

Abstract
Glycogen synthase kinase-3 (GSK-3) is linked to the pathogenesis of Alzheimer's disease (AD), senile plaques (SPs), and neurofibrillary tangles (NFTs), but the specific contributions of each of the GSK-3 α and β isoforms to mechanisms of AD have not been clarified. In this study, we sought to elucidate the role of each GSK-3α and GSK-3β using novel viral and genetic approaches. First, we developed recombinant adeno-associated virus 2/1 short hairpin RNA constructs which specifically reduced expression and activity of GSK-3α or GSK-3β. These constructs were injected intraventricularly in newborn AD transgenic (tg) mouse models of SPs (PDAPP+/−), both SPs and NFTs (PDAPP+/−;PS19+/−), or wild-type controls. We found that knockdown (KD) of GSK-3α, but not GSK-3β, reduced SP formation in PDAPP+/− and PS19+/−;PDAPP+/− tg mice. Moreover, both GSK-3α and GSK-3β KD reduced tau phosphorylation and tau misfolding in PS19+/−;PDAPP+/− mice. Next, we generated triple tg mice using the CaMKIIα-Cre (α-calcium/calmodulin-dependent protein kinase II-Cre) system to KD GSK-3α in PDAPP+/− mice for further study of the effects of GSK-3α reduction on SP formation. GSK-3α KD showed a significant effect on reducing SPs and ameliorating memory deficits in PDAPP+/− mice. Together, the data from both approaches suggest that GSK-3α contributes to both SP and NFT pathogenesis while GSK-3β only modulates NFT formation, suggesting common but also different targets for both isoforms. These findings highlight the potential importance of GSK-3α as a possible therapeutic target for ameliorating behavioral impairments linked to AD SPs and NFTs.
Introduction
Alzheimer's disease (AD) is the most common form of dementia and presents clinically with progressive memory loss and cognitive impairments. AD is characterized pathologically by extracellular senile plaques (SPs), composed of amyloid-β (Aβ) peptides derived from the proteolysis of the amyloid precursor protein (APP), and by intracellular neurofibrillary tangles (NFTs), composed of hyperphosphorylated tau protein. Glycogen synthase kinase-3 (GSK-3) is a serine/threonine kinase that has been implicated in the formation of both SPs and NFTs (Jope and Johnson, 2004; Giese, 2009). For example, GSK-3 activation modulates Aβ production (Phiel et al., 2003; Ryder et al., 2003), while Aβ activates GSK-3 (Kim et al., 2003; Akiyama et al., 2005; Ryan and Pimplikar, 2005). Additionally, GSK-3 is a principal kinase that phosphorylates tau at key residues found in AD NFT (Hanger et al., 2009). Thus, GSK-3 modulates pathways related to SP and NFT formation and it has been suggested that GSK-3 reduction may represent an attractive therapeutic target for AD (Phiel et al., 2003; Ryder et al., 2003).
There are two mammalian GSK-3 isoforms, i.e., GSK-3α and GSK-3β, each of which is encoded by a separate gene. Both are highly conserved and widely expressed serine/threonine kinases that share a high degree of homology (Woodgett, 1990). Historically, the most widely used strategy to study the effects of GSK-3 reduction in vivo has been to use pharmacological GSK-3 inhibitors. When administered to various AD transgenic (tg) mice, these inhibitors reduce hyperphosphorylated tau accumulation, Aβ production, and/or SP burden (Pérez et al., 2003; Phiel et al., 2003; Su et al., 2004; Noble et al., 2005; Serenó et al., 2009). However, available inhibitors lack specificity for GSK-3 isoforms and may also have off-target effects, which may confound experimental results. Although a few recent studies have used genetic approaches in vivo (Gómez-Sintes et al., 2007; Alon et al., 2011; Jaworski et al., 2011), they have been unable to fully distinguish which GSK-3 isoform is responsible for hyperphosphorylated tau accumulation and/or SP formation.
