

Abstract
Herpetosiphon aurantiacus Holt and Lewin 1968 is the type species of the genus Herpetosiphon, which in turn is the type genus of the family Herpetosiphonaceae, type family of the order Herpetosiphonales in the phylum Chloroflexi. H. aurantiacus cells are organized in filaments which can rapidly glide. The species is of interest not only because of its rather isolated position in the tree of life, but also because Herpetosiphon ssp. were identified as predators capable of facultative predation by a wolf pack strategy and of degrading the prey organisms by excreted hydrolytic enzymes. The genome of H. aurantiacus strain 114-95T is the first completely sequenced genome of a member of the family Herpetosiphonaceae. The 6,346,587 bp long chromosome and the two 339,639 bp and 99,204 bp long plasmids with a total of 5,577 protein-coding and 77 RNA genes was sequenced as part of the DOE Joint Genome Institute Program DOEM 2005.
Keywords: Chemoorganoheterotrophic, Gram-negative, gliding, ensheathed filaments, free-living, predator, Herpetosiphonaceae, Chloroflexi, DOEM2005
Introduction
Strain 114-95T (= ATCC 23779 = DSM 785 = CCUG 48726) is the type strain of Herpetosiphon aurantiacus, which in turn is the type species of the genus Herpetosiphon [1,2]. Because most of the species were reclassified as members of other genera in 1998 [3], only one other species currently remains in this genus: H. geysericola (Copeland 1936) Lewin 1970. The genus name, meaning gliding tube, was derived from the Greek words herpeton, gliding animal or reptile, and siphon, tube or pipe [4]. The species epithet is derived from the Neo-Latin adjective aurantiacus, meaning orange-colored [4]. Strain 114-95T was originally isolated from the slimy coating of a freshwater alga (Chara sp.) in Birch Lake, Minnesota (USA), but strains belonging to the species were also isolated from well water, cow dung, hot springs and marine shores [1]. H. aurantiacus 114-95T is capable of predation of other bacteria and can thereby destroy whole colonies [5]. It has even been suggested that Herpetosiphon spp. are capable of facultative predation by a wolf pack strategy, in which a quorum of predatory cells is required to degrade the prey organism by excreted hydrolytic enzymes [6]. Here we present a summary classification and a set of features for H. aurantiacus 114-95T, together with the description of the complete genome and its annotation.
Classification and features
A representative genomic 16S rRNA sequence of H. aurantiacus strain 114-95T was compared using NCBI BLAST [7,8] under default settings (e.g., considering only the high-scoring segment pairs (HSPs) from the best 250 hits) with the most recent release of the Greengenes database [9] and the relative frequencies of taxa and keywords (reduced to their stem [10]) were determined, weighted by BLAST scores. The most frequently occurring genera were Herpetosiphon (82.9%), Chloroflexus (9.9%), 'Kouleothrix' (4.5%), Oscillochloris (2.2%) and 'Chlorothrix' (0.5%) (46 hits in total). Regarding the 13 hits to sequences from members of the species, the average identity within HSPs was 99.8%, whereas the average coverage by HSPs was 96.9%. Regarding the two hits to sequences from other members of the genus, the average identity within HSPs was 97.8%, whereas the average coverage by HSPs was 94.2%. Among all other species, the one yielding the highest score was Herpetosiphon geysericola (NR_028694), which corresponded to an identity of 97.8% and an HSP coverage of 94.2%. (Note that the Greengenes database uses the INSDC (= EMBL/NCBI/DDBJ) annotation, which is not an authoritative source for nomenclature or classification.) The highest-scoring environmental sequence was JF098937 ('skin popliteal fossa clone ncd1008d03c1'), which showed an identity of 98.2% and an HSP coverage of 88.3%. The most frequently occurring keywords within the labels of environmental samples that yielded hits were 'soil' (4.2%), 'microbi' (4.1%), 'geyser' (3.3%), 'geotherm' (2.9%) and 'mat' (2.8%) (204 hits in total). Environmental samples that yielded hits of a higher score than the highest scoring species were not found. These keywords fit well with the ecological properties reported for strain 114-95T in the original description [1].
Figure 1 shows the phylogenetic neighborhood of H. aurantiacus in a tree based upon 16S rRNA. The sequences of the five 16S rRNA gene copies in the genome differ from each other by up to two nucleotides, and differ by up to seven nucleotides from the previously published 16S rRNA sequence (M34117), which contains 64 ambiguous base calls.
Figure 1.

Phylogenetic tree highlighting the position of H. aurantiacus relative to the other type strains within the phylum Chloroflexi. The tree was inferred from 1,350 aligned characters [11,12] of the 16S rRNA gene sequence under the maximum likelihood (ML) criterion [13]. Rooting was done initially using the midpoint method [14] and then checked for its agreement with the current classification (Table 1). The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 100 ML bootstrap replicates [15] (left) and from 1,000 maximum parsimony bootstrap replicates [16] (right) if the value is larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [17] are labeled with one asterisk, and those also listed as 'Complete and Published' with two asterisks (see [18,19] and AP012029 for Anaerolinea thermophila, CP002084 for Dehalogenimonas lykanthroporepellens, CP001337 for Chloroflexus aggregans, CP000909 C. aurantiacus, and CP000804 for Roseiflexus castenholzii).
