

Abstract
Acute activation of calcium/calmodulin-dependent protein kinase (CaMKII) in permeabilized phospholamban knockout (PLN-KO) mouse myocytes phosphorylates ryanodine receptors (RyRs) and activates spontaneous local sarcoplasmic reticulum (SR) Ca release events (Ca sparks) even at constant SR Ca load. To assess how CaMKII regulates SR Ca release in intact myocytes (independent of SR Ca content changes or PLN effects), we compared Ca sparks in PLN-KO versus mice, which also have transgenic cardiac overexpression of CaMKIIδC in the PLN-KO background (KO/TG). Compared with PLN-KO mice, these KO/TG cardiomyocytes exhibited 1), increased twitch Ca transient and fractional release (both by ∼35%), but unaltered SR Ca load; 2), increased resting Ca spark frequency (300%) despite a lower diastolic [Ca]i, which also slowed twitch [Ca]i decline (suggesting CaMKII-dependent RyR Ca sensitization); 3), elevated Ca spark amplitude and rate of Ca release (which might indicate that more RyR channels participate in a single spark); 4), prolonged Ca spark rise time (which implies that CaMKII either delays RyR closure or prolongs the time when openings can occur); 5), more frequent repetitive sparks at single release sites. Analysis of repetitive sparks from individual Ca release sites indicates that CaMKII enhanced RyR Ca sensitivity, but did not change the time course of SR Ca refilling. These results demonstrate that there are dramatic CaMKII-mediated effects on RyR Ca release that occur via regulation of both RyR activation and termination processes.
Introduction
Calcium/calmodulin-dependent protein kinase II-δ (CaMKIIδ) is a multifunctional serine/threonine kinase that has been implicated in cellular Ca regulation related to excitation-contraction (EC) coupling (1,2). Key CaMKII targets in cardiac myocytes include L-type Ca channels (3–5), Na and K channels (6,7), phospholamban (PLN (8)), and ryanodine receptors (RyR) channels (9–11). CaMKIIδ-mediated phosphorylation of L-type Ca channels causes Ca current (ICa) facilitation and enhances Ca influx at higher heart rates. PLN phosphorylation enhances sarcoplasmic reticulum (SR) Ca uptake, accelerates intracellular [Ca] ([Ca]i) decline during relaxation, and consequently increases SR Ca load (12,13). CaMKIIδ-dependent RyR phosphorylation increases RyR activity in lipid bilayers (5,9,14), increases SR Ca leak and Ca sparks in myocytes (10,15–17), and enhances fractional SR Ca release during EC coupling for a given ICa and SR Ca content (15), although not all results agree (18,19).
Transgenic (TG) mice overexpressing the CaMKIIδC splice variant of CaMKIIδ exhibit enhanced resting spontaneous SR Ca release (Ca sparks) and fractional SR Ca release, but decreased SR Ca load and lower diastolic [Ca]i (20). These TG CaMKIIδC mice also exhibit profoundly reduced Ca transients, heart failure (HF), and arrhythmias (6,20,21). In a recent study, we focused on whether PLN ablation could rescue HF and restore Ca transient amplitude in CaMKIIδC-TG mice by crossing them with PLN knockout (KO) mice (22). PLN-KO did improve Ca transients in CaMKIIδC myocytes, but the mice fared worse because of enhanced mitochondrial Ca loading and cell death. We attributed the mitochondrial Ca overload to an exacerbation of the Ca spark frequency enhancement seen in CaMKIIδC-TGs (RyR sensitization (20)) by the increase in SR Ca content caused by PLN-KO (23).
The effects of CaMKII on RyR2 function are complicated by effects of CaMKII on PLN phosphorylation and the consequently altered SR Ca content. Here, we focus on how CaMKIIδC itself alters RyR2 properties, Ca transients, and Ca sparks in myocytes lacking PLN, by comparing PLN-KO mice with CaMKIIδC overexpression (KO/TG) versus PLN-KO (KO; i.e., a different comparison from the previous rescue study (22).
We found that in the PLN-KO background CaMKIIδC increased twitch Ca transients, fractional release, and spontaneous SR Ca release (as indicated by Ca sparks), slowed twitch [Ca]i decline, left SR Ca load unaltered, and decreased diastolic [Ca]i. Detailed analysis of Ca sparks suggested that CaMKII sensitized RyR via regulation of RyR activation and the Ca spark termination process, and that SR Ca leak was sufficient to significantly slow twitch [Ca]i decline.
Materials and Methods
Generation of TG mice and myocyte isolation
TG mice overexpressing CaMKIIδC targeted to cardiac myocytes by the α-myosin heavy chain promoter (CaMKIIδC-TG) and phospholamban knockout mice (PLN-KO) were as described previously (21,24). CaMKIIδC-TG mice were crossed with PLN-KO mice to produce mice with PLN ablation either alone (KO) or with CaMKIIδC overexpression (KO/TG (22). Ca handling was assessed using 6–10-week-old KO/TG mice (n = 8) where CaMKII activity was increased threefold (22), and age-matched PLN-KO littermates (n = 8). Ventricular myocytes were isolated as reported (16). All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee.
Fluorescent measurement of [Ca]i and in vivo calibration
[Ca]i was measured using either fura-2 or indo-1 as previously reported (20,26). Fura-2 was excited at 340 ± 10 nm and 380 ± 10 nm, and emitted fluorescence was measured (535 ± 20 nm). Background-subtracted ratio (R) of fluorescence (R = F340/F380) was converted to [Ca]i using [Ca]i = Kdβ(R − Rmin)/(Rmax –R). Rmin, Rmax, and β were determined experimentally (Kd = 375 nM). SR Ca load was measured as ([Ca]i + 272 μmol/L cytosol /(1 + 673 nM /[Ca]i) + 50 μM/(1 + 375/[Ca]i). Indo-1 fluorescence was also used in some experiments excited at 365 ± 25 nm and monitored at 405 ± 10 nm using similar analysis assuming Kd = 840 nM (25). Rmin and Rmax were determined respectively in cells superfused with solutions containing either 5 mM EGTA (and nominally Ca free) or 20 mM Ca, in the presence of nonfluorescent Ca ionophore Br-A23187 (10 μM, EMD Millipore, Darmstadt, Germany).
