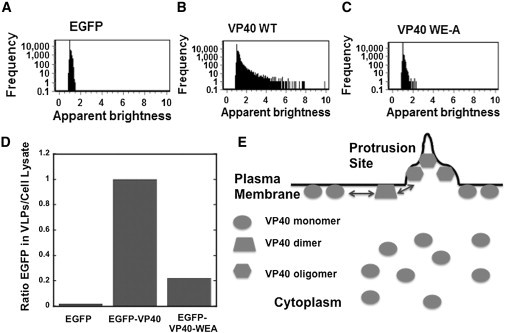
Correlation of VP40 and VP40-WEA apparent brightness and frequency as well as VLP egress. (A) Frequency versus apparent brightness plot for monomeric EGFP, (B) EGFP-VP40, and (C) EGFP-VP40 WE-A from HEK293T cells. These plots show the frequency (pixels) of VP40 oligomerization in comparison to VP40 WE-A. (D) VLPs were detected with anti-EGFP antibody to compare the release of EGFP-VP40 and EGFP-VP40-WEA VLPs. Cells transfected with EGFP were used as a control for background EGFP detection. Results are plotted as a ratio of EGFP in the VLPs/EGFP in the cell lysate. Three trials were run for each condition from two separate experiments of VLP and cell lysate collection. (E) A model proposing the localization of VP40 oligomers. Imaging data from both confocal and TIRF microscopy support the notion that VP40 is predominantly monomeric in the cytoplasm as well as when associated with the plasma membrane. Plasma membrane association of VP40 then may be able to induce the oligomerization of VP40 into dimers and trimers, which are also predominantly associated with the plasma membrane. Higher order VP40 oligomers (hexamers and octamers for instance), which have been shown to exist in infectious Ebola virions are highly enriched in membrane protrusions emanating from the plasma membrane. (See the Discussion section for a more critical discussion of the factors that may regulate VP40 assembly and oligomerization).
Discussion
Oligomerization of VP40 represents a crucial step in the assembly of the Ebola virus (22,23,37,39). Inhibition of this step of the viral life cycle has been found to downregulate the formation and release of new virions and VLPs (22,25). It has also been shown that in the absence of VP40, recruitment of viral proteins to the membrane is inhibited (17). Hence, understanding the cellular mechanisms behind VP40 oligomerization is necessary to deepen our understanding of filovirus assembly. To date, VP40 oligomerization has been studied using cell lysates or purified membranes from transfected or infected cells (18) as well as in vitro using cross-linking experiments (24,39). Although these studies have been useful in expanding the mechanism of Ebola assembly, they are not able to fundamentally address how the protein assembles within the cell in a real-time manner. Furthermore, in vitro studies that attempted to resolve oligomeric states were carried out in conditions that do not necessarily reflect the biology of the assembly process. Hence, the ability to resolve the assembly and oligomeric state of VP40 in a real-time noninvasive biologically relevant fashion is essential to moving the investigation of filovirus assembly forward. In this report, we have resolved the oligomeric state of Ebola VP40 using RICS analysis and TIRF microscopy coupled with the N&B method.