Table 1. Classification and general features of H. aurantiacus 114-95T according to the MIGS recommendations [20] and the NamesforLife database [21].
| MIGS ID | Property | Term | Evidence code |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [22] | |
| Phylum Chloroflexi | TAS [23,24] | ||
| Class Chloroflexi | TAS [23,24] | ||
| Order Herpetosiphonales | TAS [25] | ||
| Family Herpetosiphonaceae | TAS [26] | ||
| Genus Herpetosiphon | TAS [1,2,27] | ||
| Species Herpetosiphon aurantiacus | TAS [1,2] | ||
| Type strain 114-95 | TAS [1] | ||
| Gram stain | negative | TAS [1] | |
| Cell shape | cylindrical in unbranched sheathed filaments | TAS [1] | |
| Motility | gliding | TAS [1] | |
| Sporulation | not reported | ||
| Temperature range | not reported | ||
| Optimum temperature | about 30 °C | NAS | |
| Salinity | not reported | ||
| MIGS-22 | Oxygen requirement | oxic | NAS |
| Carbon source | probably carbohydrates | TAS [1] | |
| Energy metabolism | chemoorganoheterotroph | TAS [1] | |
| MIGS-6 | Habitat | diverse: coating of Chara sp., lake and well water, cow dung, hot springs, marine shores | TAS [1] |
| MIGS-15 | Biotic relationship | free living | NAS |
| MIGS-14 | Pathogenicity | none known | NAS |
| Biosafety level | 1 | TAS [28] | |
| Isolation | slimy coating of Chara sp. | TAS [1] | |
| MIGS-4 | Geographic location | Birch Lake, Minnesota, USA | TAS [1] |
| MIGS-5 | Sample collection time | 1961 | NAS |
| MIGS-4.1 | Latitude | 45.04 | |
| MIGS-4.2 | Longitude | 94.42 | |
| MIGS-4.3 | Depth | not reported | |
| MIGS-4.4 | Altitude | 390 m | NAS |
Evidence codes - TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [29].
Cells of H. aurantiacus strain 114-95T are cylindrical measuring 1-1.5 μm by 5-10 μm (Figure 2) [1]. Cells are organized in sheathed filaments of 500 μm length or more [1]. However, the existence of a sheath in the classical sense has been questioned in an analysis of the fine structure of the cells [30]. Cells of strain 114-95T stain Gram-negative, are not flagellated but are motile via gliding and divide by the formation of a transverse septum [1]. Colonies are flat, spreading, and rough, and produce an orange pigment [1]. Pigment analyses of a related strain, H. giganteus Hp a2, showed that this strain produces γ-carotene, as well as glycosylated and acyl-glycosylated derivatives of 1′-hydroxy-4-keto-gamma-carotene [31]. Strain 115-95T is catalase-positive and hydrolyzes starch, gelatine, casein and tributyrin but not cellulose [1].
Figure 2.

Scanning electron micrograph of a multicellular filament of H. aurantiacus 114-95T.
Chemotaxonomy
Data on the structure of the cell wall, quinones, cellular and polar lipids of strain 114-95T are not available, although H. giganteus Hp a2 was reported to produce menaquinones 6 and 7 [31]. Members of the Chloroflexi do not contain a lipopolysaccharide-containing outer membrane and the peptidoglycan is a variant that usually contains L-ornithine as the diamino acid [32].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing as part of the DOE Joint Genome Institute Program DOEM 2005. The genome project is deposited in the Genomes On Line Database [17] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Table 2. Genome sequencing project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Three genomic Sanger libraries: 3 kb pUC, 8 kb pMCL200 and fosmid pcc1Fos libraries. |
| MIGS-29 | Sequencing platforms | ABI3730 |
| MIGS-31.2 | Sequencing coverage | 11.0 × Sanger |
| MIGS-30 | Assemblers | phrap |
| MIGS-32 | Gene calling method | Prodigal, GenePRIMPCritica |
| INSDC ID | CP000875 (chromosome) CP000876 (plasmid HAU01) CP000877 (plasmid HAU02) |
|
| GenBank Date of Release | November 13, 2007 | |
| GOLD ID | Gc00677 | |
| NCBI project ID | 16523 | |
| Database: IMG | 2508501111 | |
| MIGS-13 | Source material identifier | DSM 785 |
| Project relevance | Biotechnology |
Strain history
The history of strain 114-95T originates with J. G. Holt, who deposited the strain in the ATCC collection in 1961, from which it was distributed to the DSMZ and the CCUG [33].
Growth conditions and DNA isolation
H. aurantiacus strain 114-95T (DSM 785) was obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ). Cells for DNA isolation were grown at 28 °C in the recommended CY liquid medium under oxic conditions with gentle shaking. DNA was isolated by the cetyl trimethylammonium bromide protocol recommended and described by the Joint Genome Institute [34]. The purity, quality and size of the bulk gDNA preparation were assessed according to DOE-JGI guidelines [34] and were consistent with JGI quality-control standards.
