

Abstract

As part of a comprehensive strategy to the welwitindolinone alkaloids possessing a bicyclo[4.3.1]decane core, we report herein concise asymmetric total syntheses of (−)-N-methylwelwitindolinone C isothiocyanate (2a), (−)-N-methylwelwitindolinone C isonitrile (2b), and (−)-3-hydroxy-N-methylwelwitindolinone C isothiocyanate (3a) from a common tetracyclic intermediate. The crucial vinyl chloride moiety was installed through electrophilic chlorination of a hydrazone, but only after adjustment of reactivity to circumvent a facile skeletal rearrangement. Selective desulfurization and oxidation of 2a provided access to 2b and 3a, respectively. Notably, this work provides corrected 1H and 13C NMR spectral data for 3a.
In the continuing search for effective new therapies to combat human disease, the field of medicine has often benefited from the discovery of novel compounds isolated from natural sources. As part of this pursuit, Moore and co-workers investigated a series of marine and terrestrial cyanobacteria and found their lipophilic extracts to display an array of cytotoxic properties.1 This biological activity was traced to a new family of polycyclic oxindole-containing alkaloids—the welwitindolinones (e.g., 1–4, Figure 1). Nearly all members of this family possess a common bicyclo[4.3.1]decane ring system set within a densely functionalized tetracyclic scaffold. Given their interesting biological activities and unique molecular architectures, the welwitindolinones have been the focus of numerous synthesis groups for nearly 15 years.2,3 Early this year, we reported the first synthesis of a bridged bicyclic member of the family, N-methylwelwitindolinone D isonitrile (4),4 and this was followed by the synthesis of dihydro-N-methylwelwitindolinone B isothiocyanate.5 Most recently, Garg and co-workers completed a remarkable synthesis of (−)-N-methylwelwitindolinone C isothiocyanate (2a).6 As part of an ongoing program aimed at the development of a comprehensive approach toward this intriguing family of alkaloids, we now report the total syntheses of (−)-N-methylwelwitindolinone C isothiocyanate (2a), (−)-N-methylwelwitindolinone C isonitrile (2b), and (−)-3-hydroxy-N-methylwelwitindolinone C isothiocyanate (3a).
Figure 1.

Welwitindolinone alkaloids.
From the outset, our objective was to design a unified strategy that would enable rapid assembly of a tetracyclic core unit, adorned in such a way as to provide avenues to all members of the bridged welwitindolinones. Toward this goal, we designed a concise sequence to forge the central bicyclo[4.3.1]decane ring system (6) through successive Lewis acid-mediated alkylative coupling and palladium-catalyzed enolate arylation transformations, starting from readily available cyclohexanone- (7) and indole-containing precursors (Scheme 1).2g,4 Having successfully employed tetracycle 6 in the synthesis of welwitindolinone D (4), we sought to use this intermediate as a key branching point from which to direct our efforts toward the subclass that possesses a vinylic chloride at C13, in particular N-methylwelwitindolinone C isothiocyanate (2a). Unique among this family of alkaloids, welwitindolinone 2a possesses the ability to antagonize the efflux activity of P-glycoprotein, the overexpression of which leads to the development of multiple-drug resistance in several types of human cancers.7 However, the studies that identified this activity were carried out prior to the isolation of welwitindolinones 3a, 3b, and 4, and as such these more highly oxidized congeners have yet to undergo biological assay. Total synthesis therefore holds the potential to provide new access to the oxidized members, and their analogues, for further screening.
Scheme 1.