Ca measurements using confocal microscopy
Ca transients and sparks were also recorded using Fluo-4 (a single wavelength Ca indicator), on a laser scanning confocal microscope (BioRad, Carl Zeiss) equipped with a 40× oil immersion objective lens (n.a. = 1.3). Fluo-4 (loaded by 30 min incubation with 10 μM of acetoxymethyl ester) was excited with the 488 nm line of an argon laser. Fluorescence was collected through a 515 nM long pass emission filter. Fluo-4 images were recorded in line scan mode with 512 pixels per line at 250 Hz. F0 refers to fluorescence at resting [Ca]i. SR Ca load was assessed by application of 10 mM caffeine in normal Tyrodes (26).
Ca sparks were analyzed using Spark Master (27). We used Ca spark detection threshold 3.8 × s.d., with human verification. Ca spark amplitudes were normalized (ΔF/F0) to fluorescence baseline (F0). Duration of Ca sparks was taken as the duration at 50% of peak (full duration at half-maximum (FDHM)). Width of Ca sparks was the spatial size at 50% of peak (full width at half-maximum (FWHM)).
Solutions and experimental protocols
Normal Tyrode's solution contained (in mM) 140 NaCl, 4 KCl, 10 HEPES, 10 Glucose, 1 MgCl2, and 1 CaCl2 (23°C). SR Ca load was evaluated by Ca transient amplitudes induced by rapid caffeine (10 mM) application, where the exponential rate constant of [Ca]i decline during caffeine exposure is an indicator of the sodium-calcium exchanger (NCX) function (26). Some myocytes from KO/TG mice were incubated for 30 min with cell permeable CaMKII inhibitor autocamtide-2 related inhibitory peptide II (AIP, EMD Millipore No. 189485).
Western blots
Cardiac homogenates from the same age groups were prepared and Western blot analysis carried out as reported (21). Antibodies used were anti-SERCA2, anti-PLN, anti-RyR (Affinity Bioreagents), and anti-NCX (monoclonal R3F1, a gift from K. D. Philipson, UCLA, Los Angeles, CA).
Results
Characterization of stimulated and caffeine-induced Ca transients
Ca transients are enhanced in PLN-KO but depressed in CaMKIIδC-TG mice (20,22). Our prior study showed that PLN-KO could rescue profound Ca transient depression seen in CaMKIIδC mice (22). Here, we extend those indo-1 Ca transient measurements, focusing on the influence of CaMKII in the PLN-KO background (a statistical comparison not previously done). Here, we carried out fura-2 Ca transient measurements (Fig. 1, A and B), demonstrating that steady-state 1 Hz twitch Ca transient amplitudes (Δ[Ca]i) are significantly higher in KO/TG versus PLN-KO (431 ± 33 nM, N = 20 vs. 343 ± 20 nM, N = 14), and this difference was also significant for Ca transients measured with Fluo-4 (Fig. 1 B) and indo-1 (not shown, 22). On the other hand, diastolic [Ca]i, assessed with the ratiometric indicator fura-2, is significantly reduced in KO/TG versus PLN-KO (122 ± 14 nM vs. 192 ± 26 nM; Fig. 1 C) and that effect was confirmed in indo-1 measurements (not shown). The lower diastolic [Ca]i in KO/TG may reflect in part the functional enhancement of the NCX function in KO/TG (see below).
Figure 1.
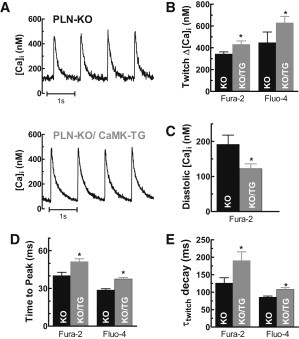
CaMKII increased twitch Δ[Ca]i transients but decreased diastolic [Ca]i in ventricular myocytes. (A) Representative steady-state twitch Δ[Ca]i during 1 Hz stimulation in KO and KO/TG myocytes using fura-2 as Ca indicator; (B) twitch Δ[Ca]i; (C) diastolic [Ca]i; (D) rise time (time to peak [Ca]i); (E) time constant τ of twitch [Ca]i decline using the indicated Ca sensors (n ≥ 10 cells in each group). ∗Significant difference between PLN-KO (KO) and CaMKIIδC-PLN-KO mice (KO/TG) at p < 0.05.
The increased twitch Δ[Ca]i amplitude in KO/TG could result from increased SR Ca load or enhanced fractional SR Ca release. SR Ca load was measured by rapid application of 10 mM caffeine (Fig. 2, A and B), and was not increased in KO/TG (using fura-2, 1449 ± 174 nM in KO/TG vs. 1554 ± 350 nM in PLN-KO; n = 10–11; p = 0.5). For fura-2 experiments, caffeine was applied at steady state (∼2 s after a twitch at 1 Hz), whereas for Fluo-4 caffeine was applied immediately after a 20 s period of Ca spark measurement. Similar results were seen with both indicators (Fig. 2 B), indicating that SR Ca load was not increased in KO/TG myocytes.
Figure 2.
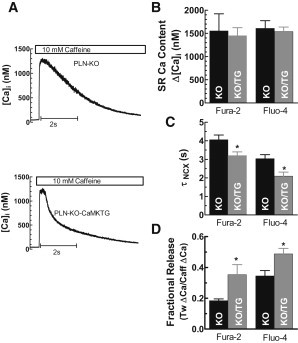
Unaltered SR Ca load, but increased NCX function in KO/TG. (A) SR Ca content assessed by caffeine-induced Fluo-4 transient amplitude (Ca sensor is indicated in each subsequent panel); (B) SR Ca content; (C) time constant of [Ca]i decline during caffeine exposure (τNCX). (D) Fractional SR Ca release (twitch/caffeine-induced Δ[Ca]i). ∗Significant difference between KO and CaMKIIδC-PLN-KO mice at p < 0.05 (n ≥ 6 for all groups).
