

Graphical abstract

Keywords: Protein kinase B, Mycobacterium tuberculosis, Aminopyrimidine, Structure–activity relationship
Abstract
A high-throughput screen against PknB, an essential serine–threonine protein kinase present in Mycobacterium tuberculosis (M. tuberculosis), allowed the identification of an aminoquinazoline inhibitor which was used as a starting point for SAR investigations. Although a significant improvement in enzyme affinity was achieved, the aminoquinazolines showed little or no cellular activity against M. tuberculosis. However, switching to an aminopyrimidine core scaffold and the introduction of a basic amine side chain afforded compounds with nanomolar enzyme binding affinity and micromolar minimum inhibitory concentrations against M. tuberculosis. Replacement of the pyrazole head group with pyridine then allowed equipotent compounds with improved selectivity against a human kinase panel to be obtained.
More than 14 million people worldwide suffer from tuberculosis (TB) which in 2009 resulted in an estimated 1.7 million deaths.1 In particular, the increasing prevalence of multi-drug resistant strains means that novel therapeutic approaches are urgently needed. PknB is one of 11 serine–threonine protein kinases (STPKs) found in Mycobacterium tuberculosis (M. tuberculosis) and has been shown to be essential for bacterial growth,2 and therefore inhibition of PknB represents a potential therapeutic approach for the treatment of TB. PknB is an attractive target as the kinase domain contains less than 30% similarity with eukaryotic kinases, which offers promise for achieving selectivity for the mycobacterial kinase over those of the human host. M. tuberculosis is unusual amongst bacteria in having a higher number of STPKs in comparison to the more common two-component signalling systems,3 and of the other STPKs present, PknA has also been found to be essential while PknG has been reported to play a crucial role in the survival of mycobacteria within macrophages.4 Previous studies have focused on the potential of PknB and PknG as drug targets in M. tuberculosis, although the majority have reported significantly lower activity against whole cells than against the purified enzymes.2,4,5 This difference may be due in part to the nature of the TB cell wall, which is recognised as difficult to permeate, particularly due to its unique mycolic acid layer.6
A high throughput screen of our compound collection against the isolated PknB enzyme was performed through measuring the effect on the in vitro phosphorylation of the substrate GarA,7 and this identified compound 1 (Fig. 1) as a 1.35 μM inhibitor of PknB, which formed the starting point for our chemistry programme.
Figure 1.
HTS hit 1.
Initial SAR exploration around 1 was performed via synthetic access from 2,4-dichloroquinazoline 3 (Scheme 1). Introduction of the aminopyrazole was performed by nucleophilic substitution at the 4-position, followed by either a Suzuki coupling to introduce an aryl group or a second nucleophilic substitution to introduce a second amino substituent at the 2-position.
Scheme 1.

Reagents and conditions: (a) POCl3, N,N-dimethylaniline, reflux; (b) 3-amino-5-R1-pyrazole, EtOH, 20 °C; (c) R2B(OH)2, Pd(PPh3)2Cl2, Et3N, MeOH, microwave, 140 °C or R2NH2, EtOH, microwave, 150 °C.
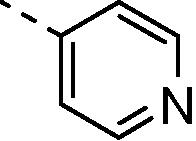
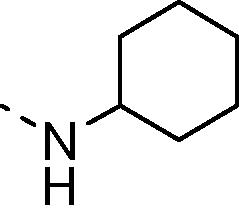
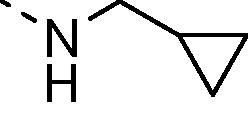
Replacement of the pyrazole methyl substituent with cyclopropyl gave a fourfold improvement in binding affinity (Table 1, 5a). The 4-pyridyl group could also be substituted by other aryl groups such as 4-fluorophenyl or phenyl while maintaining comparable potency (5b–5e). Alkyl amines were also tolerated at this position, with cycloalkyl groups such as the cyclohexyl derivative 5f giving the best IC50. However, the introduction of highly polar or charged groups was not well tolerated at this position and led to a significant loss in potency. Screening of these compounds against M. tuberculosis in an Alamar blue cellular assay8 revealed that they possessed weak or no cellular efficacy. Measured log D values for these compounds were typically in excess of 4, and measurement of their plasma–protein binding (ppb) revealed that they were highly bound (>99%), leaving only a small free fraction available to bind to the enzyme. Furthermore, the physical properties of these compounds were not considered to be well disposed for crossing the TB cell wall, as its mycolic acid layer contains barriers to the passage of hydrophobic as well as hydrophilic molecules.6
Table 1.