Genome sequencing and assembly
The genome was sequenced using a combination of 3 kb, 8 kb and fosmid DNA libraries. All general aspects of library construction and sequencing can be found at the JGI website [34]. The draft assembly contained 160 contigs in 51 scaffolds. The Phred/Phrap-/Consed software package was used for sequence assembly and quality assessment [35]. Possible mis-assemblies were corrected with Dupfinisher [36]. Gaps between contigs were closed by editing in Consed, custom priming, or PCR amplification. A total of 3,856 additional reactions and two shatter libraries were needed to close gaps and to raise the quality of the finished sequence. The error rate of the completed genome sequence is less than 1 in 100,000. Together, all libraries provided 11.0 × coverage of the genome. There are 85,815 total reactions in the final assembly.
Genome annotation
Genes were identified using Prodigal [37] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [38]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes Expert Review (IMG-ER) platform [39]
Genome properties
The genome consists of a circular chromosome (6,346,587 bp) and two circular plasmids, pHAU01 (339,639 bp) and pHAU02 (88,204 bp), respectively with an overall G+C content of 50.9% (Table 3, Figure 3, Figure 4, Figure 5 and Figure 6). These are the only plasmids that have been identified to date among the seventeen Chloroflexi strains whose genomes have been sequenced. Interestingly, the GC content of the chromosome (50.7%) is notably lower than that of pHAU01 (53.7%) or pHAU02 (53.1%). Plasmid pHAU02 is predicted to encode 71 proteins, and pHAU01 is predicted to encode 231 potential proteins, which included a variety of transposases, phage recombinases and integrases, a CRISPR and CRISPR-associated gene cluster, and a variety of predicted transcription regulators. Neither plasmid encodes a product known to be essential for cell viability. Of the 5,654 genes predicted, 5,577 were protein-coding genes, and 77 encoded RNAs; 82 pseudogenes were identified. The majority of the protein-coding genes (67.4%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.
Table 3. Genome Statistics.
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 6,785,430 | 100.00% |
| DNA coding region (bp) | 5,654,495 | 88.21% |
| DNA G+C content (bp) | 3,453,669 | 50.90% |
| Number of replicons | 3 | |
| Extrachromosomal elements | 2 | |
| Total genes | 5,654 | 100.00% |
| RNA genes | 77 | 1.36% |
| rRNA operons | 5 | |
| Protein-coding genes | 5,577 | 98.64% |
| Pseudogenes | 82 | |
| Genes with function prediction | 3,812 | 67.42% |
| Genes in paralog clusters | 505 | 8.93% |
| Genes assigned to COGs | 3,831 | 67.76% |
| Genes assigned Pfam domains | 3,766 | 66.61% |
| Genes with signal peptides | 1,071 | 18.94% |
| Genes with transmembrane helices | 1,495 | 26.44% |
| CRISPR repeats | 11 |
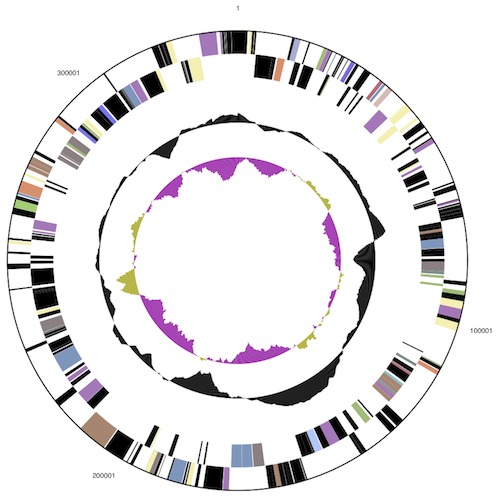
Figure 3.

Graphical circular map of the chromosome (not drawn to scale with plasmids). From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
Figure 4.
Graphical circular map of the plasmid pHAU01. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
Figure 5.
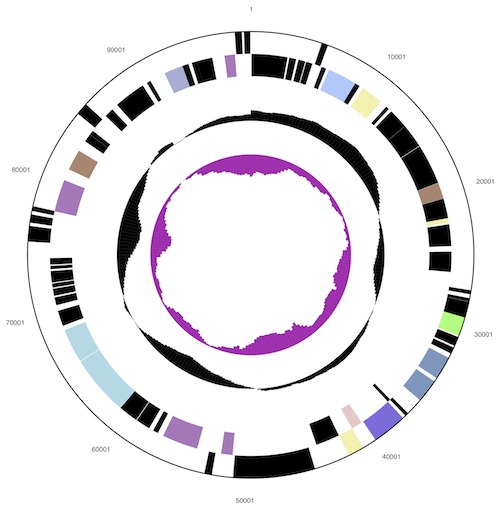
Graphical circular map of the plasmid pHAU02 (not drawn to scale with chromosome). From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
Figure 6.
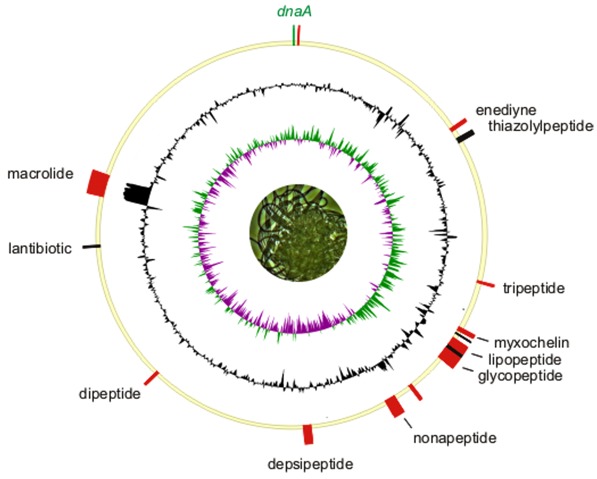
Chromosome of H. aurantiacus strain 114-95T oriented to the dnaA gene (top). The inner ring shows a normalized plot of GC skew, while the center ring shows a normalized plot of GC content. The outer circle shows the distribution of secondary metabolite gene clusters. Biosynthetic gene clusters associated with thiotemplate-based assembly (PKS, NRPS) are depicted in red and bacteriocin loci are marked in black.