On the other hand, SR Ca fractional release (the ratio of twitch/caffeine Δ[Ca]i) was significantly higher in KO/TG (see Fig. 2 D), again with both fura-2 and Fluo-4 measurements. Thus, CaMKIIδC enhances fractional SR Ca release in KO/TG, consistent with a stimulatory effect of CaMKII on RyR during EC coupling for the same SR Ca load and ICa (15).
The rate of [Ca]i decline during caffeine-induced contracture is mainly attributed to Ca extrusion by NCX (with a minor contribution by the plasma membrane Ca-ATPase and mitochondrial Ca uptake (23). Accordingly, the exponential rate constant kNCX (1/τ) reflects mainly the NCX function. Fig. 2 C shows that the time constant of [Ca]i decline during caffeine exposure (τNCX) was faster in KO/TG versus PLN-KO for both indicators, suggesting ∼35% higher NCX function in KO/TG versus PLN-KO.
Twitch [Ca]i decline is dominated by SR Ca-ATPase (SERCA) activity, especially in mouse myocytes (23). Fig. 1 E shows that the time constant of twitch [Ca]i decline (τtwitch) was slower in KO/TG (e.g., for fura-2, 190 ± 15 ms in KO/TG vs. 126 ± 20 ms in PLN-KO). We can separate the apparent SR transport component of [Ca]i decline, assuming that Ca removal is attributed to parallel function of SR and NCX. The twitch [Ca]i decline rate constant (ktwitch) is the sum of that for NCX and SR transport (ktwitch = kSR + kNCX) (28). Using kNCX from above we can infer kSR. In PLN-KO myocytes, ktwitch = 9.4 ± 0.7 s−1 and kSR = 9.1 s−1, therefore the SERCA contributes ∼97% (kSR/ktwitch) to twitch Ca removal, consistent with previous studies in this mouse model (24). Similarly, in KO/TG myocytes, ktwitch and kSR (7.46 ± 0.7 and 7.13 s−1) indicate that SERCA is still responsible for ∼95% of twitch [Ca]i decline in KO/TG. Using values for either ktwitch or kSR (i.e., correcting for kNCX) we estimate that the SERCA function is impaired by ∼22% in KO/TG in comparison with PLN-KO myocytes. Because PLN is absent in both KO/TG and PLN-KO mice, this slowing of the apparent SERCA function cannot be attributed to altered PLN expression or phosphorylation. An obvious explanation would be reduced SERCA expression in KO/TG versus PLN-KO, however SERCA expression levels were not different between these groups (Fig. 3 A). We therefore have an apparent discrepancy: slowed SR-mediated Ca clearance with unaltered SERCA expression. This issue is explored further below.
Figure 3.
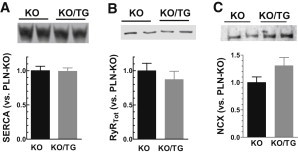
Unaltered protein expression of SERCA and RyR2 but increased NCX in cardiac homogenates. (A) SERCA2 level; (B) RyR2; (C) NCX from KO and KO/TG.
The time to peak of Ca transient is increased in KO/TG versus PLN-KO myocytes using fura-2 or Fluo-4 (Fig. 1 D) or indo-1 (not shown). Two factors could contribute to this delayed time to peak: a), slowed Ca removal flux, and b), prolonged duration of Ca release in KO/TG myocytes. This point is also examined further below.
Protein expression of SERCA, RyR, and NCX expression
Fig. 3 shows unaltered expression of SERCA2 and a tendency (not significant) toward reduced RyR protein expression in KO/TG versus in PLN-KO. Thus, the enhanced SR fractional release in KO/TG cannot be ascribed to a higher number of RyRs in KO/TG. The mean NCX expression level was 30% higher in KO/TG versus PLN-KO (P = 0.11; n = 6). Although not quite significant, this is quantitatively consistent with the 35% increase of the NCX function based on faster [Ca]i decline of caffeine-induced Ca transient (Fig. 2 C). Notably, the RyR2 phosphorylation level at Ser-2815 was significantly increased in KO/TG versus PLN-KO mice (22).
Resting Ca sparks in intact myocytes: CaMKIIδC effects
Ca sparks were measured immediately after steady state pacing at 1 Hz, and caffeine was rapidly applied 20 s later to assess the SR Ca load. Figs. 4 and 5 show that resting Ca spark frequency (CaSpF) is increased more than threefold in KO/TG versus KO myocytes (1.63 ± 0.27 vs. 0.51 ± 0.11 sparks s−1⋅100 μm−1; P < 0.05), despite the lower diastolic [Ca]i and unaltered SR Ca load (Figs. 1 C and 2 B). Spatial spread is also higher in KO/TG versus PLN-KO (2.03 ± 0.02 vs. 1.67 ± 0.03) and duration is longer in KO/TG (31.2 ± 0.1 ms vs. 25.0 ± 0.7 in PLN-KO). Ca spark amplitude is also higher in KO/TG versus PLN-KO (1.07 ± 0.01 vs. 0.74 ± 0.01 ΔF/F0), possibly due to the prolonged release duration (and/or faster maximum Ca release, see below). Multiplying Ca spark frequency, amplitude, and duration suggests ∼7-fold increase in diastolic SR Ca leak flux in KO/TG versus PLN-KO myocytes.
Figure 4.
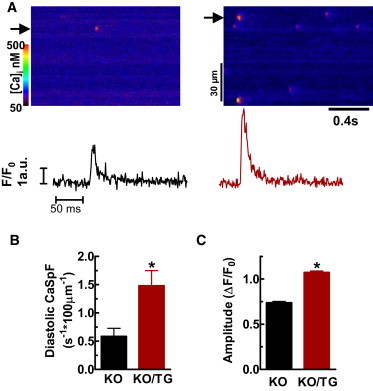
Resting Ca sparks in KO and KO/TG myocytes. (A) Representative diastolic Ca sparks recording from KO and KO/TG myocytes; (B) Ca spark frequency (n = 23, 31 myocytes from KO and KO/TG, respectively); (C) spark amplitude (n = 564, 2226 from KO and KO/TG, respectively).