SAR of disubstituted quinazolines
| Compound | R1 | R2 or NHR2 | PknB IC50/μMa |
|---|---|---|---|
| 5a | 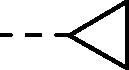 |
 |
0.322 |
| 5b |  |
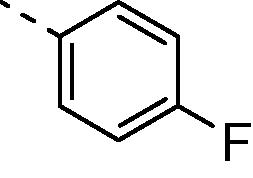 |
0.453 |
| 5c | 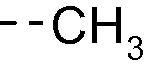 |
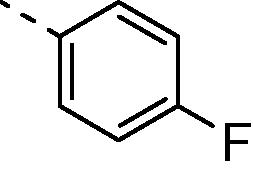 |
1.162 |
| 5d | 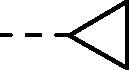 |
 |
0.223 |
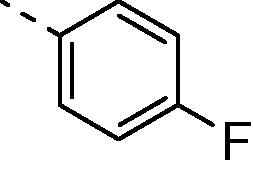
| 5e | 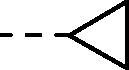 |
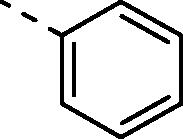 |
0.440 |
| 5f | 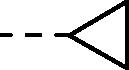 |
 |
0.107 |
| 5g | 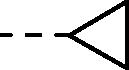 |
 |
0.428 |
Assay conditions are described in Ref. 8.
In order to improve the physical properties of the compounds with the aim of improving their cellular activity, it was decided to reduce the log D of the compounds and explore the effect of this in advancing the SAR. The quinazoline core was switched to a pyrimidine, and while retaining the cyclopropylaminopyrazole at the 4-position, a range of substituents including aryl and amino groups were introduced at the 2-position to allow exploration of the SAR (Scheme 2). This afforded compounds with improved potency against the enzyme (Table 2), and amongst the aryl variants, the 3-sulfonylphenyl compound 8d and 3-cyanophenyl derivative 8f showed the highest affinities at 86 and 87 nM, respectively. For the amino compounds, in addition to the cyclohexyl and cyclopentyl examples 8h and 8n at 84 and 115 nM, the substituted phenylethylamines 8k and 8l showed promising enzyme affinities of 74 and 64 nM, respectively. These compounds also demonstrated improved efficacy against M. tuberculosis although this was still relatively weak, with the majority of minimum inhibitory concentrations (MICs) falling in the 63–250 μM range. The most active example was the 3-sulfonamidophenyl variant 8c at 31 μM.
Scheme 2.

Reagents and conditions: (a) 3-Amino-5-cyclopropylpyrazole, DIPEA, i-PrOH, 50 °C; (b) R1B(OH)2, Pd(PPh3)2Cl2, Et3N, MeOH, microwave, 150 °C or R1NH2, DIPEA, i-PrOH, microwave, 170 °C.
Table 2.