In this study, we used two distinct approaches to evaluate the effect of GSK-3α or GSK-3β knockdown (KD) in vivo on AD-related neuropathology: a viral short hairpin RNA (shRNA) approach and a genetic approach. First, we intraventricularly delivered adeno-associated virus (AAV) encoding shRNAs directed toward GSK-3α or GSK-3β into newborn tg mice displaying SP pathology (PDAPP+/−), both SPs and NFTs (PDAPP+/−;PS19+/−), or wild-type (wt) control mice. Second, we generated a triple tg mouse model using CaMKIIα-cre (α-calcium/calmodulin-dependent protein kinase II-Cre) system to KD GSK-3α alleles in PDAPP+/− mice. Using these two models, we demonstrated that knocking down GSK-3α or GSK-3β reduces the accumulation of phosphorylated tau; however, a single GSK-3α KD was sufficient to decrease plaque formation and improve cognition in the triple tg mouse model.
Materials and Methods
Screening shRNA
Neuro 2a (N2a) cells were transfected with shRNA plasmids directed toward murine GSK-3α or GSK-3β. shRNA plasmids containing the puromycin selection marker were purchased from Origene. Forty-eight hours post-transfection, cells were treated with 5 μg/ml puromycin for 7 d. Immunoblot analysis of cell lysates led to the identification of specific GSK-3 shRNAs used in this study with the following sequences: GSK-3α shRNA: AAGGACGAGCTGTATTTGAATCTGGTGCT and GSK-3β shRNA: GGCGACCGAGAACCACCTCCTTTGCGGAG along with a scramble control shRNA: TGACCACCCTGACCTACGGCGTGCAGTGC, designated as shRNA-α, shRNA-β, and shRNA-scr, respectively.
To measure eukaryotic translation initiation factor 2α (eIF2α) response, N2a cells were transfected with shRNA-α, shRNA-β, and shRNA-scr as described above and compared with untreated N2a cells. As a positive control, N2a cells were treated with thapsigargin (300 nm; Sigma) for 1 h and cell lysates were used for immunoblotting.
AAV construction, packaging, and intraventricular injection
Recombinant AAV 2/1 (rAAV2/1) expressing shRNA used in this study was produced at the University of Pennsylvania Vector Core as described previously (Gao et al., 2006). These AAV vectors were purified by two rounds of cesium chloride–gradient centrifugation, buffer-exchanged with PBS, and concentrated using Amicon Ultra 15 centrifugal filter devices-100K (Millipore). Genome titer (genome copies per milliliter) of AAV vectors were determined by real-time PCR. Viral vector titers were as follows: 4.7 × 1012 GC/ml for shRNA-α, 3.2 × 1012 GC/ml for shRNA-β, and 2.1 × 1012 GC/ml for shRNA-scr. The recombinant genome contained AAV 2 (AAV2) inverted terminal repeats that flanked five cassettes: the U6 promoter; a 29 bp palindromic DNA sequence with a 7 bp loop and a termination (TTTTTT) sequence, the CMV promoter, cDNA ZsGreen (Clontech), and an SV-40 poly(A) sequence. Recombinant AAV2 genome was cross-packaged with AAV-1 cap proteins generating pseudotype rAAV2/1.
Mouse intraventricular injection procedure was adapted from previously described studies (Passini and Wolfe, 2001; Li and Daly, 2002). Briefly, on the day of birth (designated as P 0.5), neonatal females were cryo-anesthetized on ice for 5 min. Once anesthetized, neonates were transilluminated over a cold fiber-optic illuminator to highlight cranial landmarks and 2 μl rAAV2/1 (containing shRNA-α, shRNA-β, or shRNA-scr) was injected into each lateral ventricle using a 10 μl Hamilton syringe with a 33 gauge needle. Pups were subsequently placed on a heating pad for recovery and then returned to their home cage. Of the ∼230 total mice injected, ∼218 pups survived surgery and were tattooed 10 d later, yielding a “success rate” of ∼95%.