Table 4. Number of genes associated with the general COG functional categories.
| Code | value | %age | Description |
|---|---|---|---|
| J | 176 | 4.1 | Translation, ribosomal structure and biogenesis |
| A | 3 | 0.1 | RNA processing and modification |
| K | 370 | 8.7 | Transcription |
| L | 213 | 5.4 | Replication, recombination and repair |
| B | 2 | 0.1 | Chromatin structure and dynamics |
| D | 35 | 0.8 | Cell cycle control, cell division, chromosome partitioning |
| Y | 0 | 0.0 | Nuclear structure |
| V | 93 | 2.2 | Defense mechanisms |
| T | 393 | 9.2 | Signal transduction mechanisms |
| M | 261 | 6.1 | Cell wall/membrane/envelope biogenesis |
| N | 14 | 0.3 | Cell motility |
| Z | 0 | 0.0 | Cytoskeleton |
| W | 0 | 0.0 | Extracellular structures |
| U | 56 | 1.3 | Intracellular trafficking and secretion, and vesicular transport |
| O | 142 | 3.3 | Posttranslational modification, protein turnover, chaperones |
| C | 205 | 4.8 | Energy production and conversion |
| G | 276 | 6.5 | Carbohydrate transport and metabolism |
| E | 307 | 7.2 | Amino acid transport and metabolism |
| F | 92 | 2.2 | Nucleotide transport and metabolism |
| H | 174 | 4.1 | Coenzyme transport and metabolism |
| I | 128 | 3.0 | Lipid transport and metabolism |
| P | 172 | 4.0 | Inorganic ion transport and metabolism |
| Q | 143 | 3.4 | Secondary metabolites biosynthesis, transport and catabolism* |
| R | 647 | 15.1 | General function prediction only |
| S | 349 | 8.2 | Function unknown |
| - | 1,823 | 32.2 | Not in COGs |
* manual annotation yielded 6.4% genes in the secondary metabolites biosynthesis, transport and catabolism category.
Insights into the genome
The phylum Chloroflexi is fascinatingly diverse and includes chlorophototrophs (Chloroflexales) as well as Gram-positive spore-forming organisms (Ktedonobacteria), reductive dehalogenating bacteria (Dehalococcoidetes), obligately aerobic heterotrophic thermophiles (Thermomicrobia), obligately anaerobic heterotrophs (Anaerolineae, Caldilineae), and marine organisms for which no example has yet been cultivated (e.g., SAR202 cluster). H. aurantiacus is described as an aerobic heterotroph from the order Herpetosiphonales, one of two orders within the class Chloroflexi. All characterized members of the other order, Chloroflexales, are chlorophototrophs. These include organisms from the genera Chloroflexus, Roseiflexus, Oscillochloris, Chloronema, Chlorothrix, and Heliothrix [40]. An organism that appears to be most similar although not very closely related to members of Anaerolinea has also recently been shown to be chlorophototrophic [41]. The genome sequence strongly supports the conclusion that H. aurantiacus is not a chlorophyll-based phototroph or an autotroph. Other than genes for carotenoid biosynthesis, a trait that is very widely distributed among members of all three kingdoms of life, no genes specifically used for photosynthetic light harvesting, electron transport, or chlorophyll biosynthesis were identified in the H. aurantiacus genome. Moreover, although Chloroflexus spp. and Roseiflexus spp. possess the genetic capacity to fix bicarbonate/CO2 by the 3-hydroxypropionate cycle, the corresponding genes are absent in H. aurantiacus [40,42].
Based upon the genes for carotenogenesis that are found in the H. aurantiacus genome, one can predict potential carotenoids that might be produced by this organism. The genome encodes homologs of crtB (phytoene synthase, crtI (phytoene desaturase), crtO (carotene 4-ketolase), crtD (hydroxyneurosporene dehydrogenase) and cruA (lycopene cyclase), and this combination of enzymes should allow this organism to produce γ-carotene derivatives with a keto-group at the 4 position. Additionally, the genome encodes cruF (γ-carotene 1′,2′-hydratase), cruC (1′-OH glycosyltransferase), and cruD, (acyl transferase) [43-45]. The presence of these genes would allow H. aurantiacus to synthesize monocyclic carotenoids carrying glycosyl, or glycosyl-fatty acyl ester moieties, attached to the 1′-OH group at the ψ–end of the molecule. This biosynthetic potential is in good agreement with the carotenoid contents of H. giganteus strain Hpa2 [31] and Roseiflexus spp. strains. The former produces γ-carotene and its glycosyl and fatty acyl-glycoside ester derivatives, while the latter produce γ-carotene, 4-keto-myxocoxanthin-glucoside, and 4-keto-myxocoxanthin-glucoside fatty acyl esters [44,46].