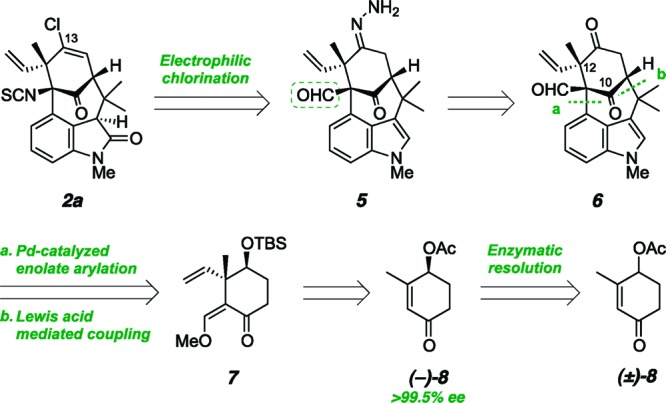
Our approach toward N-methylwelwitindolinone C isothiocyanate (2a) hinged on late-stage introduction of the vinylic chloride through electrophilic chlorination of hydrazone 5, which would be available from ketone 6. We envisioned the use of this compound, whose synthesis was established en route to N-methylwelwitindolinone D isonitrile (4), as a strategic branching point from which to target several members of the family. In order to realize an asymmetric synthesis of (−)-2a, this intermediate was ultimately prepared from enantioenriched acetate (−)-8 (>99.5% ee), which was obtained through pig liver esterase-catalyzed resolution of racemic 4-acetoxy-3-methylcyclohexenone.8
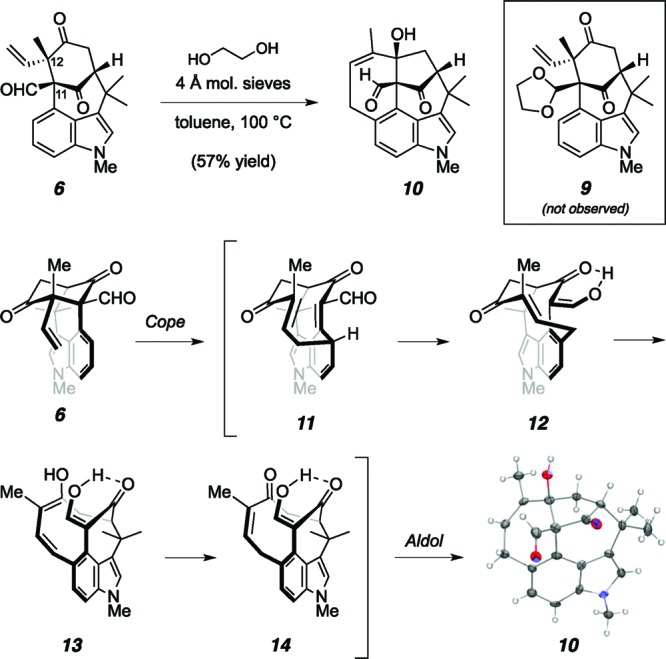
A key challenge for the synthesis of N-methylwelwitindolinone C isothiocyanate is installation of the vinyl chloride at C13.9 Our plan was to introduce the chloride through electrophilic chlorination of the C13 hydrazone.10,11 Of the three different carbonyl groups in the starting tetracycle (6), the least likely to condense with hydrazine was the bridging ketone at C10, due to its congested steric environment. So as to avoid formation of the hydrazone with the remaining carbonyl, the aldehyde, the plan was to protect this functionality as the corresponding dioxolane (9). Remarkably, when subjected to standard acetalization conditions, compound 6 gave none of the expected acetal and instead transformed cleanly into pentacycle 10. In fact, the same product was formed upon simply heating 6 in toluene at 85 °C, absent any additional reagents.12,13 A rationale for the structural reorganization is presented in Scheme 2 and involves an initial dearomatizing Cope rearrangement between the indole and the neighboring vinyl moiety to generate macrocycle 11, followed by rearomatization to provide vinylogous acid 12.14 Isomerization of the initially formed E-olefin to the required Z-olefin (14), possible through the intermediacy of dienol 13, enables an intramolecular aldol reaction leading to the product. The ease with which the Cope rearrangement takes place is ascribed to the rigidity of the bridged skeleton as well as the presence of the two electron-withdrawing groups on the benzylic carbon, which may serve to weaken the central σ bond (C11–C12).
Scheme 2.
