

Abstract
Sulfuricurvum kujiense Kodama and Watanabe 2004 is the type species of the monotypic genus Sulfuricurvum, which belongs to the family Helicobacteraceae in the class Epsilonproteobacteria. The species is of interest because it is frequently found in crude oil and oil sands where it utilizes various reduced sulfur compounds such as elemental sulfur, sulfide and thiosulfate as electron donors. Members of the species do not utilize sugars, organic acids or hydrocarbons as carbon and energy sources. This genome sequence represents the type strain of the only species in the genus Sulfuricurvum. The genome, which consists of a circular chromosome of 2,574,824 bp length and four plasmids of 118,585 bp, 71,513 bp, 51,014 bp, and 3,421 bp length, respectively, harboring a total of 2,879 protein-coding and 61 RNA genes and is a part of the Genomic Encyclopedia of Bacteria and Archaea project.
Keywords: facultatively anaerobic, microaerobic, motile, Gram-negative, sulfur-oxidizing, mesophilic, chemolithoautotrophic, Helicobacteracea, GEBA
Introduction
Strain YK-1T (= DSM 16994 = ATCC BAA-921 = JCM 11577) is the type strain of the species Sulfuricurvum kujiense, which is the type species of the monotypic genus Sulfuricurvum [1,2]. The genus name was derived from the Latin words 'sulfur' and 'curvus' meaning 'curved', yielding the Neo-Latin word 'Sulfuricurvum', the 'curved bacterium that utilizes sulfur' [1]. The species epithet is derived from the Neo-Latin word 'kujiense' (referring to Kuji, Iwate Prefecture, Japan, where the bacterium was isolated) [1]. Three more strains of the species S. kujiense were isolated from the same habitat and exhibited identical physiological characteristics with the type strain YK-1T [3]. Sulfuricurvum spp. have been detected in different groundwater environments [4,5] and in oil fields [6]. Here we present a summary classification and a set of features for S. kujiense strain YK-1T, together with the description of the complete genomic sequencing and annotation.
Classification and features
A representative genomic 16S rRNA sequence of S. kujiense YK-1T was compared using NCBI BLAST [7,8] under default settings (e.g., considering only the high-scoring segment pairs (HSPs) from the best 250 hits) with the most recent release of the Greengenes database [9] and the relative frequencies of taxa and keywords, reduced to their stem [10], were determined, weighted by BLAST scores. The most frequently occurring genus was Sulfuricurvum (100.0%) (3 hits in total). Regarding the three hits to sequences from members of the species, the average identity within HSPs was 99.1%, whereas the average coverage by HSPs was 92.9%. No hits to sequences with (other) species names were found. (Note that the Greengenes database uses the INSDC (= EMBL/NCBI/DDBJ) annotation, which is not an authoritative source for nomenclature or classification.) The highest-scoring environmental sequence was AB030609 ('groundwater clone 1061') [11], which showed an identity of 99.7% and an HSP coverage of 96.9%. The most frequently occurring keywords within the labels of all environmental samples which yielded hits were 'spring' (9.6%), 'cave' (9.4%), 'microbi' (6.9%), 'sulfid' (5.7%) and 'mat' (5.2%) (247 hits in total). These keywords suggest that habitats for S. kujiense well-matched to that supposed in the original description [1] and other publications [3,12]. Environmental samples which yielded hits of a higher score than the highest scoring species were not found.
Figure 1 shows the phylogenetic neighborhood of S. kujiense YK-1T in a 16S rRNA based tree. The sequences of the three 16S rRNA gene copies in the genome differ from each other by one nucleotide, and differ by up to two nucleotides from the previously published 16S rRNA sequence (AB053951).
Figure 1.

Phylogenetic tree highlighting the position of S. kujiense relative to the type strains of the type species of the other genera within the class Epsilonproteobacteria. The tree was inferred from 1,364 aligned characters [13,14] of the 16S rRNA gene sequence under the maximum likelihood (ML) criterion [15]. Rooting was done initially using the midpoint method [16] and then checked for its agreement with the current classification (Table 1). The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 1,000 ML bootstrap replicates [17] (left) and from 1,000 Maximum-Parsimony bootstrap replicates [18] (right) if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [19] are labeled with one asterisk, those also listed as 'Complete and Published' with two asterisks [20-24].
