

Abstract
Diet strongly affects human health, partly by modulating gut microbiome composition. We used diet inventories and 16S rDNA sequencing to characterize fecal samples from 98 individuals. Fecal communities clustered into enterotypes distinguished primarily by levels of Bacteroides and Prevotella. Enterotypes were strongly associated with long-term diets, particularly protein and animal fat (Bacteroides) versus carbohydrates (Prevotella). A controlled-feeding study of 10 subjects showed that microbiome composition changed detectably within 24 hours of initiating a high-fat/low-fiber or low-fat/high-fiber diet, but that enterotype identity remained stable during the 10-day study. Thus, alternative enterotype states are associated with long-term diet.
We coexist with our gut microbiota as mutualists, but this relationship sometimes becomes pathological, as in obesity, diabetes, atherosclerosis, and inflammatory bowel diseases (1, 2). Factors including age, genetics, and diet may influence microbiome composition (3). Of these, diet is easiest to modify and presents the simplest route for therapeutic intervention. Recently, an analysis of gut microbial communities proposed three predominant variants, or “enterotypes,” dominated by Bacteroides, Prevotella, and Ruminococcus, respectively (4). The basis for enterotype clustering is unknown but appears independent of nationality, sex, age, or body mass index (BMI).
Here, we investigated the association of dietary and environmental variables with the gut microbiota. First, in a cross-sectional analysis of 98 healthy volunteers (abbreviated “COMBO”), we collected diet information using two questionnaires that queried recent diet (“Recall”) and habitual long-term diet (food frequency questionnaire; “FFQ”). Second, 10 individuals were sequestered in a hospital environment in a controlled-feeding study (abbreviated “CAFE”) to compare high-fat/low-fiber and low-fat/high-fiber diets. Stool samples were collected (5), and DNA samples were analyzed by 454/Roche pyrosequencing (6) of 16S rDNA gene segments and, for selected samples, shotgun metagenomics (7). In CAFE, rectal biopsy samples were also collected and analyzed on days 1 and 10.
For COMBO, we used 16S ribosomal DNA (rDNA) sequence information to calculate pair-wise UniFrac distances (8) among the microbial communities. We assessed both relative abundance data (weighted analysis) and presence/absence information (unweighted analysis). Specific nutrients associated with variation in the gut microbiome for the 98 subjects were extracted, along with demographic factors (table S1). For each nutrient, we performed PERMANOVA (9) to test for nutrient microbiome association, from which we identified 72 and 97 microbiome-associated nutrients in Recall and FFQ, respectively, at a false discovery rate (FDR) of 25% (the relatively high value was used so as not to miss possible effects of diet on low-abundance bacteria). Both weighted and unweighted UniFrac identified similar nutrients, although the discrimination was sharper with unweighted UniFrac, indicating that change in community membership rather than community composition was the main factor.
For each of these nutrients, we used Spearman correlations to identify the associated bacterial genera. We considered only the 78 taxa that had abundance ≥0.2% in at least one sample and appeared in more than 10% of the samples. Figure 1 shows a heat map summarizing Spearman correlations between nutrients from the FFQ and bacterial taxa. For a given taxon, individual nutrients account for 3 to 20% of the between-subject variation in abundance.
Fig. 1.

Correlation of diet and gut microbial taxa identified in the cross-sectional COMBO analysis. Columns correspond to bacterial taxa quantified using 16S rDNA tags; rows correspond to nutrients measured by dietary questionnaire. Red and blue denote positive and negative association, respectively. The intensity of the colors represents the degree of association between the taxa abundances and nutrients as measured by the Spearman's correlations. Bacterial phyla are summarized by the color code on the bottom; lower-level taxonomic assignments specified are in fig. S1. The dots indicate the associations that are significant at an FDR of 25%. The FFQ data were used for this comparison (both FFQ and Recall dietary data are shown together in fig. S1). Columns and rows are clustered by Euclidean distance, with rows separated by the predominant nutrient category.