Figure 5.
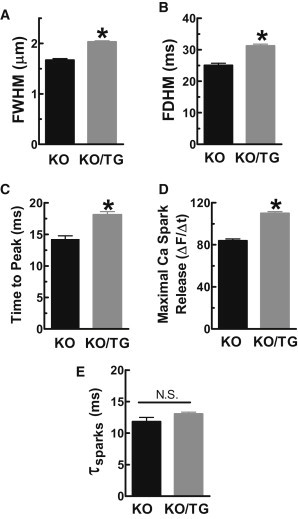
Resting Ca spark frequency and spark properties. (A) Increased FWHM; (B) prolonged FDHM; (C) longer Ca spark rise time; (D) increased maximal rate of [Ca]i rise during Ca sparks; (E) unaltered spark [Ca]i decline.
Macrosparks (FWHM >5 μm) and miniwaves (propagating Ca-induced Ca release) occasionally occurred in KO/TG (0.07 ± 0.01/s and 0.012 ± 0.01/s, respectively) but were absent in PLN-KO (not shown) and not counted in the Ca spark analysis of KO/TG mice (such that the sevenfold increase in leak is probably an underestimate). This is consistent with previously published findings that activation of endogenous CaMKII induced macrosparks and miniwaves in permeabilized PLN-KO myocytes (16). These data indicate that there is a robust CaMKII-dependent enhancement of RyR activity in intact myocytes, an effect that could initiate delayed afterdepolarizations and trigger arrhythmias.
Fig. 5 D shows that CaMKII overexpression increased the maximal rate of [Ca]i rise during Ca sparks (with unaltered SR Ca load), which would be consistent with the notion that CaMKII sensitizes RyR to [Ca]i. That is, during a Ca spark more individual RyRs might be recruited with CaMKII. The duration of the rising phase (time to peak) was significantly prolonged in KO/TG myocytes versus PLN-KO myocytes (Fig. 5 C). This may reflect longer RyR open time and/or slower termination of RyR Ca release (29) in KO/TG versus PLN-KO myocytes. This CaMKII-dependent prolongation of release might be partially responsible for the prolonged rise times of global Ca transients observed in KO/TG (Fig. 1 D). In summary, CaMKII appears to alter the kinetics of both activation and termination of elementary Ca release events.
Repetitive Ca sparks at a single release site occurred much more frequently in KO/TG versus in PLN-KO myocytes (∼10% in KO/TG versus 4% in PLN-KO of all resting spark events). One speculation is that these repetitive events reflect sites where CaMKII has phosphorylated and hence activated or sensitized certain RyRs. Two plausible explanatory mechanisms are that CaMKII: a), increases Ca sensitivity of RyR channels (30,31), or b), hastens Ca spark restitution (which depends on both local SR Ca refilling and recovery of RyR from a refractory state (32)). To test whether RyR recovery is accelerated, we analyzed consecutive sparks at a given site (those which initiate within one pixel of the first spark center; Fig. 6 A). With increasing interspark interval, the amplitude of the second spark recovers toward the amplitude of the first spark, and the relative amplitude of the second spark (Amp2/Amp1) may reflect recovery within that specific RyR cluster (Fig. 6 B). Spark amplitude recovery was not significantly different and a monoexponential fit was sufficient to describe recovery (τPLN-KO ∼67 vs. τKO/TG ∼71 ms; p = 0.61). We conclude that CaMKII overexpression does not appreciably alter refilling or restitution of Ca sparks, but instead elevates Ca sensitivity of RyR channels.
Figure 6.
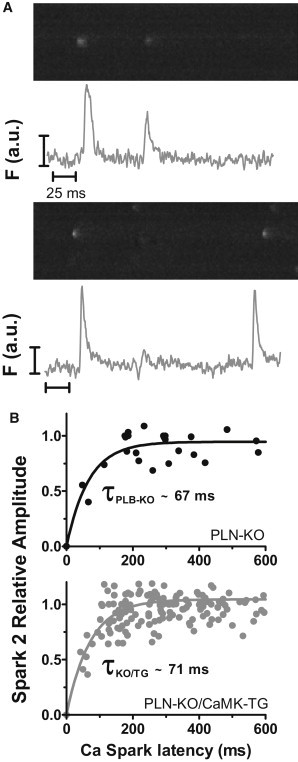
CaMKII does not alter Ca spark amplitude recovery. (A) Repetitive Ca release events from a single release site; (B) scatter plot of spark-to-spark delay versus second spark relative amplitude with a single exponential fit to the data obtained from PLN-KO (n = 29 pairs) and KO/TG (n = 196 pairs).
Ca sparks during twitch [Ca]i decline slow global [Ca]i decline
Normally, Ca sparks are not observed during the declining phase of the Ca transient, when in mouse the action potential has repolarized and Ca current does not flow. This delay in Ca spark occurrence attributed to the need for recovery of both SR Ca content and RyR from refractoriness (30,31). Despite the very rapid Ca resequestration observed in PLN-KO mice, Ca sparks were very rarely seen during twitch [Ca]i decline. In contrast such events were relatively common in KO/TG myocytes (Fig. 7 and Fig S1 A in the Supporting Material). Reopening of RyRs during the twitch Ca relaxation could slow down overall [Ca]i decline in KO/TG and thus explain the slower ktwitch and kSR discussed previously (Fig. 1 E and Fig. 7, B and C). Fig. 7 B presents typical effects of CaMKII overexpression on global Ca transients (measured with Fluo-4), consistent with results from myocytes loaded with fura-2 (Fig. 1) and indo-1 (not shown). Again, global twitch [Ca]i decline is slower in KO/TG (despite unaltered SERCA2a expression).
Figure 7.
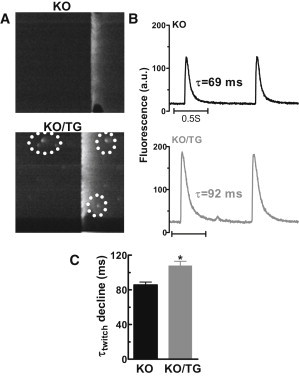
Ca sparks between Ca transients (Fluo-4 AM loaded myocytes). (A) Ca transient from PLN-KO and KO/TG myocytes, note spontaneous Ca release events during twitch Ca relaxation; (B) global Ca transients from PLN-KO and KO/TG; (C) mean time constant of twitch [Ca]i decline.