The path toward the target was expected to be facilitated by reduction of the aldehyde, which would serve the dual purpose of obviating its protection and circumventing the undesired skeletal rearrangement. In the event, treatment of tricarbonyl 6 with NaBH(OMe)3 chemoselectively reduced the aldehyde, and, as expected, the resulting alcohol (15) was found to be thermally stable, even at 100 °C (Scheme 3). We then returned our attention to introduction of the vinyl chloride. Condensation of diketone 15 with hydrazine required elevated temperatures and the presence of acetic acid but gave the desired hydrazone (16) selectively.15 We were pleased to find that exposure of the crude hydrazone to N-chlorosuccinimide in pyridine afforded vinyl chloride 17 in 61% yield. The next task was the oxidation of the indole. While this oxidation was achieved using m-CPBA, the transformation proceeded more cleanly using the milder oxidant, magnesium monoperoxyphthalate (MMPP). Additionally, in order to obtain oxindole 18 in optimum yield, we found it necessary to perform the oxidation in a mixture of trifluoroacetic acid and acetic acid (2:3). In less acidic solvents (e.g., MeOH or AcOH), products of overoxidation, such as the corresponding 3-hydroxyoxindole, were observed. Having served its role, the primary alcohol was returned to the aldehyde state in nearly quantitative yield using the Dess–Martin periodinane. We were surprised to find that condensation of aldehyde 19 with hydroxylamine provided a variable mixture of separable oximes ((3S)-20 and (3R)-20, 1.6:1 to 4.4:1),16 epimeric at C3. The ready formation of the 3S diastereomer is noteworthy, as this stereochemistry is found in at least one member of the welwitindolinone family.17 Interestingly, the mixture of oxime diastereomers ultimately proved inconsequential, as both diastereomers when subjected to our modified protocol for Kim’s oxime rearrangement4,18 converted smoothly to (−)-N-methylwelwitindolinone C isothiocyanate (2a), spectroscopically identical to the natural product.
Scheme 3.

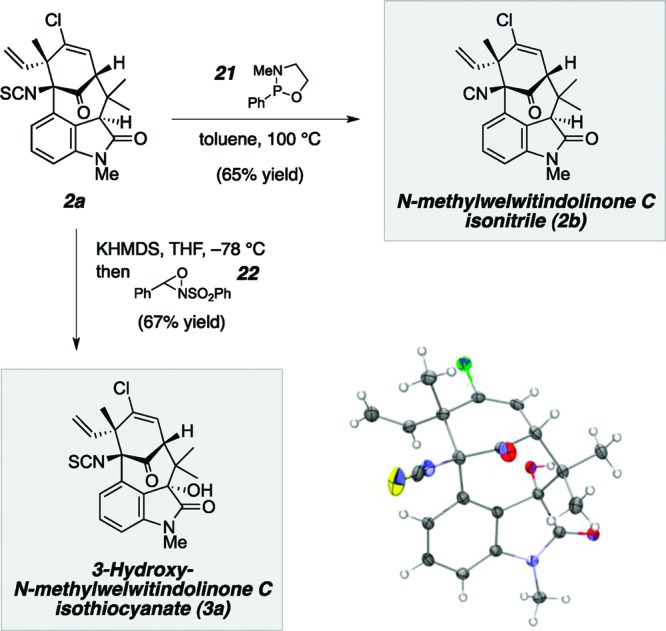
With an efficient route to N-methylwelwitindolinone C isothiocyanate (2a) at hand, our next goal was to uncover pathways by which this product could be directly converted to its siblings, N-methylwelwitindolinone C isonitrile (2b) and 3-hydroxy-N-methylwelwitindolinone C isothiocyanate (3a) (Scheme 4). Desulfurization of isothiocyanate 2a with Mukaiyama’s oxazaphospholidine (21) gave rise to the first of these targets, isonitrile 2b, in 65% yield.4,19 Conversion of 2a to hydroxyoxindole 3a posed a greater challenge, given the susceptibility of sulfur to oxidants and the need for diastereoselective oxidation. Based on prior work,1b,4 the conformation of the natural product was expected to provide some measure of diastereocontrol during oxidation. Indeed, we found that treatment of 2a with KHMDS followed by Davis’s oxaziridine (22) afforded a single compound in 67% yield possessing the exact mass of the expected 3-hydroxyoxindole derivative.20 We were puzzled to find, however, that the 1H and 13C NMR data for this compound did not correspond to those