As one of the families selected for Figure 1, Nautiliaceae (comprising the genera Caminibacter, Lebetimonas, Nautilia, Nitratifractor, Nitratiruptor and Thioreductor) did not appear as monophyletic in the tree, we conducted both unconstrained heuristic searches for the best tree under the maximum likelihood (ML) [15] and maximum parsimony (MP) criteria [18] as well as searches constrained for the monophyly of all families (for details of the data matrix see the figure caption). Our own re-implementation of CopyCat [25] in conjunction with AxPcoords and AxParafit [26] was used to determine those leaves (species) whose placement significantly deviated between the constrained and the unconstrained tree. The best-known ML tree had a log likelihood of -8,012.83, whereas the best trees found under the constraint had a log likelihood of -8,014.70. The significantly (α = 0.05) distinctly placed species were Hydrogenimonas thermophila ('Hydrogenimonaceae'), Nitratifractor salsuginis and Thioreductor micantisoli (Nautiliaceae). However, the constrained tree was not significantly worse than the globally best one in the Shimodaira-Hasegawa test as implemented in RAxML [15] (α = 0.05). The best-known MP trees had a score of 1,290, whereas the best constrained trees found had a score of 1,295 and were not significantly worse in the Kishino-Hasegawa test as implemented in PAUP* [16] (α = 0.05). (See, e.g. chapter 21 in [27] for an in-depth description of such paired-site tests.) Accordingly, the current classification of Campylobacterales (Campylobacteraceae, Helicobacteraceae, 'Hydrogenimonaceae') and Nautiliales (Nautiliaceae) is not in significant disagreement with the 16S rRNA data.
The cells of strain YK-1T are curved rods of 0.4 × 1-2 µm length (Figure 2) [1]. Spiral cells are also observed in the exponential growth phase [1]. S. kujiense cells stain Gram-negative and non spore-forming (Table 1). The organism is described as motile with one polar flagellum (not visible in Figure 2). Motility-related genes account for 5.3% of total genes in the genome (COG category N). The organism is a facultatively anaerobic chemolithoautotroph [1,3]. S. kujiense can grow only under NaCl concentrations below 1% [1,3]. A low-ion-strength medium (MBM) has been developed for growing S. kujiense [1,3]. The organism also grows in solid medium containing 1.5% Bacto-agar [1,3]. The temperature range for growth is between 10°C and 35°C, with an optimum at 25°C [1,3]. The pH range for growth is 6.0-8.0, with an optimum at pH 7.0 [1,3]. S. kujiense grows autotrophically on carbon dioxide and bicarbonate [1,3]. The organism does not utilize organic acids such as acetate, lactate, pyruvate, malate, succinate, or formate nor does it utilize methanol, glucose or glutamate [1,3]. S. kujiense is not able to ferment phenol, octane, toluene, benzene, benzoate or ascorbate [1,3]. S. kujiense uses sulfide, elemental sulfur, thiosulfate and hydrogen as electron donors, and nitrate as well as small amounts of molecular oxygen (1% in gas phase) as electron acceptors [1,3]. It does not utilize nitrite [1,3]. S. kujiense shows oxidase activity, but is catalase-negative [1,3]. The organism is of ecological interest because of its ability to utilize different sulfur species and nitrate [1,3].
Figure 2.
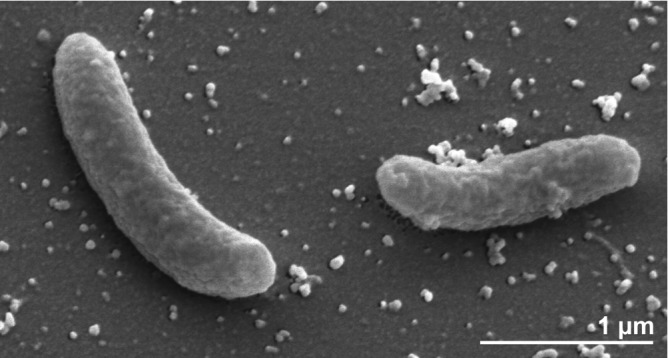
Scanning electron micrograph of S. kujiense YK-1T
Table 1. Classification and general features of S. kujiense YK-1T according to the MIGS recommendations [28] and the NamesforLife database [29].