SAR of disubstituted pyrimidines
| Compound | R1 or NHR1 | PknB IC50/μMa | M. tuberculosis MIC/μMa |
|---|---|---|---|
| 8a | 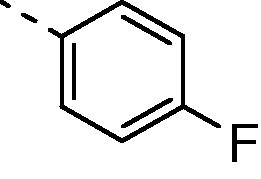 |
0.375 | >500 |
| 8b | 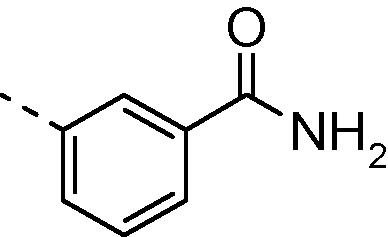 |
0.384 | 63 |
| 8c | 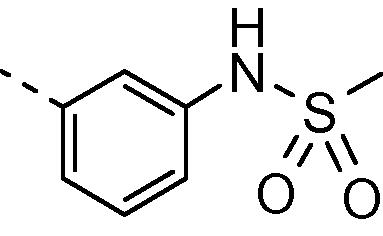 |
0.393 | 31 |
| 8d | 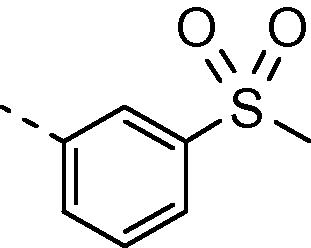 |
0.086 | 63 |
| 8e | 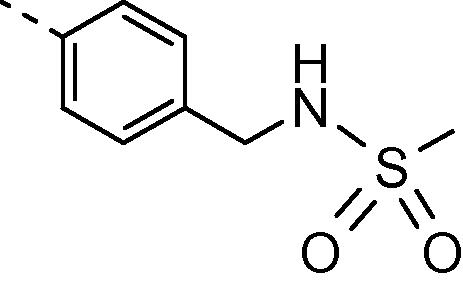 |
0.241 | 63 |
| 8f | 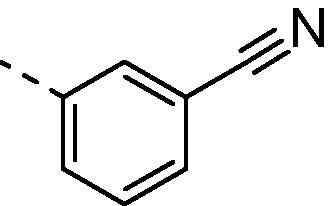 |
0.087 | 125 |
| 8g | 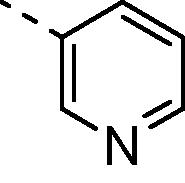 |
0.202 | 125 |
| 8h | 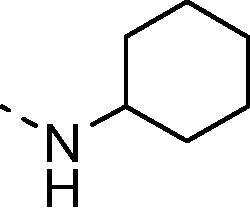 |
0.084 | 63 |
| 8i | 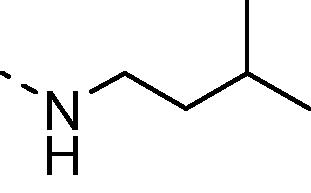 |
0.354 | 63 |
| 8j | 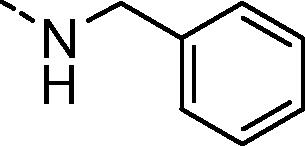 |
0.715 | 125 |
| 8k | 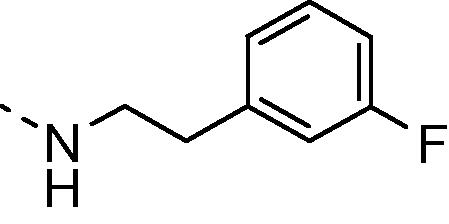 |
0.074 | 63 |
| 8l | 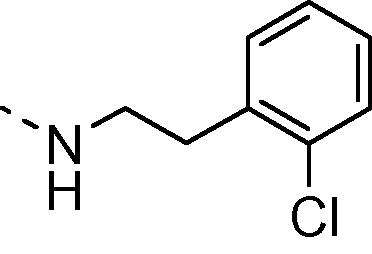 |
0.064 | 125 |
| 8m | 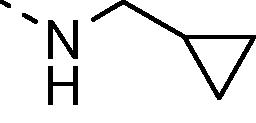 |
0.217 | 250 |
| 8n | 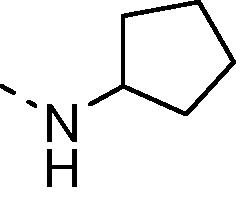 |
0.115 | 63 |
Assay conditions are described in Ref. 8.