Scanning of the H. aurantiacus genome for siderophore biosynthetic genes revealed the presence of a cluster with remarkable similarity to that of the myxobacterial iron chelator myxochelin [47]. The annotated locus (Haur_01919 – Haur_01928) contains a complete set of genes for the production of 2,3-dihydroxybenzoic acid, including open reading frames encoding isochorismate synthase (Haur_01921), isochorismate hydrolase (Haur_01923) and 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase (Haur_01919). Additional genes in the operon are proposed to be involved in siderophore transport and iron utilization. Direct comparison with the myxochelin (mxc) cluster from Myxococcus xanthus DK1622 reveals the absence of an aminotransferase gene analogous to mxcL. Since mxcL is crucial for the formation of myxochelin B [47], H. aurantiacus probably only produces the biosynthetic predecessor mxyochelin A for iron sequestration. Homologs of genes involved in gliding motility in Myxococcus xanthus [48,49] and Flavobacterium johnsoniae [50-53] were not found in the H. aurantiacus genome. The genes required for gliding in H. aurantiacus and other members of the Chloroflexi have not yet been identified.
One of the most interesting and unique findings within the H. aurantiacus genome was the abundance of gene loci involved in secondary metabolism. Chloroflexi genomes were screened via BLASTP alignment against a representative library of conserved biosynthetic enzymes, a method proven useful in the analysis of the Salinispora tropica genome [54]. Apart from genes involved in fatty acid and carotenoid biosynthesis, no hits were obtained for the chlorophototrophic Chloroflexi (Table 5). On the other hand, families of genes encoding enzymes of secondary metabolism were found to be highly overrepresented in the H. aurantiacus genome. A total of fourteen biosynthetic gene clusters were identified and annotated, 11 of which are specific for this organism (Table 6). The combined length of these clusters is estimated to be ~448.6 kb, i.e. 6.6% of the H. aurantiacus genome is dedicated to natural product assembly. The biosynthetic capacity for secondary metabolite production of H. aurantiacus is thus comparable to that of other specialist producers of secondary metabolites, such as Actinobacteria and Myxobacteria [55,56]. The distribution of the biosynthetic loci on the H. aurantiacus chromosome seems non-random, with seven of them being located between 1.8 and 2.7 Mb clockwise from the replication origin, i.e. 50% of all clusters are concentrated in a region covering only 14% of the chromosome.
Table 5. Chloroflexi genome data and biosynthetic potential.
|
Organism |
Size, Mb |
Topology |
Biosynthetic Gene Cluster |
|||
|---|---|---|---|---|---|---|
| PKS | NRPS | PKS/NRPS | Bacteriocin | |||
| H. aurantiacus strain 114-95T | 6.351 | Circular | 2 | 4 | 5 | 3 |
|
Chloroflexus aurantiacus J-10-fl Chloroflexus aggregans DSM 9485 Roseiflexus castenholzii DSM 13941 Roseiflexus sp. RS-1 |
5.26 4.68 5.72 5.80 |
Circular Circular Circular Circular |
- - - - |
- - - - |
- - - - |
- - - - |
1chromosome only
Table 6. Herpetosiphon aurantiacus strain 114-95T biosynthetic loci.
| Type # | Location on the chromosome | Features | Product | Estimated Size [kb] |
|---|---|---|---|---|
| PKS/NRPS 1 | Haur_01976- Haur_01989 | 2 PKS modules, 6 NRPS modules | Lipopeptide | 47.7 |
| 2 | Haur_02003 Haur_02024 |
1 PKS module, 11 NRPS modules, genes for the production of hydroxyphenylglycine | Glycopeptide | 69.0 |
| 3 | Haur_02147 | 1 PKS module, 1 NRPS module | Unknown | 12.3 |
| 4 | Haur_02568-Haur_02581 | 2 PKS modules, 3 NRPS modules | Depsipeptide | 35.9 |
| 5 | Haur_04184-Haur_04206 | 16 PKS modules, 7 NRPS modules, genes for the production of hydroxymalonyl-acyl carrier protein | Macrolide | 112.6 |
| PKS 1 | Haur_00021 | naringenin-chalcone synthase (type-III PKS) | Aromatic polyketide | 1.1 |
| 2 | Haur_00919-Haur_00937 | iterative type-I PKS | Enediyne | 27.5 |
| NRPS 1 | Haur_01684-Haur_01687 | 3 NRPS modules | Tripeptide | 10.0 |
| 2 | Haur_01919-Haur_01928 | 1 NRPS module | Myxochelin | 14.0 |
| 3 | Haur_02229-Haur_02262 | 9 NRPS modules | Nonapeptide | 62.6 |
| 4 | Haur_03313-Haur_3318 | 2 NRPS modules | Dipeptide | 13.3 |
|
Bacteriocin 1 2 3 |
Haur_00966-Haur_00988 Haur_01990-Haur_02002 Haur_03953-Haur_03957 |
genes for Ser/Thr dehydration and heterocycle formation LanM-like lanthionine synthetase LanM-like lanthionine synthetase |
Thiazolylpeptide Lantibiotic Lantibiotic |
24.2 11.0 7.4 |
A striking feature of secondary metabolism in H. aurantiacus, which is also observed in myxobacterial and cyanobacterial genomes, is the preponderance of mixed polyketide synthase (PKS)-non-ribosomal peptide synthetase (NRPS) and NRPS gene clusters [56,57]. On the other hand, only two solely PKS systems were identified, including a type-III PKS of unknown function, as well as an iterative type-I PKS associated with enediyne biosynthesis (Table 6 and Figure 6). Enediyne clusters have as yet only been reported from Actinobacteria [58], and the presence of such a locus on the chromosome of H. aurantiacus is suggestive of horizontal gene transfer. Evidence for the recent acquisition of biosynthetic genes was obtained in case of the cluster designated pks-nrps5. This megacluster, which spans 112.6 kb of contiguous DNA on the chromosome (Table 6), is flanked both upstream and downstream by a number of transposon fragments. An above-average GC content of ~66% as well as significant shifts of the latter in both border regions of the cluster suggest a foreign origin. The observation that horizontal gene transfer may account for the accumulation of biosynthetic genes is, to a degree, reminiscent of the evolution of the M. xanthus DK1622 genome [59].