To assess the impact of RyR reopening on twitch Ca removal (τtwitch) in KO/TG, τtwitch was plotted as a function of the frequency of independent Ca sparks occurring between twitch Ca transients. Fig. 8 A shows a positive correlation between τtwitch and these Ca sparks. Moreover, when no Ca sparks were detected between Ca transients (Y-intercept), τtwitch was ∼80 ms, similar to that in PLN-KO myocytes (see Fig. 8 D). However, when increasing numbers of Ca sparks were detected during twitches, τtwitch was progressively increased from 80 ms.
Figure 8.
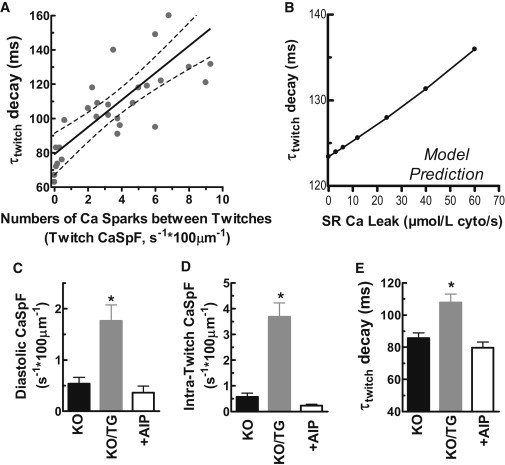
CaMKII inhibitor AIP reversed the increase of CaSpF and slowed twitch [Ca]i decline in KO/TG myocytes. (A) Time constant of twitch [Ca]i decline versus number of Ca sparks between twitch [Ca]i upstrokes; (B) computational model predictions of the impact of SR Ca leak on the τ of twitch [Ca]i decline (τtwitch); (C) CaMKII inhibitor AIP reversed higher resting CaSpF in KO/TG; (D) CaSpF between twitches; (E) delayed twitch [Ca]i relaxation in KO/TG.
To test whether a 5- to 10-fold increase in SR Ca leak is a feasible explanation for the slowed τtwitch we developed a simple Ca flux model during [Ca]i decline, like that developed by Bassani et al. (33) and previously used in PLN-KO mice ((23); see the Supporting Material and Fig. S2 and Fig. S3). We used the [Ca]i-dependence of Ca transport via the SR Ca-ATPase and NCX inferred from Figs. 1 and 2, and included a range of Ca leak from the SR from physiological (4–6 μmol/l cytosol/s (34,35)) to 10-fold higher (Fig. 8 B). Although increasing leak over this range is predicted to slow [Ca]i decline (e.g., by 12 ms) it is smaller than the mean increase in τtwitch between PLN-KO and KO/TG (22 ms; see Fig. 8 E). Thus, the slower twitch [Ca]i decline in KO/TG may at least partially be due to recurring SR Ca release events during the twitch. The SR Ca leak that is not apparent as Ca sparks might be increased by more than the sevenfold increase in spark-mediated leak, because the factors that favor nonspark versus spark-mediated leak would dominate during the Ca transient decline (lower [Ca]SR and/or refractoriness of some RyR (36)).
To further test whether these effects are CaMKII-dependent, the specific CaMKII inhibitory peptide AIP was applied to KO/TG myocytes. Pretreatment with AIP (1 μM) abolished the elevated CaSpF occurring both at rest and between twitches in KO/TG (Fig. 8, C and D, and Fig. S1 B) and accelerated twitch [Ca]i decline (Fig. 8 E and Fig. S1 C), suggesting that the observed differences are under acute regulation by CaMKII. The marked suppression of Ca sparks by AIP treatment in KO/TG myocyte occurred despite an unaltered SR Ca content based on caffeine-induced Ca transients (101 ± 5% versus cells not treated with AIP). Therefore, increased CaMKII activity sensitizes RyR, both in the resting state and during twitch Ca stimulation. Moreover, the frequent occurrence of Ca sparks (and leak) during twitch relaxation could slow the twitch [Ca]i decline and thereby contribute to triggered arrhythmias.
Discussion
Here, we assessed the effects of CaMKIIδC overexpression (as occurs in HF) on SR Ca handling in intact ventricular myocytes in the absence of PLN. The use of the PLN-KO background provided a means of avoiding complicating effects mediated by the actions of CaMKII on PLN. We confirmed the powerful activating effect of CaMKII on RyR activation in the intact myocyte environment, as previously reported in permeabilized myocytes, bilayers, and in myocytes containing native PLN (9,10,14–16,20). The use of intact myocytes allowed us to carry out a more detailed analysis of PLN independent effects of CaMKII on Ca transients and sparks.
Our initial description of the KO/TG mice (22) focused primarily on comparisons with CaMKIIδC-TG mice and the question of whether the deletion of PLN to enhance refilling of the depleted SR Ca stores would rescue the severe HF induced by cardiac TG expression of CaMKIIδC alone. The remarkable outcome was that KO/TG mice had restored individual myocyte Ca transients and contraction, but showed more severe HF and decreased survival than CaMKIIδC mice. Experimental evidence supported the notion that there were increases in mitochondrial Ca loading and cell death, secondary to a further increase in Ca spark frequency (beyond that already seen in CaMKIIδC mice). This remarkable level of Ca spark activity was suggested to be due to a synergistic effect of direct CaMKII stimulated RyR2 gating (via RyR2 phosphorylation) combined with the higher SR Ca load attained in PLN-KO mice (which intrinsically increases Ca sparks). Notably, in resting ventricular myocytes Ca spark frequency is not due to Ca entry (total block of ICa and Na/Ca exchange has no acute affect), but is rather due to stochastic RyR2 opening that depends on [Ca]i [Ca]SR and RyR2 state (30). Here, we focus on how CaMKIIδC alters the RyR function in PLN-KO mice where SR content was coincidentally unaltered, by specifically examining the effects of CaMKII overexpression in PLN-KO. It is important to also note that we studied the KO/TG mice at a relatively early age, when the full HF phenotype had not yet developed (e.g., upregulated NCX, but unaltered SERCA2 or RyR2 expression). Moreover, the affects that we attribute to CaMKII (enhanced Ca spark activity and slowed [Ca]i decline) are acutely prevented by CaMKII inhibition with the AIP peptide (Fig. 8, C–E).