reported in the literature for the natural product,1b suggesting that the oxidation had taken place from the opposite face or perhaps some other transformation had occurred. To definitively establish the identity of the oxidation product, a single-crystal X-ray analysis was carried out, and the results showed our synthetic material to possess the very same chemical structure reported by Moore and co-workers for 3-hydroxy-N-methylwelwitindolinone C isothiocyanate (3a). The discrepancy between our spectral data and those reported for the natural product implied that either the structure was mis-assigned or the data were incorrectly reported. In order to resolve this issue, we requested new NMR data for the authentic sample corresponding to 3a and found that, indeed, the newly recorded 1H and 13C NMR spectra were different from those in the original published work and fully matched those of our synthetic material.21−23
Scheme 4.
In summary, we have completed concise syntheses of three members of the welwitindolinone C group of alkaloids. The synthesis of N-methylwelwitindolinone C isothiocyanate (2a) was achieved in 12% overall yield through a longest linear sequence of 16 steps from commercially available starting materials. This compound was then transformed directly into two additional members of the welwitindolinone C group, isonitrile 2b and 3-hydroxyoxindole 3a, thereby enabling correction of the reported spectral data for 3a. With the completion of this work, we have successfully employed our unified synthetic strategy toward members of each of the three structural subclasses comprising the bridged welwitindolinone alkaloids.
Acknowledgments
Generous financial support from the National Cancer Institute of the NIH (R01 CA101438) is gratefully acknowledged. We thank Dr. Ian Steele for X-ray crystallographic structure determination and Prof. Thomas Hemscheidt and Mr. Wesley Y. Yoshida of the University of Hawaii at Manoa for obtaining NMR spectra of an authentic sample of 3a.
Supporting Information Available
Experimental details, characterization data, NMR spectra, and X-ray crystal data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Stratmann K.; Moore R. E.; Bonjouklian R.; Deeter J. B.; Patterson G. M. L.; Shaffer S.; Smith C. D.; Smitka T. A. J. Am. Chem. Soc. 1994, 116, 9935. [Google Scholar]; b Jimenez J. I.; Huber U.; Moore R. E.; Patterson G. M. L. J. Nat. Prod. 1999, 62, 569. [DOI] [PubMed] [Google Scholar]
- a Konopelski J. P.; Deng H.; Schiemann K.; Keane J. M.; Olmstead M. M. Synlett 1998, 1105. [Google Scholar]; b Wood J. L.; Holubec A. A.; Stoltz B. M.; Weiss M. M.; Dixon J. A.; Doan B. D.; Shamji M. F.; Chen J. M.; Heffron T. P. J. Am. Chem. Soc. 1999, 121, 6326. [Google Scholar]; c Deng H.; Konopelski J. P. Org. Lett. 2001, 3, 3001. [DOI] [PubMed] [Google Scholar]; d Jung M. E.; Slowinski F. Tetrahedron Lett. 2001, 42, 6835. [Google Scholar]; e López-Alvarado P.; García-Granda S.; Àlvarez-Rúa C.; Avendaño C. Eur. J. Org. Chem. 2002, 1702. [Google Scholar]; f Ready J. M.; Reisman S. E.; Hirata M.; Weiss M. M.; Tamaki K.; Ovaska T. V.; Wood J. L. Angew. Chem., Int. Ed. 2004, 43, 1270. [DOI] [PubMed] [Google Scholar]; g MacKay J. A.; Bishop R. L.; Rawal V. H. Org. Lett. 2005, 7, 3421. [DOI] [PubMed] [Google Scholar]; h Baudoux J.; Blake A. J.; Simpkins N. S. Org. Lett. 2005, 7, 4087. [DOI] [PubMed] [Google Scholar]; i Greshock T. J.; Funk R. L. Org. Lett. 2006, 8, 2643. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Lauchli R.; Shea K. J. Org. Lett. 2006, 8, 5287. [DOI] [PubMed] [Google Scholar]; k Xia J.; Brown L. E.; Konopelski J. P. J. Org. Chem. 2007, 72, 6885. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Richter J. M.; Ishihara Y.; Masuda T.; Whitefield B. W.; Llamas T.; Pohjakallio A.; Baran P. S. J. Am. Chem. Soc. 2008, 130, 17938. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Boissel V.; Simpkins N. S.; Bhalay G.; Blake A. J.; Lewis W. Chem. Commun. 2009, 1398. [DOI] [PubMed] [Google Scholar]; n Boissel V.; Simpkins N. S.; Bhalay G. Tetrahedron Lett. 2009, 50, 3283. [Google Scholar]; o Tian X.; Huters A. D.; Douglas C. J.; Garg N. K. Org. Lett. 2009, 11, 2349. [DOI] [PubMed] [Google Scholar]; p Trost B. M.; McDougall P. J. Org. Lett. 2009, 11, 3782. [DOI] [PMC free article] [PubMed] [Google Scholar]; q Brailsford J. A.; Lauchli R.; Shea K. J. Org. Lett. 2009, 11, 5330. [DOI] [PMC free article] [PubMed] [Google Scholar]; r Freeman D. B.; et al. Tetrahedron 2010, 66, 6647. [DOI] [PMC free article] [PubMed] [Google Scholar]; s Heidebrecht R. W. Jr.; Gulledge B.; Martin S. F. Org. Lett. 2010, 12, 2492. [DOI] [PMC free article] [PubMed] [Google Scholar]; t Ruiz M.; López-Alvarado P.; Menéndez J. C. Org. Biomol. Chem. 2010, 8, 4521. [DOI] [PubMed] [Google Scholar]; u Bhat V.; MacKay J. A.; Rawal V. H. Org. Lett. 2011, 13, 3214. [DOI] [PMC free article] [PubMed] [Google Scholar]; v Bhat V.; MacKay J. A.; Rawal V. H. Tetrahedron 2011, 67, 10097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Two elegant syntheses of the unique, cyclobutane-containing member of the family, welwitindolinone A isonitrile, have been reported:; a Baran P. S.; Richter J. M. J. Am. Chem. Soc. 2005, 127, 15394. [DOI] [PubMed] [Google Scholar]; b Reisman S. E.; Ready J. M.; Hasuoka A.; Smith C. J.; Wood J. L. J. Am. Chem. Soc. 2006, 128, 1448. [DOI] [PubMed] [Google Scholar]
- Bhat V.; Allan K. M.; Rawal V. H. J. Am. Chem. Soc. 2011, 133, 5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat V.; Rawal V. H. Chem. Commun. 2011, 47, 9705. [DOI] [PubMed] [Google Scholar]
- Huters A. D.; Quasdorf K. W.; Styduhar E. D.; Garg N. K. J. Am. Chem. Soc. 2011, 133, 15797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Smith C. D.; Zilfou J. T.; Stratmann K.; Patterson G. M. L.; Moore R. E. Mol. Pharmacol. 1995, 47, 241. [PubMed] [Google Scholar]; b Zhang X.; Smith C. D. Mol. Pharmacol. 1996, 49, 288. [PubMed] [Google Scholar]
- Polla M.; Frejd T. Tetrahedron 1991, 47, 5883. [Google Scholar]