First, we demonstrated that VP40 has different dynamic properties in the cytosol and on the membrane. Using RICS, we determined the diffusion coefficient of EGFP-VP40 is reduced 22-fold on the membrane compared to the cytosol. The decrease in diffusion coefficient could certainly be attributed to membrane association, which is interesting as VP40 binds the lipid PS with nanomolar affinity (E. Adu-Gyamfi and R. V. Stahelin, unpublished results) and PS is enriched at the inner leaflet of the plasma membrane. In addition, other regulatory factors such as VP40-protein interactions or VP40-cytoskeleton interactions at the plasma membrane should also be considered. Intensity derivative analysis, which computes changes in intensity of the diffusing fluorophores at the pixel level, demonstrated that on the membrane the VP40 intensity increases as a function of time despite any photobleaching effects. However, under the same conditions EGFP-VP40 intensity in the cytosol remained constant. These data suggested VP40 assembles on the plasma membrane. Consistent with this notion VP40 has previously been shown to undergo a conformational change upon membrane binding (37), which has been proposed to trigger its oligomerization (10). Motivated by this suggestion and the observation that aggregates of VP40 were found to be associated with detergent-resistant membranes (18) we studied the localization of VP40 oligomers on the plasma membrane and throughout the cell. Our results show that although oligomers of VP40 were detected on the plasma membrane a significant proportion of plasma membrane-associated VP40 was monomeric. This is not particularly surprising if the monomers are continuously being recruited from the cytosol (Fig. 5 E) to serve as building blocks for self-multimerization into hexamers required for viral egress (25) and octamers required for RNA binding (22,24). How then is VP40 recruited to the plasma membrane? Nanomolar affinity of VP40 for PS is certainly one possibility. Additionally, VP40 may also have significant affinity for other lipids or proteins enriched at the inner leaflet of the plasma membrane. In addition, transport of VP40 on vesicles of similar composition to the plasma membrane is also a possibility.
We also observed that oligomers (hexamers, octamers, and larger oligomers) were found enriched at the tips of the cells in filamentous protruding structures (Fig. 4, I and J). Filamentous protrusion sites are consistent with sites of viral egress for Ebola (17) and Marburg virus (40). In further support of localization of VP40 oligomers in sites of membrane protrusion, VP40-WEA, which abrogates oligomerization diminished detectable membrane protrusion sites. To the best of our knowledge larger oligomers of VP40 have not been previously reported and the nature and role of these larger oligomers in VP40-mediated assembly remain unknown. Large oligomers have been observed to be essential to the membrane deforming activity of lipid binding domains such as BAR (41) and ENTH (42) domains and could play a role in Ebola-mediated egress through formation of a lattice as proposed for other lipid enveloped viruses (28). It may also suggest that VP40 molecules in the cell are organized into larger structures that are beyond the resolution detectable by cross-linking assays. However, it is possible these large oligomers do not have significant mechanistic consequences in the Ebola life cycle and clearly further analysis of these structures will be necessary. Nonetheless, we believe our analysis is suitable for detecting the presence of VP40 oligomers in cells and offers superior detection and resolution compared to previous analysis from cell lysates and in vitro cross-linking assays (18,22). First, our analysis takes into account oligomerization within the cell in real time while avoiding the use of harsh conditions such as detergent solubilization and excessive heat that may disrupt oligomers. Second, in vivo or in vitro cross-linking may result in nonspecific cross-links further complicating the analysis. Third, the inherent sensitivity of our methods makes it possible to obtain high spatial resolution images elucidating the distribution of oligomers in the cells at different time points. One drawback to this assay is the necessary tagging of VP40 with EGFP. However, we demonstrate EGFP-VP40 robustly produces VLPs offering the ability to study single-molecule interactions with fluorescently labeled host cell factors in the future.
In conclusion, we have detected the oligomeric states of Ebola VP40 in a biologically relevant and noninvasive system using TIRF microscopy that selectively excites membrane resident molecules. We found that VP40 oligomers are found primarily in filamentous plasma membrane protrusion sites. Interestingly, the majority of VP40 proteins on the membrane are monomers suggesting that monomeric VP40 is recruited from the cytosol or trafficked on transport vesicles to serve as building blocks for multimerization. VP40-mediated oligomerization then either induces or is localized to plasma membrane protrusion sites, where viral egress occurs (Fig. 5 E). The main determinant of VP40-mediated oligomerization is still unknown and it is tempting to propose that high affinity of VP40 for an enriched plasma membrane component such as PS is sufficient to induce oligomerization. Indeed, PS is able to induce significant oligomerization of VP40 in an in vitro cross-linking assay compared to other anionic lipids (E. Adu-Gyamfi and R. V. Stahelin unpublished results). Clearly, the investigation of specific VP40-lipid and VP40-protein interactions at the plasma membrane is of utmost interest as these interactions may serve as pharmacological targets to inhibit Ebola budding and egress. In closing, application of single molecule studies using both confocal and TIRF microscopy was essential to investigating the real-time assembly and egress of Ebola VP40 as similarly demonstrated for HIV (43–45). These techniques hold much promise for building a spatio temporal map of the host cell factors required for VP40-mediated assembly and egress on the plasma membrane.