CaMKIIδC enhances Ca spark frequency and amplitude at unaltered SR Ca load
Diastolic Ca spark frequency was significantly increased threefold by CaMKIIδC expression in PLN-KO mice, despite an unaltered SR Ca load (Figs. 2 and 4). Although the SR Ca load tended to be slightly (but insignificantly) lower in the KO/TG, as might be predicted by the increased leak (37,38), it was relatively maintained. Our simple model predicted that a 10-fold increase in leak would reduce SR Ca by ∼6% (within the range of our measurements). The relative maintenance of SR Ca likely occurs because SR Ca uptake is so fast in the PLN-KO myocytes, and because net SR Ca depletion occurs only when leak becomes significant compared to the uptake rate (38). Indeed, PLN-KO myocytes actually maintain elevated SR Ca load, despite a higher SR Ca leak rate at that higher load (16,23). Thus, we observed high Ca spark frequency at high SR Ca load in PLN-KO, as might be induced by catecholamines, but without the complication of adrenergic downstream effects. In this case the superposition of CaMKII overexpression may broadly mimic the RyR activating effects seen in HF, whether due to upregulation and activation of CaMKII or other potential modifiers (e.g., oxidation or catecholaminergic polymorphic ventricular tachycardia mutant RyR2; (39)). Oxidative effects can also alter RyR2 gating, but the effects here were acutely blocked by CaMKII inhibition (Fig. 8). Thus, if our observed RyR2 alterations are due to oxidation effects, they may be mediated via CaMKII oxidation (40).
Not only were Ca sparks more frequent in KO/TG versus PLN-KO mice, they were also larger in amplitude, with greater maximal rate of rise, time to peak, FDHM, and FWHM (Figs. 4 and 5). This is most consistent with an increase in the number of RyRs recruited during a Ca spark (note that total RyR expression and SR Ca load were unaltered). There could also be delayed termination of SR Ca release flux mediated by the termination threshold moving to a lower free intra-SR [Ca] (41), consistent with CaMKII-dependent higher fractional SR Ca release during the twitch ((15), Fig. 2 D, again despite unaltered SR Ca load). Thus, CaMKII seems to sensitize RyR activation and delay the release termination processes during both Ca sparks and EC coupling.
Furthermore, the increased resting CaSpF occurred despite a lower diastolic [Ca]i; accordingly, the ∼sevenfold apparent increase in SR Ca leak previously estimated may underestimate the true magnitude of the CaMKII effect on RyR2. It is not obvious why diastolic [Ca]i is lower in the KO/TG myocytes, but this might be an adaptive change to limit myocyte Ca overload and could be caused in part by the higher expression and function of NCX in these myocytes (Figs. 2 C and 3 C). Note that the upregulation of NCX function seen here is similar to that seen in CaMKIIδC mice versus wild-type (WT) mice (20) and may reflect in part CaMKII effects on NCX expression (42).
Twitch Ca transients in KO/TG myocytes also reach a higher peak and exhibit a prolonged time to peak [Ca]i (Fig. 1). This is consistent with the previous Ca spark characteristics (e.g., more RyRs activated and longer SR Ca release flux). The surprising thing from a Ca flux-balance standpoint is that this higher Ca transient amplitude is maintained in the steady state; instead of driving SR Ca load down via increased extrusion via NCX (43). We cannot rule out the possibility that ICa is slightly elevated in the KO/TG versus PLN-KO (as we found in CaMKIIδC versus WT (20)). However, the higher Ca transient amplitude observed in these mice would limit Ca entry due to stronger Ca-dependent inactivation; it is also possible that the SR Ca uptake is so fast in the PLN-KO mouse compared to NCX (>30 times (23)) that the NCX has less opportunity to extrude Ca during [Ca]i decline than in normal myocytes.
SR Ca refilling rate and restitution of Ca sparks
Despite the apparent prolongation of SR Ca release during Ca sparks, most of the prolongation in FDHM is attributable to prolongation in time to peak rather than [Ca]i decline (Fig. 5). The data for repetitive sparks from a single Ca release site show that Ca spark amplitude recovers with a similar ∼70 ms time constant in both KO/TG and PLN-KO myocytes (Fig. 6). Notably, this amplitude recovery time includes both restoration of SR content and also recovery from RyR refractoriness. SR refilling seems unaltered because Ca spark amplitude recovery was unaltered. We speculate, but cannot prove, that KO/TG RyRs recover faster from refractoriness, resulting in many second events (even before refilling) in the KO/TG myocytes (only 3 events at <170 ms latency in PLN-KO, but 35 such events in the KO/TG myocytes). This could simply be one manifestation of the CaMKII-induced increase in RyR excitability discussed previously, and would be consistent with the overall increase in Ca spark frequency. One of the two prior quantitative studies on Ca spark restitution used ryanodine pretreatment to promote repetitive Ca sparks at individual sites (32), but ryanodine could influence recovery time (∼100 ms). Our recovery time course (τ∼70 ms) is similar to recovery of sensitivity of Ca sparks induced by ultraviolet-laser flash photolysis of caged Ca, as also determined in PLN-KO mouse myocytes (∼60 ms (44)).
Of note, the specific peptide inhibitor of CaMKII, AIP, abrogated the increased resting CaSpF, the enhanced CaSpF during and between twitch Ca transients, and the slowed [Ca]i decline (Fig. 8 and Fig. S1). The acute reversibility of CaMKII effects on RyR, as seen in our prior studies comparing young CaMKIIδC and WT mice and direct CaMKII activation in permeabilized PLN-KO myocytes (16,20), indicates that these are acute CaMKII effects, rather than secondary to long-term developmental effects in the mice.