Generation of tg mice
Using the abovementioned approach, three mouse lines were generated: (1) heterozygous PDAPP (PDAPP+/−), (2) heterozygous PS19;PDAPP (PS19+/−;PDAPP+/−), and (3) control wt (B6C3 background strain). The PDAPP mouse model is well described, and was engineered to encode a human APP minigene carrying the V717F mutation driven by the PDGF promoter (Games et al., 1995). The PS19 model harbors the T34 isoform of tau with one N-terminal insert and four microtubule binding repeats (1N4R) encoding the P301S mutation driven by the mouse prion promoter (Yoshiyama et al., 2007). F1 hybrid PS19+/−;PDAPP+/− and PDAPP+/− were generated by crossing parental strains PS19+/− and homozygous PDAPP. F1 hybrids wt were generated by mating parental strains heterozygous PS19+/− and WT mice. Only female F1 hybrids were used in experimental groups.
Using the abovementioned lines, five cohorts of mice were generated for this study: (1) 4-month-old wt mice, (2) 11-month-old wt mice, (3) 11-month-old PDAPP+/− mice, (4) 11-month-old PDAPP+/−;PS19+/− mice, and (5) wt survival group. In each cohort, a separate group of mice received one of four injection paradigms: rAAV containing shRNA-α, shRNA-β, shRNA-scr, or noninjected control. Each group consisted of 15 mice except for the survival cohort (n = 10/group).Together, a grand total of 20 experimental groups and 280 mice were used.
Conditional knock-out generation
GSK-3α conditional knock-out (cko) mice were generated using the Cre-loxP system through a contract with Taconic. To produce heterozygous GSK-3α floxed (GSK-3αflox/−) mice, the targeting vector based on a 9.9 kb genomic fragment from the GSK-3α gene encompassing exons 1–11 and surrounding sequences was obtained from the C57BL/6J RP23 Bacterial Artificial Chromosomes Library and was modified by inserting a loxP site and an FRT-flanked neomycin resistance gene in intron 1 and a loxP site in intron 4, as well as a ZsGreen cassette at its 3′ end (see Fig. 5A). Thirty micrograms of linearized DNA vector were electroporated using a Bio-Rad Gene Pulser at 240 V and 500 μF into 1 × 107 cells of the C57BL/6N embryonic stem (ES) cell lines. Only nonfluorescent clones suggesting the absence of the ZsGreen region were selected on day 8. After expansion and freezing clones in nitrogen, 176 clones were analyzed by using 5′, 3′, and neomycin probes after digestion by KpnI, EcoRI, AflIII, PflFI, and BglII restriction enzymes in standard Southern blotting techniques (Fig. 5A–D). Proper homologous recombination was identified in four of the clones, and clone B-C9 was used for blastocyst injection.
Figure 5.

Generation of GSK-3α fx mice, confirmation and characterization of GSK-3α cko mice. cko GSK-3α mice generation. A, Restriction maps (from top to bottom) of the genomic locus, targeting vector, homologous alleles, cko allele, and a constitutive KO allele. After proper homologous recombination, exons 2–4 of targeted allele were flanked by loxP sites with the addition of a neomycin (neo) resistant marker flanked by FRT sites and in the absence of ZsGreen. Nonfluorescent selected clones were analyzed by Southern blot. Probes (blue rectangles) were located at the 5′ and 3′ ends and in the neo fragment. Genomic DNA was digested with AflIII for the 5′ (B) and Neo probe (D) or KpnI for the 3′ probe (C). All four clones were positive for proper homologous recombination and ES clone BC-09 was selected. After Flp-mediated deletion, a conditional KO allele was generated without Neo through mating with a Cre-deleter mouse line. Using this Cre-loxP system, wt or cko genotypes (n = 10–15 mice/group) were generated (GSK-3α wt, GSK-3α cko, GSK-3α wt;PDAPP, and GSK-3α cko;PDAPP) and analyzed at 17 months of age. GSK-3α cko was confirmed by IHC using GSK-3αβ-specific antibodies. E, Representative images counterstained with hematoxylin show that in the CA1 region of hippocampus, GSK-3α cko produced a mosaic pattern of neuronal GSK-3α expression and significantly reduced GSK-3α but not GSK-3β immunoreactivity. Scale bar, 100 μm. Neurons with reduced GSK-3 expression are highlighted by black circles and neurons expressing normal GSK-3 levels are highlighted by dashed black circles. F, GSK-3α cko was also confirmed by Western blot from hippocampal lysates (n = 4 mice/group) and densitometric analysis showing a significant reduction in GSK-3α (G) (67%, t(5) = 9, p = 0.0003) but not in GSK-3β levels (H) (t(3) = 0.4912, p = 0.6570). Data show mean ± SEM; ***p < 0.001 compared with αwt. GSK-3α wt, αwt; GSK-3α cko, αcko.