| MIGS ID | Property | Term | Evidence code |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [30] | |
| Phylum Proteobacteria | TAS [31] | ||
| Class Epsilonproteobacteria | TAS [32,33] | ||
| Order Campylobacterales | TAS [32,34] | ||
| Family Helicobacteraceae | TAS [32,35] | ||
| Genus Sulfuricurvum | TAS [1] | ||
| Species Sulfuricurvum kujiense | TAS [1] | ||
| Type strain YK-1 | TAS [1] | ||
| Gram stain | negative | TAS [1] | |
| Cell shape | curved rods | TAS [1] | |
| Motility | motile | TAS [1] | |
| Sporulation | none | TAS [1] | |
| Temperature range | 10°C–35°C | TAS [1] | |
| Optimum temperature | 25°C | TAS [1] | |
| Salinity | below 1% NaCl; best without NaCl | TAS [1] | |
| MIGS-22 | Oxygen requirement | anaerobic, microaerobic | TAS [1] |
| Carbon source | carbon dioxide, bicarbonate | TAS [1] | |
| Energy metabolism | chemolithoautotroph | TAS [1] | |
| MIGS-6 | Habitat | groundwater | TAS [3,11] |
| MIGS-15 | Biotic relationship | free-living | NAS |
| MIGS-14 | Pathogenicity | none | NAS |
| Biosafety level | 1 | TAS [36] | |
| Isolation | drain water from an underground crude-oil storage cavity | TAS [3,11] | |
| MIGS-4 | Geographic location | Kuji in Iwate, Japan | TAS [1] |
| MIGS-5 | Sample collection time | March 1999 | TAS [3,11] |
| MIGS-4.1 | Latitude | 40.19 | NAS |
| MIGS-4.2 | Longitude | 141.78 | NAS |
| MIGS-4.3 | Depth | not reported | |
| MIGS-4.4 | Altitude | sea level | NAS |
Evidence codes - NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [37].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [38], and is part of the Genomic Encyclopedia of Bacteria and Archaea project [39]. The genome project is deposited in the Genomes On Line Database [19] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Table 2. Genome sequencing project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Three genomic libraries: one 454 pyrosequence standard library, one 454 PE library (8.7 kb insert size), one Illumina library |
| MIGS-29 | Sequencing platforms | Illumina GAii, 454 GS FLX Titanium |
| MIGS-31.2 | Sequencing coverage | 357.4 × Illumina; 51.1 × pyrosequence |
| MIGS-30 | Assemblers | Newbler version 2.3, Velvet, phrap version SPS - 4.24 |
| MIGS-32 | Gene calling method | Prodigal 1.4, GenePRIMP |
| INSDC ID | CP002355 (chromosome) CP002356-9 (plasmids SULKU01-04) |
|
| Genbank Date of Release | October 7, 2011 (chromosome and plasmids) | |
| GOLD ID | Gc01552 | |
| NCBI project ID | 43399 | |
| Database: IMG-GEBA | 649633097 | |
| MIGS-13 | Source material identifier | DSM 16994 |
| Project relevance | Tree of Life, GEBA |
Growth conditions and DNA isolation
S. kujiense strain YK-1T, DSM 16994, was grown anaerobically in DSMZ medium 1020 (MBM medium) [40] at 25°C. DNA was isolated from 0.5-1 g of cell paste using MasterPure Gram-positive DNA purification kit (Epicentre MGP04100) following the standard protocol as recommended by the manufacturer with modification st/DL for cell lysis as described in Wu et al. 2009 [39]. DNA is available through the DNA Bank Network [41].