Acknowledgments
This work was supported by the National Institutes of Health (AI081077) and the Notre Dame Center for Rare and Neglected Diseases to R.V.S. Additionally, this work was supported by the Indiana University School of Medicine-South Bend Imaging and Flow Cytometry Core Facility to R.V.S. E.A.G. is supported by a Notre Dame Eck Institute for Global Health graduate training fellowship. E.G. and M.A.D acknowledges support of National Institutes of Health grants: P41-RRO3155 and P50-GM076516 and the Keck's Foundation grant 44769549507.
Supporting Material
References
- 1.Bowen E.T., Lloyd G., Vella E.E. Viral haemorrhagic fever in southern Sudan and northern Zaire. Preliminary studies on the aetiological agent. Lancet. 1977;1:571–573. doi: 10.1016/s0140-6736(77)92001-3. [DOI] [PubMed] [Google Scholar]
- 2.Elliott L.H., Kiley M.P., McCormick J.B. Descriptive analysis of Ebola virus proteins. Virology. 1985;147:169–176. doi: 10.1016/0042-6822(85)90236-3. [DOI] [PubMed] [Google Scholar]
- 3.Johnson K.M., Lange J.V., Murphy F.A. Isolation and partial characterisation of a new virus causing acute haemorrhagic fever in Zaire. Lancet. 1977;1:569–571. doi: 10.1016/s0140-6736(77)92000-1. [DOI] [PubMed] [Google Scholar]
- 4.Ksiazek T.G., Rollin P.E., Peters C.J. Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J. Infect. Dis. 1999;179(Suppl 1):S177–S187. doi: 10.1086/514321. [DOI] [PubMed] [Google Scholar]
- 5.Sullivan N.J., Martin J.E., Nabel G.J. Correlates of protective immunity for Ebola vaccines: implications for regulatory approval by the animal rule. Nat. Rev. Microbiol. 2009;7:393–400. doi: 10.1038/nrmicro2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahanty S., Bray M. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect. Dis. 2004;4:487–498. doi: 10.1016/S1473-3099(04)01103-X. [DOI] [PubMed] [Google Scholar]
- 7.Harty R.N. No exit: targeting the budding process to inhibit filovirus replication. Antiviral Res. 2009;81:189–197. doi: 10.1016/j.antiviral.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Panchal R.G., Reid S.P., Bavari S. Identification of an antioxidant small-molecule with broad-spectrum antiviral activity. Antiviral Res. 2012;93:23–29. doi: 10.1016/j.antiviral.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 9.Warren T.K., Warfield K.L., Bavari S. Antiviral activity of a small-molecule inhibitor of filovirus infection. Antimicrob. Agents Chemother. 2010;54:2152–2159. doi: 10.1128/AAC.01315-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartlieb B., Weissenhorn W. Filovirus assembly and budding. Virology. 2006;344:64–70. doi: 10.1016/j.virol.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 11.Olejnik J., Ryabchikova E., Mühlberger E. Intracellular events and cell fate in filovirus infection. Viruses. 2011;3:1501–1531. doi: 10.3390/v3081501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feldmann H., Volchkov V.E., Klenk H.D. The glycoproteins of Marburg and Ebola virus and their potential roles in pathogenesis. Arch. Virol. Suppl. 1999;15:159–169. doi: 10.1007/978-3-7091-6425-9_11. [DOI] [PubMed] [Google Scholar]