Ten to fifteen targeted C57BL/6N.tac ES cells were microinjected into each blastocyst obtained after superovulation of BALB/c females and 8 injected blastocysts were transferred to each uterine horn of a 2.5 days postcoitum, pseudopregnant Naval Medical Research Institute female mouse. Highly chimeric mice were bred to female C57BL/6 mice with Flp-Deleter recombinase gene to remove the Neo selection marker. Germline transmission was identified by the presence of black C57BL/6 offspring.
Triple tg mice were generated by crossing the GSK-3αflox/− mice with CaMKIIα-cre (C57BL/6 background) (Tsien et al., 1996) and PDAPP lines. Four crosses resulted in triple tg mice GSK-3αflox/flox;CaMKIIα-cre+/−;PDAPP+/− that were subsequently mated with GSK-3αflox/flox mice, generating four genotypes: GSK-3αflox/flox;CaMKIIα-cre−/−;PDAPP−/− (GSK-3α wt), GSK-3αflox/flox;CaMKIIα-cre+/− PDAPP−/− (GSK-3α cko), GSK-3αflox/flox;CaMKIIα-cre−/−;PDAPP+/− (GSK-3α wt;PDAPP), and GSK-3αflox/flox;CaMKIIα-cre+/−;PDAPP+/− (GSK-3α cko;PDAPP). Each of the four groups consisted of n = 10–15 mice that were analyzed at 17 months of age.
Histology and brain preparation
Mice were anesthetized and transcardially perfused with PBS, pH 7.0, in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Brains were surgically removed and the right hemisphere was fixed in 4% neutral buffered formalin (NBF), while the left hemisphere was dissected into various brain regions and frozen at −80°C for biochemical analysis. To maintain consistency of dissection and to facilitate analysis brains were coronally sliced starting with an initial cut parallel to the olfactory tract as it bends toward the median eminence. Bisection at this point generated anterior and posterior cortical regions which were fixed in NBF and paraffin embedded. Paraffin blocks were sectioned at 6 μm exhaustively. Tissue sections were stained using a Polymer horseradish peroxidase detection system (Biogenex) and automatically stained using the i6000 Automated Staining System (Biogenex) as previously described (Hurtado et al., 2010).
Antibodies used in histology
Viral distribution was assessed using rabbit polyclonal antibody (pAb) ZsGreen (Clontech). ShRNA off-target effects were identified by activated microglia using ionized calcium binding adaptor molecule 1 (iba1) rabbit pAb (019-19741; Wako Chemicals), and activated astrocytes using glial fibrillary acidic protein (GFAP) rabbit pAb (Dako). GSK-3 expression was monitored through the use of a GSK-3α rabbit pAb (9338) and GSK-3β rabbit mouse monoclonal antibody (mAb) (27C10; Cell Signaling Technology). To track the distribution of Aβ plaques, we used mouse mAb Nab228 which binds to 1–11 in Aβ peptides (Lee et al., 2006). Anti-tau antibodies used in this study include: AT8 mouse mAb specific for phosphorylated Ser202/Thr205 (Innogenetics), 12E8 mouse mAb specific for phosphorylated S262 (gift from P. Seubert, Elan Pharma), AT180 mouse mAb specific for phosphorylated Thr231 (Innogenetics), and anti-tau mouse mAb T14, which is specific for human tau (Yoshiyama et al., 2007). Additionally we used Alz50 and MC1 mouse mAb (a gift from Peter Davies) to monitor conformational tau. Fibrillary tau lesions were detected using Thioflavine-S/Lipofuscin (ThS) autofluorescence quenching protocols as described by Yoshiyama et al. (2007).