Genome sequencing and assembly
The genome was sequenced using a combination of Illumina and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website [42]. Pyrosequencing reads were assembled using the Newbler assembler (Roche). The initial Newbler assembly consisting of 18 contigs in two scaffolds was converted into a phrap [43] assembly by making fake reads from the consensus, to collect the read pairs in the 454 paired end library. Illumina GAii sequencing data (788.0 Mb) was assembled with Velvet [44] and the consensus sequences were shredded into 1.5 kb overlapped fake reads and assembled together with the 454 data. The 454 draft assembly was based on 124.3 Mb 454 draft data and all of the 454 paired end data. Newbler parameters are -consed -a 50 -l 350 -g -m -ml 20. The Phred/Phrap/Consed software package [43] was used for sequence assembly and quality assessment in the subsequent finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution [43], Dupfinisher [45], or sequencing cloned bridging PCR fragments with subcloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J.-F. Chang, unpublished). A total of 85 additional reactions were necessary to close gaps and to raise the quality of the finished sequence. Illumina reads were also used to correct potential base errors and increase consensus quality using a software Polisher developed at JGI [46]. The error rate of the completed genome sequence is less than 1 in 100,000. Together, the combination of the Illumina and 454 sequencing platforms provided 408.5 × coverage of the genome. The final assembly contained 368,924 pyrosequence and 27,990,437 Illumina reads.
Genome annotation
Genes were identified using Prodigal [47] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [48]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [49].
Genome properties
The genome consists of a 2,574,824 bp long circular chromosome with a G+C content of 45% and four circular plasmids of 3,421 bp, 51,014 bp, 71,513 bp and 118,585 bp length, respectively (Table 3 and Figure 3). Of the 2,879 genes predicted, 2,818 were protein-coding genes, and 61 RNAs; 20 pseudogenes were also identified. The majority of the protein-coding genes (67.9%) were assigned with a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.
Table 3. Genome Statistics.
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 2,819,357 | 100.00 |
| DNA coding region (bp) | 2,623,121 | 93.04 |
| DNA G+C content (bp) | 1,256,420 | 44.56 |
| Number of replicons | 5 | |
| Extrachromosomal elements | 4 | |
| Total genes | 2,879 | 100.00 |
| RNA genes | 61 | 2.12 |
| rRNA operons | 3 | |
| Protein-coding genes | 2,818 | 97.88 |
| Pseudo genes | 20 | 0.69 |
| Genes with function prediction | 1,964 | 67.87 |
| Genes in paralog clusters | 1,264 | 43.90 |
| Genes assigned to COGs | 2,129 | 73.95 |
| Genes assigned Pfam domains | 2,100 | 72.94 |
| Genes with signal peptides | 926 | 32.16 |
| Genes with transmembrane helices | 633 | 21.99 |
| CRISPR repeats | 0 |
Figure 3.

Graphical map of the chromosome (plasmids not shown). From bottom to center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
Table 4. Number of genes associated with the general COG functional categories.
| Code | Value | %age | Description |
|---|---|---|---|
| J | 154 | 6.4 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0.0 | RNA processing and modification |
| K | 119 | 5.0 | Transcription |
| L | 126 | 5.3 | Replication, recombination and repair |
| B | 0 | 0.0 | Chromatin structure and dynamics |
| D | 33 | 1.4 | Cell cycle control, cell division, chromosome partitioning |
| Y | 0 | 0.0 | Nuclear structure |
| V | 46 | 1.9 | Defense mechanisms |
| T | 283 | 11.8 | Signal transduction mechanisms |
| M | 177 | 7.4 | Cell wall/membrane/envelope biogenesis |
| N | 127 | 5.3 | Cell motility |
| Z | 0 | 0.0 | Cytoskeleton |
| W | 0 | 0.0 | Extracellular structures |
| U | 96 | 4.0 | Intracellular trafficking, secretion, and vesicular transport |
| O | 102 | 4.3 | Posttranslational modification, protein turnover, chaperones |
| C | 168 | 7.0 | Energy production and conversion |
| G | 73 | 3.1 | Carbohydrate transport and metabolism |
| E | 129 | 5.4 | Amino acid transport and metabolism |
| F | 57 | 2.4 | Nucleotide transport and metabolism |
| H | 107 | 4.5 | Coenzyme transport and metabolism |
| I | 40 | 1.7 | Lipid transport and metabolism |
| P | 134 | 5.6 | Inorganic ion transport and metabolism |
| Q | 21 | 0.9 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 229 | 9.6 | General function prediction only |
| S | 175 | 7.3 | Function unknown |
| - | 750 | 26.1 | Not in COGs |
Acknowledgements
We would like to gratefully acknowledge the help of Maren Schröder (DSMZ) for growing S. kujiense cultures. This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, UT-Battelle and Oak Ridge National Laboratory under contract DE-AC05-00OR22725, as well as German Research Foundation (DFG) INST 599/1-2.