- 13.Dessen A., Volchkov V., Weissenhorn W. Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J. 2000;19:4228–4236. doi: 10.1093/emboj/19.16.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jasenosky L.D., Neumann G., Kawaoka Y. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J. Virol. 2001;75:5205–5214. doi: 10.1128/JVI.75.11.5205-5214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neumann G., Ebihara H., Kawaoka Y. Ebola virus VP40 late domains are not essential for viral replication in cell culture. J. Virol. 2005;79:10300–10307. doi: 10.1128/JVI.79.16.10300-10307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Licata J.M., Johnson R.F., Harty R.N. Contribution of ebola virus glycoprotein, nucleoprotein, and VP24 to budding of VP40 virus-like particles. J. Virol. 2004;78:7344–7351. doi: 10.1128/JVI.78.14.7344-7351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noda T., Ebihara H., Kawaoka Y. Assembly and budding of Ebola virus. PLoS Pathog. 2006;2:e99. doi: 10.1371/journal.ppat.0020099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panchal R.G., Ruthel G., Aman M.J. In vivo oligomerization and raft localization of Ebola virus protein VP40 during vesicular budding. Proc. Natl. Acad. Sci. USA. 2003;100:15936–15941. doi: 10.1073/pnas.2533915100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Licata J.M., Simpson-Holley M., Harty R.N. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol. 2003;77:1812–1819. doi: 10.1128/JVI.77.3.1812-1819.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reynard O., Nemirov K., Volchkov V.E. Conserved proline-rich region of Ebola virus matrix protein VP40 is essential for plasma membrane targeting and virus-like particle release. J. Infect. Dis. 2011;204(Suppl 3):S884–S891. doi: 10.1093/infdis/jir359. [DOI] [PubMed] [Google Scholar]
- 21.Yamayoshi S., Noda T., Kawaoka Y. Ebola virus matrix protein VP40 uses the COPII transport system for its intracellular transport. Cell Host Microbe. 2008;3:168–177. doi: 10.1016/j.chom.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoenen T., Biedenkopf N., Becker S. Oligomerization of Ebola virus VP40 is essential for particle morphogenesis and regulation of viral transcription. J. Virol. 2010;84:7053–7063. doi: 10.1128/JVI.00737-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Timmins J., Schoehn G., Weissenhorn W. Oligomerization and polymerization of the filovirus matrix protein VP40. Virology. 2003;312:359–368. doi: 10.1016/s0042-6822(03)00260-5. [DOI] [PubMed] [Google Scholar]
- 24.Gomis-Rüth F.X., Dessen A., Weissenhorn W. The matrix protein VP40 from Ebola virus octamerizes into pore-like structures with specific RNA binding properties. Structure. 2003;11:423–433. doi: 10.1016/S0969-2126(03)00050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoenen T., Volchkov V., Weissenhorn W. VP40 octamers are essential for Ebola virus replication. J. Virol. 2005;79:1898–1905. doi: 10.1128/JVI.79.3.1898-1905.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen T.L., Schoehn G., Bavari S. An all-atom model of the pore-like structure of hexameric VP40 from Ebola: structural insights into the monomer-hexamer transition. J. Struct. Biol. 2005;151:30–40. doi: 10.1016/j.jsb.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 27.McCarthy S.E., Johnson R.F., Harty R.N. Role for amino acids 212KLR214 of Ebola virus VP40 in assembly and budding. J. Virol. 2007;81:11452–11460. doi: 10.1128/JVI.00853-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hurley J.H., Boura E., Różycki B. Membrane budding. Cell. 2010;143:875–887. doi: 10.1016/j.cell.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shnyrova A.V., Ayllon J., Frolov V.A. Vesicle formation by self-assembly of membrane-bound matrix proteins into a fluidlike budding domain. J. Cell Biol. 2007;179:627–633. doi: 10.1083/jcb.200705062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dalal R.B., Digman M.A., Gratton E. Determination of particle number and brightness using a laser scanning confocal microscope operating in the analog mode. Microsc. Res. Tech. 2008;71:69–81. doi: 10.1002/jemt.20526. [DOI] [PubMed] [Google Scholar]