Nutrients of the same food groups from Recall and FFQ tended to cluster together (fig. S1A). The nutrients from fat versus plant products and fiber showed inverse associations with microbial taxa (Spearman ρ = −0.68, P < 0.0001). Inverse associations were also seen with amino acids and proteins versus carbohydrates (Spearman ρ = −0.73, P < 0.0001) and with fat versus carbohydrates (Spearman ρ = −0.61, P = 0.0001). Phyla positively associated with fat but negatively associated with fiber were predominantly Bacteroidetes and Actinobacteria, whereas Firmicutes and Proteobacteria showed the opposite association. However, within each phylum, not all lower-level taxa demonstrated similar correlations with dietary components (fig. S1B). Taxa correlated with BMI also correlated with fat and percent calories from saturated fatty acids (fig. S1B and table S1).
Following the suggestion by Arumugam et al. (4) that the human gut microbiome can be partitioned into enterotypes, we investigated whether the 98 COMBO samples partitioned into clusters that were detectably associated with dietary or demographic data (Fig. 2). Several methods for data processing and clustering were compared (fig. S2). In one analytical approach (weighted UniFrac, no lane masking; fig. S2), partitioning around medoids (PAM) analysis favored partitioning into three clusters, although with quite low support (silhouette score 0.2) suggesting that clustering could be due to chance. Comparison to the three genera specified by Arumugam et al. (4) showed that relatively high levels of the genera Bacteroides and Prevotella distinguished two of the clusters, whereas the third showed slightly higher levels of Ruminococcus. However, most methods showed two clusters, with stronger support (Fig. 2; Jensen-Shannon distance, silhouette score 0.66), in which the Bacteroides enterotype was fused with the less well distinguished Ruminococcus enterotype. As described below, dietary effects primarily distinguish the Prevotella enterotype from the Bacteroides enterotype.
Fig. 2.

Clustering of gut microbial taxa into entero-types is associated with long-term diet. (A) Clustering in the COMBO cross-sectional study using Jensen-Shannon distance. The left panel shows that the data are most naturally separated into two clusters by the PAM method. The x axis shows cluster number; the y axis shows silhouette width, a measure of cluster separation (12). The right panel shows the clustering on the first two principal components. (B) Proportions of bacterial taxa characteristic of each enterotype. Boxes represent the interquartile range (IQR) and the line inside represents the median. Whiskers denote the lowest and highest values within 1.5 × IQR. (C) The association of dietary components with each enterotype. The strength and direction of each association, as measured by the means of the standardized nutrient measurements, is shown by the color key at the lower right. Enterotype is shown at the right. Red indicates greater amounts, blue lesser amounts of each nutrient in each enterotype (complete lists of nutrients are in table S2). Columns were clustered by Euclidean distance.
At an FDR of 5%, six genera differed between the Prevotella and Bacteroides enterotypes (fig. S3). The Bacteroides enterotype was distinguished by the additional presence of Alistipes and Parabacteroides (phylum Bacteroidetes). The Prevotella enterotype was distinguished by the additional presence of Paraprevotella (phylum Bacteroidetes) and Catenibacterium (phylum Firmicutes) (fig. S3). The enterotype clustering was driven primarily by the ratio of the two dominant genera, Prevotella to Bacteroides, which defines a gradient across the two enterotypes (fig. S5).
At an FDR of 25%, nutrients from the long-term FFQ but not the short-term Recall questionnaire were associated with enterotype composition, indicating that long-term diet strongly correlates with enterotype (the relatively high FDR was used to avoid excessively strict filtering and to visualize the full pattern). The Bacteroides entero-type was highly associated with animal protein, a variety of amino acids, and saturated fats (Fig. 2C), which suggests that meat consumption as in a Western diet characterized this enterotype. The Prevotella enterotype, in contrast, was associated with low values for these groups but high values for carbohydrates and simple sugars, indicating association with a carbohydrate-based diet more typical of agrarian societies. Self-reported vegetarians (n = 11) showed enrichment in the Prevotella enterotype (27% Prevotella enterotype versus 10% Bacteroides enterotype; P = 0.13). The one self-reported vegan was in the Prevotella enterotype. No significant associations were seen with demographic data at this FDR.