References
- 1.Kodama Y, Watanabe K. Sulfuricurvum kujiense gen.nov., sp. nov., a facultatively anaerobic, chemolithoautotrophic, sulfur-oxidizing bacterium isolated from an underground crude-oil storage cavity. Int J Syst Evol Microbiol 2004; 54:2297-2300 10.1099/ijs.0.63243-0 [DOI] [PubMed] [Google Scholar]
- 2.Euzeby J. List of bacterial names with standing in nomenclature: A folder available on the Internet. Int J Syst Bacteriol 1997; 47:590-592 10.1099/00207713-47-2-590 [DOI] [PubMed] [Google Scholar]
- 3.Kodama Y, Watanabe K. Isolation and characterization of a sulfur-oxidizing chemolithotroph growing on crude oil under anaerobic conditions. Appl Environ Microbiol 2003; 69:107-112 10.1128/AEM.69.1.107-112.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell BJ, Engel AS, Porter ML, Takai K. The versatile ε-proteobacteria: key players in sulphidic habitats. Nat Rev Microbiol 2006; 4:458-468 10.1038/nrmicro1414 [DOI] [PubMed] [Google Scholar]
- 5.Porter ML, Engel AS. Diversity of uncultured Epsilonproteobacteria from terrestrial sulfidic caves and springs. Appl Environ Microbiol 2008; 74:4973-4977 10.1128/AEM.02915-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hubert CR, Oldenburg TBP, Fustic M, Gray ND, Larter SR, Penn K, Rowan AK, Seshadri R, Sherry A, Swainsbury R, et al. Massive dominance of Epsilonproteobacteria in formation waters from a Canadian oil sands reservoir containing severely biodegraded oil. [PubMed]. Environ Microbiol 2011; (In press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altschul SF, Gish W, Miller W, Myers E, Lipman D. Basic local alignment search tool. [PubMed]. J Mol Biol 1990; 215:403-410 [DOI] [PubMed] [Google Scholar]
- 8.Korf I, Yandell M, Bedell J. BLAST, O'Reilly, Sebastopol, 2003. [Google Scholar]
- 9.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069-5072 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter MF. An algorithm for suffix stripping. Program: electronic library and information systems 1980; 14:130-137.
- 11.Watanabe K, Watanabe K, Kodama Y, Syutsubo K, Harayama S. Molecular characterization of bacterial populations in petroleum-contaminated groundwater discharged from underground crude oil storage cavities. Appl Environ Microbiol 2000; 66:4803-4809 10.1128/AEM.66.11.4803-4809.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haaijer SC, Harhangi HR, Meijerink BB, Strous M, Pol A, Smolders AJ, Verwegen K, Jetten MS, Op den Camp HJ. Bacteria associated with iron seeps in a sulfur-rich, neutral pH, freshwater ecosystem. ISME J 2008; 2:1231-1242 10.1038/ismej.2008.75 [DOI] [PubMed] [Google Scholar]
- 13.Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452-464 10.1093/bioinformatics/18.3.452 [DOI] [PubMed] [Google Scholar]
- 14.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. [PubMed]. Mol Biol Evol 2000; 17:540-552 [DOI] [PubMed] [Google Scholar]
- 15.Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web-servers. Syst Biol 2008; 57:758-771 10.1080/10635150802429642 [DOI] [PubMed] [Google Scholar]
- 16.Hess PN, De Moraes Russo CA. An empirical test of the midpoint rooting method. Biol J Linn Soc Lond 2007; 92:669-674 10.1111/j.1095-8312.2007.00864.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184-200 10.1007/978-3-642-02008-7_13 [DOI] [PubMed] [Google Scholar]
- 18.Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4.0 b10. Sinauer Associates, Sunderland, 2002. [Google Scholar]
- 19.Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markowitz VM, Kyrpides NC. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38:D346-D354 10.1093/nar/gkp848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sikorski J, Munk C, Lapidus A, Ngatchou Djao OD, Lucas S, Glavina Del Rio T, Nolan M, Tice H, Han C, Cheng JF, et al. Complete genome sequences of Sulfurimonas autotrophica type strain (OK 10T). Stand Genomic Sci 2010; 3:194-202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baar C, Eppinger M, Raddatz G, Simon J, Lanz C, Klimmek O, Nandakumar R, Gross R, Rosinus A, Keller H, et al. Complete genome sequence and analysis of Wolinella succinogenes. Proc Natl Acad Sci USA 2003; 