Further improvements to the physical properties of the compounds were sought, and a basic amine side chain was introduced to give compounds with a lower log D. The 6-position was chosen as a suitable point for appending this side-chain as modelling studies suggested that this region of the molecule was solvent exposed, so would be amenable to the introduction of this group. A range of basic side chains were introduced into this position, initially with a phenyl group fixed at the 2-position (Scheme 3). The products, shown in Table 3 demonstrated sub-100 nM enzyme potency and generally improved MIC values, and compound 11e containing the (R)-3-dimethylaminopyrrolidine basic side-chain displayed an IC50 of 53 nM against the enzyme and a 16 μM MIC against M. tuberculosis. Oxygen-linked basic side chains were also well tolerated, for example compound 11f, which showed an IC50 of 56 nM against the enzyme and 31 μM MIC against the Mycobacterium.
Scheme 3.

Reagents and conditions: (a) 3-Amino-5-cyclopropylpyrazole, DIPEA, NaI, DMA, 100 °C; (b) R1R2NH, n-BuOH, microwave, 150–170 °C or R1OH, t-BuOK, THF, microwave, 160 °C.
Table 3.
SAR of trisubstituted pyrimidines
| Compound | NR1R2 or OR1 | PknB IC50/μMa | M. tuberculosis MIC/μMa | m Log Db |
|---|---|---|---|---|
| 11a | 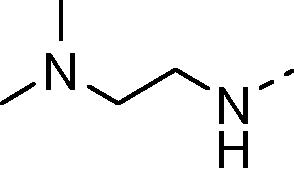 |
0.088 | 125 | 3.7 |
| 11b | 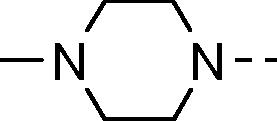 |
0.121 | 63 | 3.6 |
| 11c | 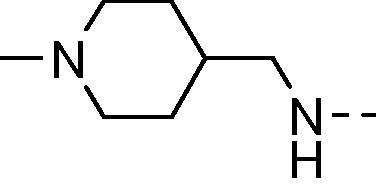 |
0.055 | 31 | 2.3 |
| 11d | 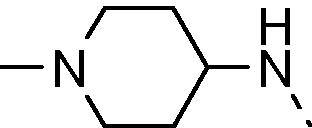 |
0.073 | 31 | 2.3 |
| 11e | 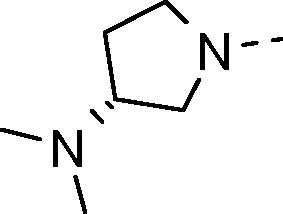 |
0.053 | 16 | 3.8 |
| 11f | 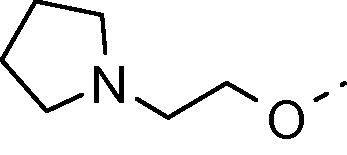 |
0.056 | 31 | 3.4 |
Assay conditions are described in Ref. 8.
Log D values were measured by BioFocus.
Considering the enhanced enzyme binding affinity that had resulted from the presence of a 3-cyano or 3-sulfonyl group on the phenyl ring in the disubstituted variants (Table 2), these substituents were introduced into the trisubstituted pyrimidines aiming for a corresponding increase in enzyme potency and activity against M. tuberculosis. Starting with 4,6-dichloro-2-(methylthio)pyrimidine 12, the cyclopropylaminopyrazole group and the basic amine side-chain were sequentially introduced under nucleophilic displacement conditions. This was followed by a Liebeskind–Srogl coupling9 under microwave heating on the S-methyl pyrimidine 14 to install the desired aryl group (Scheme 4). However, the resultant compounds displayed modest gains in enzyme affinity (approximately twofold for 15b and 15c) and no significant improvement in activity against M. tuberculosis (Table 4), which remained between 31 and 63 μM.
Scheme 4.

Reagents and conditions: (a) 3-Amino-5-cyclopropylpyrazole, Et3N, NaI, DMA, microwave, 150 °C; (b) R1R2NH, n-BuOH, microwave, 170 °C; (c) R3PhB(OH)2, Pd(PPh3)4, copper(I) thiophene carboxylate, dioxane, microwave, 150 °C.
Table 4.