CaMKIIδC enhances Ca spark frequency sufficiently to slow twitch [Ca]i decline
Previous work has not demonstrated the ability of increased SR Ca leak to slow Ca transient decline. Indeed, even exposure to 0.5 mM caffeine (which is a strong RyR activator/sensitizer) did not alter twitch Ca transient amplitude or kinetics (43). Thus, our observation of slowed twitch [Ca]i decline, directly attributable to CaMKII-dependent SR Ca leak occurring during that time, is consistent with a very profound RyR activating effect of CaMKII, and that might also shorten RyR restitution.
Notably, there is normally a quiescent period immediately during and after a twitch during which time Ca sparks are suppressed, presumably due to RyR refractoriness (30). This normal refractoriness could be protective with respect to SR Ca release-induced arrhythmias, analogous to the way the long cardiac action potential protects the heart from reentrant and tetanic activation. The loss of this RyR refractoriness via CaMKII (like severe Ca overload; (30)) could thus contribute to triggered arrhythmias, and CaMKIIδC-TG mice are indeed at increased risk of arrhythmias (6).
CaMKII has been shown to be upregulated in both human and animal HF (45). In a well-studied arrhythmogenic rabbit HF model, more CaMKII is associated with RyR2, and the enhanced RyR-mediated diastolic SR Ca leak measured in these HF myocytes was blocked by CaMKII inhibitors KN-93 and AIP (but not by the PKA inhibitor H89) (17,46). These outcomes provide compelling evidence that CaMKII upregulation increases Ca leak via RyR, and could directly contribute to slowed twitch [Ca]i decline, decreased free [Ca]SR, and SR Ca load in HF (26) as well as for consequent mitochondrial Ca overload and cell death (22). Indeed, CaMKII inhibitors can prevent cardiac arrhythmias (47,48) and CaMKIIδ knockout has been shown to inhibit pathological hypertrophy and HF in response to pressure overload (49,50).
Acknowledgments
We thank Drs. Lothar Blatter and Sabine Huke for discussions.
This work was supported by National Institutes of Health R37-HL30077 (to D.M.B.) and P01-HL80101 (to D.M.B.; J.H.B.) and Fondation Leducq (to D.M.B.).
Supporting Material
References
- 1.Hook S.S., Means A.R. Ca(2+)/CaM-dependent kinases: from activation to function. Annu. Rev. Pharmacol. Toxicol. 2001;41:471–505. doi: 10.1146/annurev.pharmtox.41.1.471. [DOI] [PubMed] [Google Scholar]
- 2.Bers D.M., Grandi E. CaMKII regulation of cardiac ion channels. J. Cadriovasc. Res. 2009;54:180–187. doi: 10.1097/FJC.0b013e3181a25078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuan W., Bers D.M. Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin-dependent protein kinase. Am. J. Physiol. 1994;267:H982–H993. doi: 10.1152/ajpheart.1994.267.3.H982. [DOI] [PubMed] [Google Scholar]
- 4.Xiao R.P., Cheng H., Lakatta E.G. Dual regulation of Ca2+/calmodulin-dependent kinase II activity by membrane voltage and by calcium influx. Proc. Natl. Acad. Sci. USA. 1994;91:9659–9663. doi: 10.1073/pnas.91.20.9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu Y., Colbran R.J., Anderson M.E. Calmodulin kinase is a molecular switch for cardiac excitation-contraction coupling. Proc. Natl. Acad. Sci. USA. 2001;98:2877–2881. doi: 10.1073/pnas.051449198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner S., Dybkova N., Maier L.S. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner S., Hacker E., Maier L.S. Ca/calmodulin kinase II differentially modulates potassium current. Circ. Arrhythm. Electrophysiol. 2009;2:285–294. doi: 10.1161/CIRCEP.108.842799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simmerman H.K., Collins J.H., Jones L.R. Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J. Biol. Chem. 1986;261:13333–13341. [PubMed] [Google Scholar]
- 9.Witcher D.R., Kovacs R.J., Jones L.R. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J. Biol. Chem. 1991;266:11144–11152. [PubMed] [Google Scholar]
- 10.Currie S., Loughrey C.M., Smith G.L. Calcium/calmodulin-dependent protein kinase IIdelta associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biochem. J. 2004;377:357–366. doi: 10.1042/BJ20031043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez P., Bhogal M.S., Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J. Biol. Chem. 2003;278:38593–38600. doi: 10.1074/jbc.C301180200. [DOI] [PubMed] [Google Scholar]
- 12.Bassani J.W., Yuan W., Bers D.M. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am. J. Physiol. 1995;268:C1313–C1319. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- 13.DeSantiago J., Maier L.S., Bers D.M. Frequency-dependent acceleration of relaxation in the heart depends on CaMKII, but not phospholamban. J. Mol. Cell. Cardiol. 2002;34:975–984. doi: 10.1006/jmcc.2002.2034. [DOI] [PubMed] [Google Scholar]
- 14.Wehrens X.H., Lehnart S.E., Marks A.R. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 2004;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 15.Li L., Satoh H., Bers D.M. The effects of Ca2+-calmodulin-dependent protein kinase II on cardiac excitation-contraction coupling in ferret ventricular myocytes. J. Physiol. 1997;501:17–32. doi: 10.1111/j.1469-7793.1997.017bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo T., Zhang T., Bers D.M. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ. Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 17.Curran J., Hinton M.J., Shannon T.R. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ. Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 18.Lokuta A.J., Rogers T.B., Valdivia H.H. Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation-dephosphorylation mechanism. J. Physiol. 1995;487:609–622. doi: 10.1113/jphysiol.1995.sp020904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang D., Zhu W.Z., Cheng H. Ca2+/calmodulin kinase II-dependent phosphorylation of ryanodine receptors suppresses Ca2+ sparks and Ca2+ waves in cardiac myocytes. Circ. Res. 2007;100:399–407. doi: 10.1161/01.RES.0000258022.13090.55. [DOI] [PubMed] [Google Scholar]