Even though thiotemplate-based chemistry involving PKSs and NRPSs appears to be the predominant theme in natural product biosynthesis by H. aurantiacus, genomic analyses also revealed the molecular basis for the assembly and posttranslational modification of ribosomally encoded peptides [60,61]. Furthermore, various pathways to specific biosynthetic building blocks, such as the rare amino acid L-p-hydroxyphenylglycine [62] and the polyketide extender unit hydroxymalonyl-acyl carrier protein, were identified. The pathways are located adjacent to PKS and NRPS genes, which suggest that functional crosstalk is very likely to occur.
To date, the secondary metabolome of Herpetosiphon spp. has not been explored beyond a single report on a structurally complex, natural product, siphonazole, which likely derives from a mixed PKS-NRPS assembly line [63]. The genome sequence of H. aurantiacus strain 114-95T now certainly provides a rationale for further studies of the chemistry and biology of this versatile microorganism.
Acknowledgements
The work conducted by the U.S. Department of Energy Joint Genome Institute was supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 and was also supported by NSF grant MCB-0523100 to D.A.B. M.N. gratefully acknowledges the German Federal Ministry of Education and Research (BMBF 0315591A) for supporting research involving genomics within the GenoMik-Transfer framework.
References
- 1.Holt JG, Lewin RA. Herpetosiphon aurantiacus gen. et sp. n., a new filamentous gliding organism. J Bacteriol 1968; 95:2407-2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225-420 10.1099/00207713-30-1-225 [DOI] [PubMed] [Google Scholar]
- 3.Sly LI, Taghavi M, Fegan M. Phylogenetic heterogeneity within the genus Herpetosiphon: transfer of the marine species Herpetosiphon cohaerens, Herpetosiphon nigricans and Herpetosiphon persicus to the genus Lewinella gen. nov. in the Flexibacter-Bacteroides-Cytophaga phylum. Int J Syst Bacteriol 1998; 48:731-737 10.1099/00207713-48-3-731 [DOI] [PubMed] [Google Scholar]
- 4.Euzéby JP. List of bacterial names with standing in nomenclature: A folder available on the Internet. Int J Syst Bacteriol 1997; 47:590-592 10.1099/00207713-47-2-590 [DOI] [PubMed] [Google Scholar]
- 5.Quinn GR. Skerman VBD. Herpetosiphon—nature’s scavenger? Curr Microbiol 1980; 4:57-62 10.1007/BF02602893 [DOI] [Google Scholar]
- 6.Jurkevitch E. Predatory behaviors in bacteria—diversity and transitions. Microbe 2007; 2:67-73 [Google Scholar]
- 7.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Bascic local alignment search tool. J Mol Biol 1990; 215:403-410 [DOI] [PubMed] [Google Scholar]
- 8.Korf I, Yandell M, Bedell J. BLAST, O'Reilly, Sebastopol, 2003. [Google Scholar]
- 9.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069-5072 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter MF. An algorithm for suffix stripping. Program: electronic library and information systems 1980; 14:130-137.