- 31.Digman M.A., Dalal R., Gratton E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 2008;94:2320–2332. doi: 10.1529/biophysj.107.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Digman M.A., Wiseman P.W., Gratton E. Stoichiometry of molecular complexes at adhesions in living cells. Proc. Natl. Acad. Sci. USA. 2009;106:2170–2175. doi: 10.1073/pnas.0806036106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ross J.A., Digman M.A., Jameson D.M. Oligomerization state of dynamin 2 in cell membranes using TIRF and number and brightness analysis. Biophys. J. 2011;100:L15–L17. doi: 10.1016/j.bpj.2010.12.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reynard O., Reid S.P., Volchkov V.E. Unconventional secretion of Ebola virus matrix protein VP40. J. Infect. Dis. 2011;204(Suppl 3):S833–S839. doi: 10.1093/infdis/jir305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moens P.D., Gratton E., Salvemini I.L. Fluorescence correlation spectroscopy, raster image correlation spectroscopy, and number and brightness on a commercial confocal laser scanning microscope with analog detectors (Nikon C1) Microsc. Res. Tech. 2010;74:377–388. doi: 10.1002/jemt.20919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rossow M.J., Sasaki J.M., Gratton E. Raster image correlation spectroscopy in live cells. Nat. Protoc. 2010;5:1761–1774. doi: 10.1038/nprot.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scianimanico S., Schoehn G., Weissenhorn W. Membrane association induces a conformational change in the Ebola virus matrix protein. EMBO J. 2000;19:6732–6741. doi: 10.1093/emboj/19.24.6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Digman M.A., Brown C.M., Gratton E. Paxillin dynamics measured during adhesion assembly and disassembly by correlation spectroscopy. Biophys. J. 2008;94:2819–2831. doi: 10.1529/biophysj.107.104984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruigrok R.W.H., Schoehn G., Weissenhorn W. Structural characterization and membrane binding properties of the matrix protein VP40 of Ebola virus. J. Mol. Biol. 2000;300:103–112. doi: 10.1006/jmbi.2000.3822. [DOI] [PubMed] [Google Scholar]
- 40.Kolesnikova L., Bohil A.B., Becker S. Budding of Marburgvirus is associated with filopodia. Cell. Microbiol. 2007;9:939–951. doi: 10.1111/j.1462-5822.2006.00842.x. [DOI] [PubMed] [Google Scholar]
- 41.Yin Y., Arkhipov A., Schulten K. Simulations of membrane tubulation by lattices of amphiphysin N-BAR domains. Structure. 2009;17:882–892. doi: 10.1016/j.str.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoon Y., Tong J., Cho W. Molecular basis of the potent membrane-remodeling activity of the epsin 1 N-terminal homology domain. J. Biol. Chem. 2010;285:531–540. doi: 10.1074/jbc.M109.068015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baumgärtel V., Ivanchenko S., Lamb D.C. Live-cell visualization of dynamics of HIV budding site interactions with an ESCRT component. Nat. Cell Biol. 2011;13:469–474. doi: 10.1038/ncb2215. [DOI] [PubMed] [Google Scholar]
- 44.Jouvenet N., Simon S.M., Bieniasz P.D. Visualizing HIV-1 assembly. J. Mol. Biol. 2011;410:501–511. doi: 10.1016/j.jmb.2011.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jouvenet N., Zhadina M., Simon S.M. Dynamics of ESCRT protein recruitment during retroviral assembly. Nat. Cell Biol. 2011;13:394–401. doi: 10.1038/ncb2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.