A short-term controlled-feeding experiment (CAFE) was carried out to test the stability of the gut microbiome and the observed nutrient-microbiome associations. Ten subjects were sequestered and randomized to high-fat/low-fiber or low-fat/high-fiber diets and were then sampled over 10 days (Fig. 3). Analysis of 16S tag data from stool samples showed that intersubject variation was by far the predominant source of variance in the data (10). Figure 3A shows sharp clustering of the microbiome sequence data by individual in unweighted UniFrac, emphasizing that distinctive lineages are present in each subject. Over 10 days of controlled feeding, there was no reduction in UniFrac distances for stool or biopsy samples between individuals fed the same diet, demonstrating that a short-term identical diet does not overcome intersubject variation.
Fig. 3.
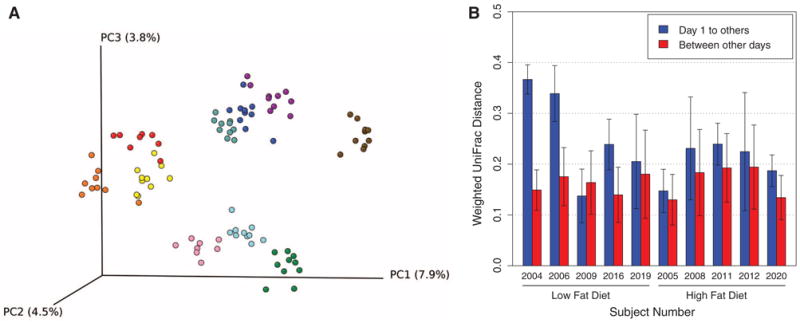
Changes in bacterial communities during controlled feeding. Ten subjects were randomized to high-fat/low-fiber or low-fat/high-fiber diets, and microbiome composition was monitored longitudinally for 10 days by sequencing 16S rDNA gene tags (CAFE study). (A) Cluster diagram–based principal coordinates analysis using unweighted UniFrac. Colors indicate samples from each individual. (B) Day 1 samples are outliers compared to all other days, indicating change in the gut microbiome within 24 hours of initiating controlled feeding. In this analysis, weighted UniFrac distances between samples are compared within subjects in two groups. The first collection of distances compares the day 1 samples to days 2 to 10; the second group compares samples from all days to all others excluding day 1, indicating rapid change (P = 0.0003, 10,000 permutations). Error bars indicate 1 SD of the distances.
Remarkably, changes in microbiome composition were detectable within 24 hours of initiating controlled feeding. For each individual sampled, the first sampling day represented an outlier (Fig. 3B; P = 0.0003, 10,000 permutations), indicating rapid change. Similar results were seen in the unweighted analysis (P = 0.0002). The taxa affected differed among individuals.
The relationship of changes in microbiome composition to the transit time of material through the gut was also investigated. Subjects swallowed x-ray–opaque markers at the start of the study, allowing quantification of transit time by abdominal x-ray. Transit time was faster with the high-fiber diet (2 to 4 days) than with the high-fat diet (2 to 7 days; P = 0.02; two-sided Wilcoxon rank sum test), as expected. All patients retained at least one of the 24 markers 48 hours after the start of the experimental diet. Thus, the changes in microbiome composition, which occurred within 24 hours, were faster than clearance of residual material from the gut.
To probe metabolic functionality during the CAFE study, we also analyzed changes in total gene content using shotgun metagenomics. We compared stool samples from day 1 and day 10 (1.05 × 106 sequence reads total). Sequence reads were annotated for function using the KEGG database (11), then interrogated to assess the taxa and classes of genes present. No significant changes in proportions among archaea, bacteria, and eukaryotes were detected, and bacterial taxa inferred from shotgun metagenomic data paralleled the 16S rDNA data (fig. S4). We investigated gene groups that changed significantly between day 1 and 10 and differed between the high-fat and high-fiber groups. To control for between-subject variability, we used the day 1 samples as within-subject controls, and subtracted each subject's day 1 functional category counts from day 10 samples from that same subject. Functional categories that differentiated diets included bacterial secretion system (P = 0.01, t test), protein export (P = 0.022), and lipoic acid metabolism (P = 0.045), thus indicating bacterial functions potentially involved in responding to these dietary changes.