100:11690-11695 10.1073/pnas.1932838100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sikorski J, Lapidus A, Copeland A, Glavina Del Rio T, Nolan M, Lucas S, Chen F, Tice H, Cheng JF, Saunders E, et al. Complete genome sequence of Sulfurospirillum deleyianum type strain (5175T). Stand Genomic Sci 2010; 2:149-157 10.4056/sigs.671209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pati A, Gronow S, Lapidus A, Copeland A, Glavina Del Rio T, Nolan M, Lucas S, Tice H, Cheng JF, Han C, et al. Complete genome sequence of Arcobacter nitrofigilis type strain (CIT). Stand Genomic Sci 2010; 2:300-308 10.4056/sigs.912121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson I, Sikorski J, Zeytun A, Nolan M, Lapidus A, Lucas S, Hammon N, Deshpande S, Cheng JF, Tapia R, et al. Complete genome sequence of Nitratifractor salsuginis type strain (E9I37-1T). Stand Genomic Sci 2011; 4:322-330 10.4056/sigs.1844518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meier-Kolthoff JP, Auch AF, Huson DH, Göker M. COPYCAT: Co-phylogenetic Analysis tool. Bioinformatics 2007; 23:898-900 10.1093/bioinformatics/btm027 [DOI] [PubMed] [Google Scholar]
- 26.Stamatakis A, Auch A, Meier-Kolthoff JP, Göker M. AxPcoords & Parallel AxParafit: Statistical co-phylogenetic analyses on thousands of taxa. BMC Bioinformatics 2007; 8:405 10.1186/1471-2105-8-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Felsenstein J. Inferring phylogenies. Sinauer Associates Inc., Sunderland, Massachusetts, 2004. [Google Scholar]
- 28.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garrity G. NamesforLife. BrowserTool takes expertise out of the database and puts it right in the browser. Microbiol Today 2010; 37:9 [Google Scholar]
- 30.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576-4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B, Springer, New York, 2005, p. 1. [Google Scholar]
- 32.Validation List No 107. List of new names and new combinations previously effectively, but not validly, published. [PubMed]. Int J Syst Evol Microbiol 2006; 56:1-6 10.1099/ijs.0.64188-0 [DOI] [PubMed] [Google Scholar]
- 33.Garrity GM, Bell JA, Lilburn T. Class V. Epsilonproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 1145. [Google Scholar]
- 34.Garrity GM, Bell JA, Lilburn T. Order I. Campylobacterales ord. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 1145. [Google Scholar]
- 35.Garrity GM, Bell JA, Lilburn T. Family II. Helicobacteraceae fam. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 1168. [Google Scholar]
- 36.BAuA. Classification of bacteria and archaea in risk groups. TRBA 466. p. 227. Bundesanstalt für Arbeitsschutz und Arbeitsmedizin, Germany. 2010. [Google Scholar]
- 37.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175-182 10.1016/j.syapm.2010.03.003 [DOI] [PubMed] [Google Scholar]
- 39.Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056-1060 10.1038/nature08656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.List of growth media used at DSMZ: http//www.dsmz.de/catalogues/catalogue-microorganisms/culture-technology/list-of-media-for-microorganisms.html
- 41.Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, Berendsohn WG, Wägele JW. The DNA Bank Network: the start from a German initiative. Biopreserv Biobank 2011; 9:51-55 10.1089/bio.2010.0029 [DOI] [PubMed] [Google Scholar]
- 42.JGI website. http://www.jgi.doe.gov/
- 43.The Phred/Phrap/Consed software package. http://www.phrap.com
- 44.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821-829 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han C, Chain P. Finishing repeat regions automatically with Dupfinisher. In: Proceeding of the 2006 international conference on bioinformatics & computational biology. Arabnia HR, Valafar H (eds), CSREA Press. June 26-29, 2006: 141-146. [Google Scholar]
- 46.Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008. [Google Scholar]
- 47.Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455-457 10.1038/nmeth.1457 [DOI] [PubMed] [Google Scholar]
- 49.Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271-2278 10.1093/bioinformatics/btp393 [DOI] [PubMed] [Google Scholar]