Immunohistochemical quantification
Aβ deposits, phospho-tau, Iba1, and GFAP immunoreactive (IR) burden were quantified by a researcher blinded to experimental conditions as previously described (Carroll et al., 2011). Images from immunolabeled sections were captured and digitized using a video capture system (Nikon camera coupled to an Olympus DP71 upright microscope). Using NIH ImageJ software, the areas of interest were drawn freehand (i.e., entire hippocampus). Afterward, digital grayscale images were converted into binary positive/negative data using a constant threshold limit. The percentage of positive pixels (i.e., IR area) was quantified for each image to generate IR “load” values (i.e., percentage area occupied by the IR product). For phospho-tau burden, images of AT180- or 12E8-stained sections of CA3 regions close to bregma −3.28 mm were measured. For Aβ deposit quantification; images of Nab228-stained sections of hippocampus were more anterior (bregma −1.95).
Sandwich ELISA analysis
Aβ 40 and Aβ 42 levels were detected using Aβ sandwich ELISA protocols as previously described (Lee et al., 2003). Briefly, brain regions were sonicated in radioimmunoprecipitation assay (RIPA) buffer (0.5% sodium deoxycholate, 0.1% SDS, 1% NP-40, 5 mm EDTA in TBS, pH 8.0) containing protease inhibitors (1 μg/ml pepstatin A, leupeptin, l-1-tosylamido-2-phenylethyl chloromethyl ketone, 1-chloro-3-tosylamido-7-amino-2-heptanone, soybean trypsin inhibitor, and 0.5 mm phenylmethanesulfonyl fluoride) at 6 μl/mg of tissue followed by centrifugation at 100,000 g for 20 min at 4°C. The resulting pellet was further extracted by sonication with 70% formic acid (FA) at 1 μl/mg tissue followed by a second identical centrifugation. Both RIPA and FA lysates were assayed by sandwich ELISA using Ban50 (anti-Aβ1–10) as a capturing antibody and end-specific BC05 and BA27 antibodies to distinguish Aβ 40 and Aβ 42, respectively.
Western blots
To measure soluble proteins, hippocampi and N2a cells were homogenized in RIPA buffer in the presence of protease inhibitors and briefly sonicated. Samples were centrifuged at 100,000 g for 30 min at 4°C, and protein concentration was measured by bicinchoninic acid assay. Samples were electrophoresed on 10% Tris-glycine acrylamide gels and transferred to a nitrocellulose or polyvinylidene fluoride membrane. Immunoblots were probed with the following antibodies: GSK-3αβ mouse mAb (Calbiochem), eIF2α (9722) rabbit pAb, phospho-eIF2α (Ser51) (119A11) rabbit mAb (Cell Signaling Technology), C terminus of APP (5685) rabbit pAb (Lee et al., 2005), PhAT, a phospho rabbit pAb raised to residues 665–673 containing phospho-Thr668 at the C terminus of APP (Lee et al., 2005), and GAPDH mouse mAb (Advanced Immunochemical) as a loading control. Immunoblots were then exposed to species-specific horseradish peroxidase-conjugated anti-IgG antibodies (Santa Cruz Biotechnology) and visualized by enhanced chemiluminescence (PerkinElmer) using a Fuji Imager. Images were quantified using Multi Gauge version 3.2 FujiFilm software.
GSK-3 kinase activity assay
Frozen hippocampal sections were sonicated 20 times at 1 s pulses, on ice at 6 μl/mg of tissue in homogenization buffer (D-PBS w/ Ca2+, Mg2+), pH 7.3 (Invitrogen), containing protease and phosphates inhibitors (complete, Mini, EDTA-free Protease Inhibitor Cocktail, and PhosSTOP) (Roche Applied Science). Samples were centrifuged at 100,000 × g for 30 min at 4°C and supernatant protein concentrations were determined using a Bradford assay (Bio-Rad). Fifty micrograms of lysate was used for immunoprecipitation (IP) for GSK-3α or GSK-3β. IP was performed using 35 μl of Protein A/G PLUS-Agarose slurry (Santa Cruz Biotechnology), 50 μg of lysate, and 2 μl of GSK-3α- or GSK-3β-specific antibodies (Cell Signaling Technology). IP was incubated overnight at 4°C on a rotator and subsequently washed and resuspended in homogenization buffer. Resuspended bead slurry was used for GSK-3 kinase activity assay. The activity assay was performed in kinase buffer (25 mm Tris-HCl, pH 7.5, 0.5 mm EGTA, 75 mm NaCl, 0.015%, Brij-35, 0.05% β-mercaptoethanol, pH 7.4, and 1 μm cAMP-dependent protein kinase inhibitor tide) and ATP buffer (10 mm MgCl2, 0.1 mm adenosine 5′-triphosphate, 3MBq/ml adenosine 5′-triphosphate [γ−32P]), with the GSK-3 substrate, phospho-glycogen synthase peptide-2 (Millipore). Reactions were performed at 37°C for 15 min. 32P-labeled peptide was absorbed on P81 phosphocellulose paper (GE Healthcare) and washed several times in 75 mm phosphoric acid followed by an acetone wash and briefly dried. Quantitation was performed in a β-scintillation counter, 1450 MicroBeta TriLux (PerkinElmer).