- 20.Maier L.S., Zhang T., Bers D.M. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 2003;92:904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 21.Zhang T., Maier L.S., Brown J.H. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 2003;92:912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 22.Zhang T., Guo T., Brown J.H. Phospholamban ablation rescues sarcoplasmic reticulum Ca2+ handling but exacerbates cardiac dysfunction in CaMKIIδC transgenic mice. Circ. Res. 2010;106:354–362. doi: 10.1161/CIRCRESAHA.109.207423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L., Chu G., Bers D.M. Cardiac myocyte calcium transport in phospholamban knockout mouse: relaxation and endogenous CaMKII effects. Am. J. Physiol. 1998;274:H1335–H1347. doi: 10.1152/ajpheart.1998.274.4.H1335. [DOI] [PubMed] [Google Scholar]
- 24.Luo W., Grupp I.L., Kranias E.G. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ. Res. 1994;75:401–409. doi: 10.1161/01.res.75.3.401. [DOI] [PubMed] [Google Scholar]
- 25.Ginsburg K.S., Bers D.M. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J. Physiol. 2004;556:463–480. doi: 10.1113/jphysiol.2003.055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo T., Ai X., Bers D.M. Intra-sarcoplasmic reticulum free [Ca2+] and buffering in arrhythmogenic failing rabbit heart. Circ. Res. 2007;101:802–810. doi: 10.1161/CIRCRESAHA.107.152140. [DOI] [PubMed] [Google Scholar]
- 27.Picht E., Zima A.V., Bers D.M. SparkMaster: automated calcium spark analysis with ImageJ. Am. J. Physiol. Cell Physiol. 2007;293:C1073–C1081. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- 28.Pogwizd S.M., Qi M., Bers D.M. Upregulation of Na(+)/Ca(2+) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ. Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 29.Terentyev D., Viatchenko-Karpinski S., Györke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: mechanism for hereditary arrhythmia. Proc. Natl. Acad. Sci. USA. 2003;100:11759–11764. doi: 10.1073/pnas.1932318100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Satoh H., Blatter L.A., Bers D.M. Effects of [Ca2+]i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. Am. J. Physiol. 1997;272:H657–H668. doi: 10.1152/ajpheart.1997.272.2.H657. [DOI] [PubMed] [Google Scholar]
- 31.Picht E., DeSantiago J., Bers D.M. Cardiac alternans do not rely on diastolic sarcoplasmic reticulum calcium content fluctuations. Circ. Res. 2006;99:740–748. doi: 10.1161/01.RES.0000244002.88813.91. [DOI] [PubMed] [Google Scholar]
- 32.Sobie E.A., Song L.S., Lederer W.J. Local recovery of Ca2+ release in rat ventricular myocytes. J. Physiol. 2005;565:441–447. doi: 10.1113/jphysiol.2005.086496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bassani J.W.M., Bassani R.A., Bers D.M. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J. Physiol. 1994;476:279–293. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shannon T.R., Pogwizd S.M., Bers D.M. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ. Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- 35.Zima A., Bovo E., Blatter L.A. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J. Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato D., Bers D.M. How does stochastic ryanodine receptor-mediated Ca leak fail to initiate a Ca spark? Biophys. J. 2011;101:2370–2379. doi: 10.1016/j.bpj.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venetucci L.A., Trafford A.W., Eisner D.A. Na/Ca exchange: regulator of intracellular calcium and source of arrhythmias in the heart. Ann. N. Y. Acad. Sci. 2007;1099:315–325. doi: 10.1196/annals.1387.033. [DOI] [PubMed] [Google Scholar]
- 38.Ginsburg K.S., Weber C.R., Bers D.M. Control of maximum sarcoplasmic reticulum Ca load in intact ferret ventricular myocytes. Effects of thapsigargin and isoproterenol. J. Gen. Physiol. 1998;111:491–504. doi: 10.1085/jgp.111.4.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dybkova N., Sedej S., Maier L.S. Overexpression of CaMKIIδc in RyR2R4496C+/− knock-in mice leads to altered intracellular Ca2+ handling and increased mortality. J. Am. Coll. Cardiol. 2011;57:469–479. doi: 10.1016/j.jacc.2010.08.639. [DOI] [PubMed] [Google Scholar]
- 40.Erickson J.R., Jr., Joiner M.L., Anderson M.E. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zima A.V., Picht E., Blatter L.A. Partial inhibition of sarcoplasmic reticulum ca release evokes long-lasting ca release events in ventricular myocytes: role of luminal ca in termination of ca release. Biophys. J. 2008;94:1867–1879. doi: 10.1529/biophysj.107.114694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mani S.K., Egan E.A., Menick D.R. beta-Adrenergic receptor stimulated Ncx1 upregulation is mediated via a CaMKII/AP-1 signaling pathway in adult cardiomyocytes. J. Mol. Cell. Cardiol. 2010;48:342–351. doi: 10.1016/j.yjmcc.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trafford A.W., Díaz M.E., Eisner D.A. Modulation of CICR has no maintained effect on systolic Ca2+: simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J. Physiol. 2000;522:259–270. doi: 10.1111/j.1469-7793.2000.t01-2-00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szentesi P., Pignier C., Niggli E. Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+-induced Ca2+ release refractoriness in heart muscle. Circ. Res. 2004;95:807–813. doi: 10.1161/01.RES.0000146029.80463.7d. [DOI] [PubMed] [Google Scholar]
- 45.Zhang T., Brown J.H. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc. Res. 2004;63:476–486. doi: 10.1016/j.cardiores.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 46.Ai X., Curran J.W., Pogwizd S.M. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 47.Mazur A., Roden D.M., Anderson M.E. Systemic administration of calmodulin antagonist W-7 or protein kinase A inhibitor H-8 prevents torsade de pointes in rabbits. Circulation. 1999;100:2437–2442. doi: 10.1161/01.cir.100.24.2437. [DOI] [PubMed] [Google Scholar]
- 48.Wu Y., Temple J., Anderson M.E. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 49.Backs J., Backs T., Olson E.N. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. USA. 2009;106:2342–2347. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ling H., Zhang T., Heller Brown J. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J. Clin. Invest. 2009;119:1230–1240. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.