- For select methods to generate vinyl chlorides from ketones, see:; a Isaacs N. S.; Kirkpatrick D. J. Chem. Soc., Chem. Commun. 1972, 443. [Google Scholar]; b Burton G.; Elder J. S.; Fell S. C. M.; Stachulski A. V. Tetrahedron Lett. 1988, 29, 3003. [Google Scholar]; c Su W.; Jin C. Org. Lett. 2007, 9, 993. [DOI] [PubMed] [Google Scholar]; Vinyl chlorides from enol triflates:; d Shirakawa E.; Imazaki Y.; Hayashi T. Chem. Commun. 2009, 5088. [DOI] [PubMed] [Google Scholar]; e Shen X.; Hyde A. M.; Buchwald S. L. J. Am. Chem. Soc. 2010, 132, 14076. [DOI] [PMC free article] [PubMed] [Google Scholar]; Vinyl chlorides from enol phosphates:; f Kamei K.; Maeda N.; Tatsuoka T. Tetrahedron Lett. 2005, 46, 229. [Google Scholar]
- a Mori H.; Wada S.; Tsuneda K. Chem. Pharm. Bull. 1963, 11, 684. [DOI] [PubMed] [Google Scholar]; b Mori H.; Tsuneda K. Chem. Pharm. Bull. 1963, 11, 1413. [DOI] [PubMed] [Google Scholar]
- See ref (2v) for attempted conversion of the enol triflate of 6 to the corresponding vinyl chloride using transition metal catalysis.
- See Supporting Information for details.
- A related rearrangement was observed in the course of a Ru- or Pd-mediated reaction of the C13 enol triflate. See ref (2v).
- For examples of dearomatizing Cope rearrangements that do not involve an oxy-Cope, see:; a Marvell E. N.; Lin C. J. Am. Chem. Soc. 1978, 100, 877. [Google Scholar]; b Lambert J. B.; Fabricius D. M.; Hoard J. A. J. Org. Chem. 1979, 44, 1480. [Google Scholar]
- It is worth noting that this reaction displays a striking sensitivity to the presence of trace quantities of adventitious reagents, presumed to be trace transition metals. For example, using glassware or stir bars that had been previously employed in the laboratory resulted in complete reduction of the C20–C21 olefin, presumably through oxidation of hydrazine to diimide and/or transition metal-mediated hydrogenation wherein hydrazine acts as a hydrogen donor.
- Submission of either oxime diastereomer to the condensation reaction conditions regenerated an equilibrium ratio (2.4:1 (3S)-20:(3R)-20) of the two diastereomeric oximes.
- One of the six 3(H)-oxindole-containing welwitindolinone alkaloids, 3-epi-welwitindolinone B isothiocyanate, possesses the 3S stereochemistry. See ref (1a).
- a Kim J. N.; Ryu E. K. Tetrahedron Lett. 1993, 34, 8283. [Google Scholar]; b Kim J. N.; Jung K. S.; Lee H. J.; Son J. S. Tetrahedron Lett. 1997, 38, 1597. [Google Scholar]
- Mukaiyama T.; Yokota Y. Bull. Chem. Soc. Jpn. 1965, 38, 858. [Google Scholar]
- The same compound was formed in 45% yield when 2a was treated with 50% aq KOH, P(OEt)3, and Bu4NCl in toluene under an ambient atmosphere. For a similar procedure, see:Sano D.; Nagata K.; Itoh T. Org. Lett. 2008, 10, 1593. [DOI] [PubMed] [Google Scholar]
- A preserved sample of 3a from Prof. Moore’s chemical collection was located by Prof. Thomas Hemscheidt, University of Hawaii at Manoa, and new NMR spectra were obtained for this compound by Mr. Wesley Y. Yoshida, at the same institution.
- It appears that the NMR data originally reported by Moore et al. in ref (1b) are in error and should be replaced by the data included in the Supporting Information of the present paper.
- We have recently succeeded in oxidizing N-methylwelwitindolinone C isonitrile (2b) to 3-hydroxy-N-methylwelwitindolinone C isonitrile (3b) in 55% yield (unoptimized) using the oxidation procedure described above (viz., 2a→3a). See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.