Behavioral analysis
Open field.
To assess general activity, locomotion, and anxiety, mice were tested in the open field as adapted as previously described (Boyce-Rustay and Holmes, 2006; Carroll et al., 2007). The apparatus was a square arena (40 × 40 × 35 cm) with black Plexiglas walls and white floor that was evenly illuminated to ∼95 lux. Mice were individually placed in the center and allowed to freely explore for 13 min while the trial was videotaped. Subsequent video scoring was completed by an observer blind to treatment groups using the SMARTv25 video-tracking software (Panlab, S.LU.). Total distance moved was automatically calculated by the tracking software as a direct measure of locomotion and activity.
Barnes maze.
Mice were tested for hippocampal-dependent spatial memory on the Barnes maze (San Diego Instruments) using a previously described protocol from our laboratory (Brunden et al., 2010). Briefly, mice were tested over >6 d. On training days 1–3, mice were exposed to the Barnes maze for three 2.5 min trials, 15 min apart. On testing days 4–6, they were exposed to two 2.5 min trials. On all 6 d, only the two last trials were scored. Mice were tested on their ability to learn and remember a fixed position of an escape compartment, and their performance was scored for strategy and latency to find the escape compartment. Strategies were defined as (1) spatial (mouse navigates directly to the escape hole with both first intent deviation and total errors < or = 3), (2) serial (mouse systematically searches of consecutive holes with no center crosses), and (3) random (no strategy at all) as previously described (O'Leary et al., 2011).
Statistical analysis
The majority of data analysis was completed using one-way ANOVA followed by Bonferroni's post hoc test. ELISA was assessed using the Kruskal–Wallis analysis and Dunn's post hoc test. Barnes maze search strategy was analyzed using a χ2 and latency with a two-way ANOVA. Life-span studies were analyzed using the Kaplan–Meier survival curve.
Results
Identification, development, and implementation of rAAV2/1 shRNA targeting GSK-3α and GSK-3β
Four hairpin shRNA constructs were designed using a proprietary Origene algorithm to target GSK-3α or GSK-3β and they were evaluated by transfection into N2a cells along with a scrambled hairpin control to determine which shRNA yielded the highest expression (Fig. 1A,B). Due to high sequence homology of GSK-3 isoforms, each hairpin was assessed for both GSK-3α and GSK-3β expression. In the case of GSK-3α, since shRNA GSK-3α #1 showed no target reduction and #4 displayed cellular toxicity (t(4) = 1.459, p = 0.2182 and >50%, respectively), both were eliminated from the study (Fig. 1A). The GSK-3α #2 and #3 of both shRNAs showed a significant GSK-3α reduction (82 and 74%, respectively, p < 0.001) compared with untreated cells (F(2,6) = 54.82, p < 0.0001) without affecting GSK-3β expression. Since shRNA GSK-3α #2 showed higher KD levels, it was chosen for further development (Fig. 1B). In the case of GSK-3β, shRNAs GSK-3β #3 and #4 displayed cellular toxicity (>50%) and were therefore eliminated from consideration. ShRNA GSK-3β #1 and #2 showed similar levels of GSK-3β reduction (65 and 57%, respectively, p < 0.001) and little toxicity compared with untreated cells (F(2,5) = 298.5, p < 0.0004). However, in shRNA GSK-3β #1, we observed a nonsignificant trend toward GSK-3α reduction (17%, p = 0.34), therefore shRNA GSK-3β #2 was selected for further development. No alterations in GSK-3 expression were observed with scramble shRNA control (F(3,4) = 1.125, p = 0.4386).