We next assessed the response of enterotypes to the controlled feeding regimen. Each of the samples from the 10 subjects was assigned to an enterotype category on the basis of their microbiome distances to the medoids (12) of the enterotype clusters as defined in the COMBO data. All subjects started in the Bacteroides enterotype (high protein and fat). None switched stably to the Prevotella (carbohydrate) enterotype over the duration of the study. A single specimen scored in the Prevotella (carbohydrate) enterotype but reverted by the time of the next sample. Thus, over the 10 days of the dietary intervention, we did not see stable switching between the two enterotype groups characterized by the dietary extremes, despite feeding of a low-fat/high-fiber diet to half the subjects.
Finally, several factors were significantly correlated with microbiome composition but not with enterotype partitioning. Examples included BMI, red wine, and aspartame consumption (7). Thus, not all associations between host and microbiota are captured in the enterotype distinctions.
Comparison of long-term and short-term dietary data showed that only the long-term diet was correlated with enterotype clustering in the cross-sectional study. In the interventional study, changes were significant and rapid, but the magnitude of the changes was modest and not sufficient to switch individuals between the enterotype clusters associated with protein/fat and carbohydrates. Thus, our data indicate that long-term diet is particularly strongly associated with enterotype partitioning. The dietary associations seen here parallel a recent study comparing European children, who eat a typical Western diet high in animal protein and fat, to children in Burkina Faso, who eat high-carbohydrate diets low in animal protein (13). The European microbiome was dominated by taxa typical of the Bacteroides enterotype, whereas the African microbiome was dominated by the Prevotella enterotype, the same pattern seen here. There are, of course, many differences between Europe and Burkina Faso that might influence the gut microbiome, but dietary differences provide an attractive potential explanation. Having confirmed enterotype partitioning and established the association with dietary patterns, it will be important to determine whether individuals with the Bacteroides enterotype have a higher incidence of diseases associated with a Western diet, and whether long-term dietary interventions can stably switch individuals to the Prevotella entero-type. If an enterotype is ultimately shown to be causally related to disease, then long-term dietary interventions may allow modulation of an individual's enterotype to improve health.
Supplementary Material
Acknowledgments
Supported by NIH grants UH2 DK083981 (F.D.B., J.D.L., and G.D.W.) and RO1 AI39368 (G.D.W.); Penn Genome Frontiers Institute; Penn Digestive Disease Center grant P30 DK050306; Joint Penn-CHOP Center for Digestive, Liver, and Pancreatic Medicine grants S10RR024525, UL1RR024134, and K24-DK078228; and the Howard Hughes Medical Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources, National Institutes of Health, or Pennsylvania Department of Health. Accession numbers (Sequence Read Archive): for the CAFE study, SRX021237, SRX021236, SRX020587, SRX020379, and SRX020378 (metagenomic); for the COMBO study, SRX020773, SRX020770, and SRX089367.
References and Notes
- 1.Hooper LV, Gordon JI. Science. 2001;292:1115. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 2.Emminger A, Kahmann E, Savage DS. Cancer Lett. 1977;2:273. doi: 10.1016/s0304-3835(77)80032-3. [DOI] [PubMed] [Google Scholar]
- 3.Gill SR, et al. Science. 2006;312:1355. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arumugam M, et al. Nature. 2011;473:174. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu GD, et al. BMC Microbiol. 2010;10:206. doi: 10.1186/1471-2180-10-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Margulies M, et al. Nature. 2005;437:376. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.See supporting material on Science Online.
- 8.Lozupone C, Hamady M, Knight R. BMC Bioinformatics. 2006;7:371. doi: 10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McArdle BH, Anderson MJ. Ecology. 2001;82:290. [Google Scholar]
- 10.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Nature. 2006;444:1022. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 11.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. Nucleic Acids Res. 2004;32(database issue):D277. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rousseeuw PJ. J Comput Appl Math. 1987;20:53. [Google Scholar]
- 13.De Filippo C, et al. Proc Natl Acad Sci USA. 2010;107:14691. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.