SAR of trisubstituted pyrimidines with functionalised phenyl ring portion
| Compound | NR1R2 | R3 | PknB IC50/μMa | M. tuberculosis MIC/μMa |
|---|---|---|---|---|
| 15a | 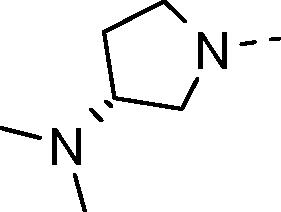 |
CN | 0.056 | 63 |
| 15b | 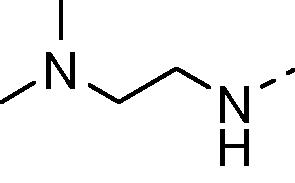 |
CN | 0.039 | 63 |
| 15c | 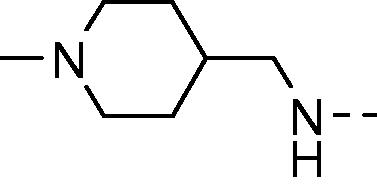 |
CN | 0.023 | 31 |
| 15d | 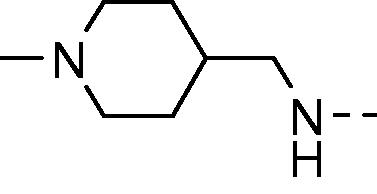 |
SO2Me | 0.040 | 63 |
Assay conditions are described in Ref. 8.
Despite the general improvement in MIC values obtained through introduction of the basic side chain to the pyrimidine core, efficacy against M. tuberculosis remained weaker than was desired. In an effort to improve the cellular activity, it was decided to replace the aminopyrazole head group with an aminopyridine group. This allowed the hydrogen bond donor count to be reduced, which was considered desirable for improving permeability through the M. tuberculosis cell wall. It also offered the potential for improved kinase selectivity by reducing the reliance on the binding energy gained at the kinase hinge region. These compounds were prepared following a similar sequence to that described in Scheme 4, replacing the aminopyrazole with the appropriate aminopyridine but using a palladium-catalysed amination for introduction of the aminopyridine instead of nucleophilic displacement. Initial variants showed a significant loss in binding affinity, but further exploration of the SAR revealed that pyridines carrying a substituent at the 4-position were optimal for enzyme inhibition and this allowed the IC50 to be restored to the double-digit nanomolar range (Table 5). However, in common with the aminopyrazoles, MIC values against M. tuberculosis remained in the micromolar region. Compound 16c displayed the most promising activity with a MIC of 8 μM.
Table 5.
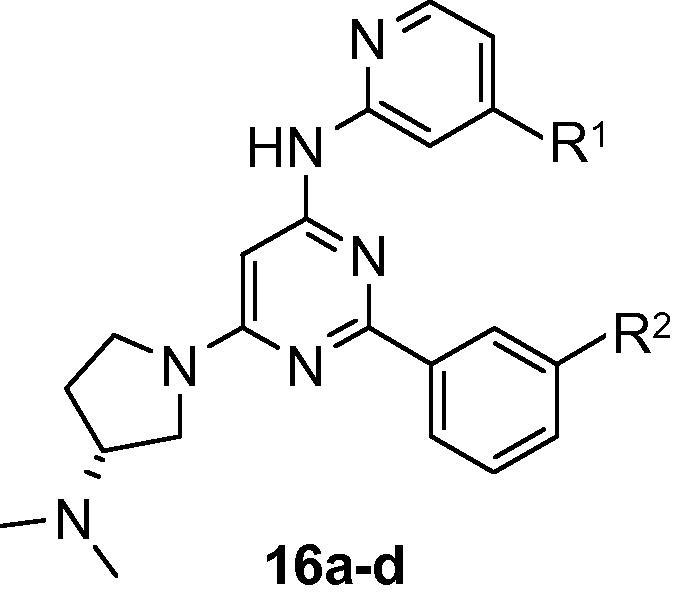
| Compound | R1 | R2 | PknB IC50/μMa | M. tuberculosis MIC/μMa |
|---|---|---|---|---|
| 16a | H | CN | 1.99 | 31 |
| 16b | OMe | H | 1.13 | 31 |
| 16c | Cl | H | 0.176 | 8 |
| 16d | CF3 | CN | 0.040 | 16 |
Assay conditions are described in Ref. 10.