Figure 1.
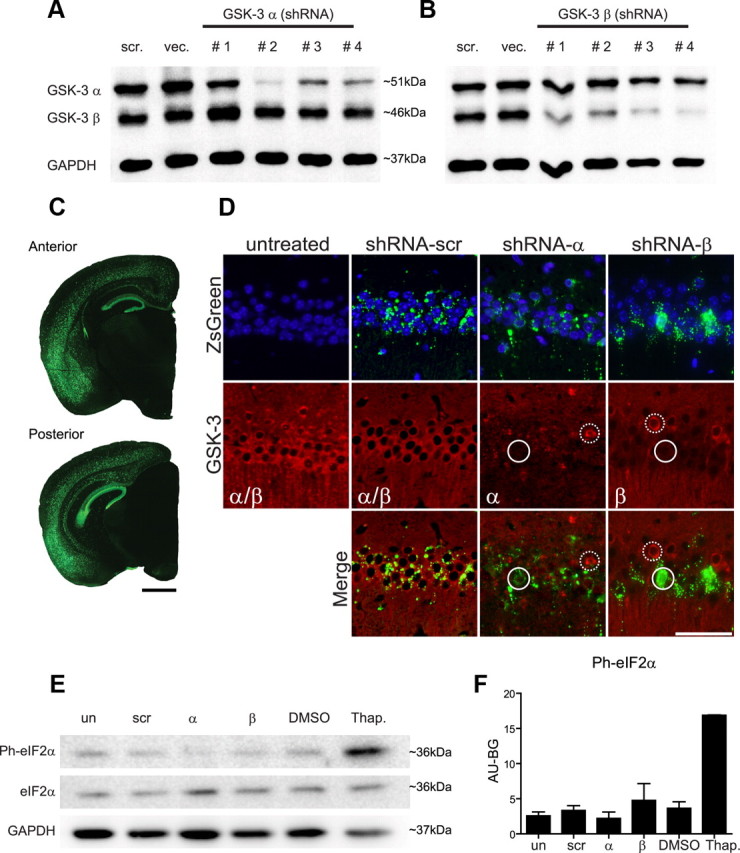
shRNA selection, rAAV distribution and expression, and lack of evidence of off-target effects. N2a cells were transfected with hairpin constructs and selected with puromycin for 7d. As controls, N2a cells were transfected with an empty vector (VEC) or a scrambled hairpin control (scr). A, Representative Western blot of GSK-3α shRNA #1–4 and (B) GSK-3β shRNA #1–4 demonstrates that shRNA GSK-3α #2 showed the highest KD level (F(2,6) = 54.82, p < 0.0001), and GSK-3β #2 showed a GSK-3β reduction without affecting GSK-3α levels (F(2,5) = 298.5, p < 0.0004). Wt neonates (n = 15/group) were uninjected or injected with shRNA-scr, shRNA-α, or shRNA-β and evaluated at 4 months of age to confirm rAAV distribution and targeted-gene KD. C, Representative images of both anterior and posterior brain after shRNA-scr treatment shows rAAV distribution stained for ZsGreen (green) within hippocampal regions and deeper cortical layers in a rostrocaudal pattern. Scale bar, 1 mm. D, Representative immunofluorescence images of the CA1 region of hippocampus from each of the three shRNA treatment groups and uninjected controls are shown immunostained for ZsGreen or for both GSK-3αβ isoforms (red) and counterstained with DAPI (blue). Merging GSK-3 and ZsGreen expression reveals that expression of rAAV2/1 directly leads to reduction of targeted GSK-3 isoforms (highlighted by white circles), whereas neurons lacking rAAV2/1 show no loss of GSK-3 expression (highlighted by dashed white circles). Scale bar, 50 μm. To analyze possible off-target effects, N2a cells were treated with shRNA-scr, shRNA-α, and shRNA-β. DMSO and thapsigargin (Thap) treatment were used as negative and positive controls, respectively (E). Representative Western blot and (F) densitometric analysis of phosphorylated eIF2α (Ph-eIF2α) normalized to GAPDH reveals no significant alterations (F(4,5) = 0.6255, p = 0.6650). Untreated, un; shRNA-scr-treated cells, scr; shRNA-α-treated cells, α; and shRNA-β-treated cells, β.