- 11.Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452-464 10.1093/bioinformatics/18.3.452 [DOI] [PubMed] [Google Scholar]
- 12.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540-552 [DOI] [PubMed] [Google Scholar]
- 13.Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 2008; 57:758-771 10.1080/10635150802429642 [DOI] [PubMed] [Google Scholar]
- 14.Hess PN, De Moraes Russo CA. An empirical test of the midpoint rooting method. Biol J Linn Soc Lond 2007; 92:669-674 10.1111/j.1095-8312.2007.00864.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184-200 10.1007/978-3-642-02008-7_13 [DOI] [PubMed] [Google Scholar]
- 16.Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4.0 b10. Sinauer Associates, Sunderland, 2002. [Google Scholar]
- 17.Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Kyrpides NC. The genomes on line database (GOLD) in 2009: Status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38:D346-D354 10.1093/nar/gkp848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu D, Raymond J, Wu M, Chatterji S, Ren Q, Graham JE, Bryant DA, Robb F, Colman A, Tallon LJ, et al. Complete genome sequence of the aerobic CO-oxidizing thermophile Thermomicrobium roseum. PLoS ONE 2009; 4:e4207 10.1371/journal.pone.0004207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pati A, LaButti K, Pukall R, Nolan M, Glavina Del Rio T, Tice H, Cheng JF, Lucas S, Chen F, Copeland A, et al. Complete genome sequence of Sphaerobacter thermophilus type strain (S 6022T). Stand Genomic Sci 2010; 2:49-56 10.4056/sigs.601105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garrity G. NamesforLife. BrowserTool takes expertise out of the database and puts it right in the browser. Microbiol Today 2010; 37:9 [Google Scholar]
- 22.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms. Proposal for the domains Archaea and Bacteria. Proc Natl Acad Sci USA 1990; 87:4576-4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hugenholtz P, Stackebrandt E. Reclassification of Sphaerobacter thermophilus from the subclass Sphaerobacteridae in the phylum Actinobacteria to the class Thermomicrobia (emended description) in the phylum Chloroflexi (emended description). Int J Syst Evol Microbiol 2004; 54:2049-2051 10.1099/ijs.0.03028-0 [DOI] [PubMed] [Google Scholar]
- 24.Garrity GM, Holt JG. Phylum BVI. Chloroflexi phy. nov. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 427-446. [Google Scholar]
- 25.Castenholz RW. Order II. 'Herpetosiphonales'. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 444. [Google Scholar]
- 26.Boone DR, Castenholz RW. Family I. 'Herpetosiphonaceae'. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 445. [Google Scholar]
- 27.Lewin RA, Leadbetter ER. Genus Herpetosiphon Holt and Lewin 1968, 2408; emend. mut. char. In: Buchanan RE, Gibbons NE (eds), Bergey's Manual of Determinative Bacteriology, Eighth Edition, The Williams and Wilkins Co., Baltimore, 1974, p. 107-109. [Google Scholar]
- 28.BAuA Classification of bacteria and archaea in risk groups. TRBA 2005; 466:205 [Google Scholar]
- 29.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reichenbach H, Golecki JR. The fine structure of Herpetosiphon, and a note on the taxonomy of the genus. Arch Microbiol 1975; 102:281-291 10.1007/BF00428379 [DOI] [PubMed] [Google Scholar]
- 31.Kleinig H, Reichenbach H. Carotenoid glucosides and menaquinones from the gliding bacterium Herpetosiphon giganteus Hp a2. Arch Microbiol 1977; 112:307-310 10.1007/BF00413098 [DOI] [PubMed] [Google Scholar]
- 32.Jürgen UJ, Meißner JD, Reichenbach H, Weckesser J. L-ornithine containing peptidoglycan-polysaccharide complex from the cell wall of the gliding bacterium Herpetosiphon aurantiacus. FEMS Microbiol Lett 1989; 60:247-250 10.1016/0378-1097(89)90404-7 [DOI] [Google Scholar]
- 33.Verslyppe B, De Smet W, De Baets B, De Vos P, Dawyndt P. Make Histri: reconstructing the exchange history of bacterial and archaeal type strains. Syst Appl Microbiol 2011; 34:328-336 10.1016/j.syapm.2011.01.004 [DOI] [PubMed] [Google Scholar]
- 34.The Joint Genome Institute http://www.jgi.doe.gov/www.jgi.doe.gov
- 35.Phrap and Phred for Windows. MacOS, Linux, and Unix. www.phrap.com
- 36.Cliff S. Han, Patrick Chain. 2006. Finishing repeat regions automatically with Dupfinisher. Proceeding of the 2006 international conference on bioinformatics & computational biology. Edited by Hamid R. Arabnia & Homayoun Valafar, CSREA Press. June 26-29, 2006: 141-146. [Google Scholar]
- 37.Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455-457 10.1038/nmeth.1457 [DOI] [PubMed] [Google Scholar]
- 39.Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271-2278 10.1093/bioinformatics/btp393 [DOI] [PubMed] [Google Scholar]
- 40.Bryant DA, Liu Z, Li T, Zhao F, Garcia Costas AM, Klatt CG, Ward DM, Frigaard NU, Overmann J. Comparative and functional genomics of anoxygenic green bacteria from the taxa Chlorobi, Chloroflexi, and Acidobacteria In: Advances in Photosynthesis and Respiration, Vol. 33, Functional Genomics and Evolution of Photosynthetic Systems, (Burnap, R. L. and Vermaas, W., eds.), pp. 47-102, Springer, Dordrecht, The Netherlands. [Google Scholar]