The selectivity of the inhibitors was examined against other M. tuberculosis kinases and this revealed that they typically showed cross-affinity with PknF, although they displayed only modest inhibition of PknG. Aminopyrimidines featuring appended pyrazole groups have been reported in the literature as inhibitors of a number of human kinases,10 so achieving selectivity in these compounds was therefore a key consideration and their kinase selectivity against a 76-member human kinase panel was examined. Pleasingly compound 16c, containing the aminopyridine head group motif, showed an improvement in selectivity relative to 11e, which contains the aminopyrazole head group (Fig. 2). This is consistent with an expected gain in selectivity that may occur through reducing the number of H-bonds that can be formed with the kinase hinge region. Docking studies were performed using Glide™ (Schrödinger Inc.) on both the aminopyrazole and aminopyridine head group variants using the PknB crystal structure (ref: 2FUM)12 from the PDB, and the predicted binding modes of compounds 15c and 16d are shown in Figure 3. It can be seen that the aminopyrazoles can form up to 3 hydrogen bonds with the hinge region of the kinase, and the appended cyclopropyl group can form a face-on lipophilic interaction with the methionine gatekeeper residue. The cyano group on the phenyl ring points in the direction of a lysine residue. By contrast, in the aminopyridine variants two hydrogen bonds with the hinge region can be formed, and the 4-substituent on the pyridine head group is orientated in the direction of the gatekeeper.
Figure 2.

Kinase selectivity heat map comparing compounds 11e (top) and 16c (bottom) against a 76-member human kinase panel, screened at 10 μM inhibitor concentration. [Key: red: >90% inhibition; yellow: 70–90% inhibition; green: <70% inhibition.11]
Figure 3.

Example aminopyrazole 15c (left) and aminopyridine 16d (right) head group compounds docked into the PknB active site.
In summary, the starting point HTS hit was optimised for potency against PknB, and initial modifications showed promising gains in enzyme binding affinity but little or no cellular activity against M. tuberculosis. Switching from the quinazoline to a pyrimidine core and introduction of a basic side chain improved the physical properties of the molecules and resulted in improved cellular activities, in the double-digit micromolar region. While further optimisation gave improvements in enzyme binding affinity, this did not translate into improved efficacy against M. tuberculosis. Replacement of the aminopyrazole head group with an aminopyridine led to an initial loss in binding affinity, but this was regained following optimisation, and selectivity against a human kinase panel was improved. Although the best of these compounds still only showed whole cell MICs in the 10 μM region, this level of activity is commensurate with that reported for a number of other heterocyclic compounds which have shown activity against M. tuberculosis.13 Possible reasons for the significant difference in enzyme and whole cell activity were investigated, including poor cell wall permeability, the role of efflux pumps, protein binding in the assay media and inhibitor specificity, and this work is described in a separate paper.8
Acknowledgements
We thank Dony Patel, Tim Nott and Vicky Spivey for production of PknB enzyme and GarA substrate. This work was supported by Grants from the MRCT development gap fund (award A853-0058) and the MRC (U117585867 and U117584228).
References and notes
- 1.WHO Report 2010, Global Tuberculosis Control, WHO Press, Geneva.[http://whqlibdoc.who.int/publications/2010/9789241564069_eng.pdf, Accessed: 29 July 2011].