- 41.Klatt CG, Wood JM, Rusch DB, Bateson MM, Hamamura N, Heidelberg JF, Grossman AR, Bhaya D, Cohan FM, Kühl M, et al. Community ecology of hot spring cyanobacterial mats: predominant populations and their functional potential. ISME J 2011; 5:1262-1278 10.1038/ismej.2011.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klatt CG, Bryant DA, Ward DM. Comparative genomics provides evidence for the 3-hydroxypropionate autotrophic pathway in filamentous anoxygenic phototrophic bacteria and in hot spring microbial mats. Environ Microbiol 2007; 9:2067-2078 10.1111/j.1462-2920.2007.01323.x [DOI] [PubMed] [Google Scholar]
- 43.Maresca JA, Bryant DA. Identification of two genes encoding new carotenoid-modifying enzymes in the green sulfur bacterium Chlorobium tepidum. J Bacteriol 2006; 188:6217-6223 10.1128/JB.00766-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maresca JA, Graham JE, Bryant DA. Carotenoid biosynthesis in chlorophototrophs: the biochemical and genetic basis for structural diversity. Photosynth Res 2008; 97:121-140 10.1007/s11120-008-9312-3 [DOI] [PubMed] [Google Scholar]
- 45.Graham JE, Bryant DA. The biosynthetic pathway for the synthesis of the myxol-2′-fucoside in the cyanobacterium Synechococcus sp. strain PCC 7002. J Bacteriol 2009; 191:3292-3300 10.1128/JB.00050-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takaichi S, Maoka T, Yamada M, Matsuura K, Hikawa Y, Hanada S. Absence of carotenes and presence of a tertiary methoxy group in a carotenoid from a thermophilic filamentous photosynthetic bacterium Roseiflexus castenholzii. Plant Cell Physiol 2001; 42:1355-1362 10.1093/pcp/pce172 [DOI] [PubMed] [Google Scholar]
- 47.Gaitatzis N, Kunze B, Müller R. Novel insights into siderophore formation in myxobacteria. ChemBioChem 2005; 6:365-374 10.1002/cbic.200400206 [DOI] [PubMed] [Google Scholar]
- 48.Nan B, Zusman DR. Uncovering the mystery of gliding motility in Myxobacteria. Annu Rev Genet 2011; 45:21-39 10.1146/annurev-genet-110410-132547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luciano J, Agrebi R, Le Gall AV, Wartel M, Fiegna F, Ducret A, Brochier-Armanet C, Mignot T. Emergence and modular evolution of a novel motility machinery in bacteria. PLoS Genet 2011; 7:e1002268 10.1371/journal.pgen.1002268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rhodes RG, Samarasam MN, Shrivastava A, van Baaren JM, Pochiraju S, Bollampalli S, McBride MJ. Flavobacterium johnsoniae gldN and gldO are partially redundant genes required for gliding motility and surface localization of SprB. [P] J Bacteriol 2010; 192:1201-1211 10.1128/JB.01495-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rhodes RG, Nelson SS, Pochiraju S, McBride MJ. Flavobacterium johnsoniae sprB is part of an operon spanning the additional gliding motility genes sprC, sprD and sprF. J Bacteriol 2011; 193:599-610 10.1128/JB.01203-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rhodes RG, Pucker HG, McBride MJ. Development and use of a gene deletion strategy for Flavobacterium johnsoniae to identify the redundant gliding motility genes remF, remG, remH, and remI. J Bacteriol 2011; 193:2418-2428 10.1128/JB.00117-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rhodes RG, Samarasam MN, Van Groll EJ, McBride MJ. Mutations in Flavobacterium johnsoniae sprE result in defects in gliding motility and protein secretion. J Bacteriol 2011; 193:5322-5327 10.1128/JB.05480-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc Natl Acad Sci USA 2007; 104:10376-10381 10.1073/pnas.0700962104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nett M, Ikeda H, Moore BS. Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat Prod Rep 2009; 26:1362-1384 10.1039/b817069j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wenzel SC, Müller R. The impact of genomics on the exploitation of the myxobacterial secondary metabolome. Nat Prod Rep 2009; 26:1385-1407 10.1039/b817073h [DOI] [PubMed] [Google Scholar]
- 57.Nett M, König GM. The chemistry of gliding bacteria. Nat Prod Rep 2007; 24:1245-1261 10.1039/b612668p [DOI] [PubMed] [Google Scholar]
- 58.Van Lanen SG, Shen B. Biosynthesis of enediyne antitumor antibiotics. Curr Top Med Chem 2008; 8:448-459 10.2174/156802608783955656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goldman BS, Nierman WC, Kaiser D, Slater SC, Durkin AS, Eisen JA, Ronning CM, Barbazuk WB, Blanchard M, Field C, et al. Evolution of sensory complexity recorded in a myxobacterial genome (2006). Proc Natl Acad Sci USA 2006; 103:15200-15205 10.1073/pnas.0607335103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Begley M, Cotter PD, Hill C, Ross RP. Identification of a novel two-peptide lantibiotic, lichenicidin, following rational genome mining for LanM proteins. Appl Environ Microbiol 2009; 75:5451-5460 10.1128/AEM.00730-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brown LCW, Acker MG, Clardy J, Walsh CT, Fischbach MA. Thirteen posttranslational modifications convert a 14-residue peptide into the antibiotic thiocillin. Proc Natl Acad Sci USA 2009; 106:2549-2553 10.1073/pnas.0900008106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kastner S, Müller S, Natesan L, König GM, Guthke R, Nett M. 4-Hydroxyphenylglycine biosynthesis in Herpetosiphon aurantiacus – a case of gene duplication and catalytic divergence. submitted [DOI] [PubMed]
- 63.Nett M, Erol Ö, Kehraus S, Köck M, Krick A, Eguereva E, Neu E, König GM. Siphonazole, an unusual metabolite from Herpetosiphon sp. Angew Chem Int Ed Engl 2006; 45:3863-3867 10.1002/anie.200504525 [DOI] [PubMed] [Google Scholar]