- 2.Fernandez P., Saint-Joanis B., Barilone N., Jackson M., Gicquel B., Cole S.T., Alzari P.M. J. Bacteriol. 2006;188:7778. doi: 10.1128/JB.00963-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Cole S.T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D. Nature. 1998;393:537. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]; (b) Av-Gay Y., Everett M. Trends Microbiol. 2000;8:238. doi: 10.1016/s0966-842x(00)01734-0. [DOI] [PubMed] [Google Scholar]; (c) Narayan A., Sachdeva P., Sharma K., Saini A.K., Tyagi A.K., Singh Y. Physiol. Genomics. 2007;29:66. doi: 10.1152/physiolgenomics.00221.2006. [DOI] [PubMed] [Google Scholar]; (d) Molle V., Kremer L. Mol. Microbiol. 2010;75:1064. doi: 10.1111/j.1365-2958.2009.07041.x. [DOI] [PubMed] [Google Scholar]
- 4.Walburger A., Koul A., Ferrari G., Nguyen L. Science. 2004;304:1800. doi: 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
- 5.(a) Hegymegi-Barakonyi B., Szekely R., Varga Z., Kiss R., Borbely G., Nemeth G. Curr. Med. Chem. 2008;15:2760. doi: 10.2174/092986708786242886. [DOI] [PubMed] [Google Scholar]; (b) Szekely R., Waczek F., Szabadkai I., Nemeth G., Hegymegi-Barakonyi B., Eros D. Immunol. Lett. 2008;116:225. doi: 10.1016/j.imlet.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Brennan P.J., Nikaido H. Annu. Rev. Biochem. 1995;64:29. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 7.Villarino A., Duran R., Wehenkel A., Fernandez P., England P., Brodin P., Cole S.T., Zimny-Arndt U., Jungblut P.R., Cerveñansky C., Alzari P.M. J. Mol. Biol. 2005;350:953. doi: 10.1016/j.jmb.2005.05.049. [DOI] [PubMed] [Google Scholar]
- 8.Lougheed K.E.A., Osborne S.A., Saxty B., Whalley D., Chapman T., Bouloc N., Chugh J., Nott T.J., Patel D., Spivey V., Kettleborough C.A., Bryans J.S., Taylor D.L., Smerdon S.J., Buxton R.S. Tuberculosis. 2011;91:277. doi: 10.1016/j.tube.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liebeskind L.S., Srogl J. Org. Lett. 2002;4:979. doi: 10.1021/ol0200091. [DOI] [PubMed] [Google Scholar]
- 10.(a) Humphries P.S., Lafontaine J.A., Agree C.S., Alexander D., Chen P., Do Q.-Q.T. Bioorg. Med. Chem. Lett. 2009;19:2099. doi: 10.1016/j.bmcl.2009.03.023. See for example: [DOI] [PubMed] [Google Scholar]; (b) Ioannidis S., Lamb M.L., Wang T., Almeida L., Block M.H., Davies A.M. J. Med. Chem. 2011;54:262. doi: 10.1021/jm1011319. [DOI] [PubMed] [Google Scholar]; (c) Lin W.-H., Hsieh S.-Y., Yen S.-C., Chen C.-T., Yeh T.-K., Hsu T. Bioorg. Med. Chem. 2011;19:4173. doi: 10.1016/j.bmc.2011.06.016. [DOI] [PubMed] [Google Scholar]; (d) Harrington E.A., Bebbington D., Moore J., Rasmussen R.K., Ajose-Adeogun A.O., Nakayama T. Nat. Med. 2004;10:262. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 11.Kinase selectivity profiling was carried out at the National Centre for Protein Kinase Profiling in the MRC Protein Phosphorylation Unit at the University of Dundee.
- 12.Wehenkel A., Fernandez P., Bellinzoni M., Catherinot V., Barilone N., Labesse G., Jackson M., Alzari P.M. FEBS Lett. 2006;580:3018. doi: 10.1016/j.febslet.2006.04.046. (PDB ref: 2FUM) [DOI] [PubMed] [Google Scholar]
- 13.(a) Chhabria M.T., Jani M.H. Eur. J. Med. Chem. 2009;44:3837. doi: 10.1016/j.ejmech.2009.04.002. See for example: [DOI] [PubMed] [Google Scholar]; (b) Wilkinson B.L., Bornaghi L.F., Wright A.D., Houston T.A., Poulsen S.-A. Bioorg. Med. Chem. Lett. 2007;17:1355. doi: 10.1016/j.bmcl.2006.11.079. [DOI] [PubMed] [Google Scholar]; (c) Zhou B., He Y., Zhang X., Xu J., Luo Y., Wang Y. PNAS. 2010;107:4573. doi: 10.1073/pnas.0909133107. [DOI] [PMC free article] [PubMed] [Google Scholar]