Pseudotype rAAV2/1 viruses containing hairpin sequences for GSK-3α #2, GSK-3β #2, and a scrambled shRNA (designated as shRNA-α, shRNA-β, and shRNA-scr, respectively) were produced and injected into newborn wt mice (see Material and Methods). To evaluate AAV distribution, wt mice were examined at 4 months postinjection by monitoring ZsGreen expression which was found predominately in hippocampal regions CA1–CA3, the granular cell layer of the dentate gyrus, and with more variability in deeper cortical layers four through six along with cortical amygdala regions (Fig. 1C). Overall, cortical regions in close proximity to the lateral ventricles showed higher transduction than regions further away. Minor or no ZsGreen expression was detected in basal ganglia, thalamus, brainstem, or cerebellum.
To confirm that rAAV2/1 vectors expressing shRNA silenced the target genes, we double-labeled mouse sections with ZsGreen and GSK-3α or GSK-3β antibodies (Fig. 1D). We observed that in shRNA-α-injected and shRNA-β-injected wt mice, cells with ZsGreen staining showed a concomitant reduction in GSK-3α or GSK-β protein expression, respectively, whereas neurons lacking ZsGreen did not. In shRNA-scr-injected mice, ZsGreen was not associated with a reduction in either GSK-3 isoform. Together, these results validate that rAAV2/1 vectors expressing shRNA lead to direct silencing of the target genes in mouse brain.
To determine potential off-target effects of rAAV2/1 infection, we assessed whether our double-stranded RNA (dsRNA) would evoke an interferon response. dsRNA is a common intermediate produced during virus infection in mammalian cells. Its binding motifs activate protein kinase R (PKR) which phosphorylates eIF2α, leading to global inhibition of cellular protein synthesis and viral replication (Sledz et al., 2003; Sledz and Williams, 2005). To test if hairpins would elicit an antiviral defense, we examined phosphorylation of elF2α in N2a cells transfected with shRNA-α, shRNA-β, or shRNA-scr plasmids. Cells treated with dimethylsulfoxide (DMSO) and thapsigargin were used as a negative and positive control, respectively. No significant change in eIF2α phosphorylation state was observed in shRNA-treated versus shRNA-untreated cells, suggesting that shRNA presence did not prompt an off-target PKR-eIF2α response (Fig. 1E,F). Second, to investigate whether the injection or the presence of the different shRNAs in the CNS produced an immune response or brain injury in mice, we analyzed the activation of astrocytes and microglia in the hippocampus of wt mice at 4 months of age (n = 9 per group). No statistical differences in astrocytosis (F(3,32) = 1.934, p = 0.144) or activated microglia (F(3,32) = 0.7316, p = 0.5408) in the shRNA-treated mice were observed, suggesting that shRNA presence did not abnormally activate the immune system (data not shown).
GSK-3 expression and survival analysis in shRNA-α-injected and shRNA-β-injected wt mice
To further define the extent of GSK-3α versus GSK-3β KD by specific shRNAs, we examined the distribution, protein levels, and kinase activity of both GSK-3 isoforms. First, immunohistochemistry (IHC) confirmed that silencing GSK-3α significantly reduced GSK-3α immunoreactivity but had no effect on GSK-3β levels and vice versa in the hippocampus of 4-month-old wt mice (Fig. 2A). Moreover, we did not detect any alterations in GSK-3α or GSK-3β staining in untreated or scramble-treated mice (Fig. 2A).
Figure 2.

Confirmation of GSK-3 KD. ShRNA-treated and noninjected wt mice were evaluated at 4 months of age to confirm targeted-gene KD. A, Representative IHC images of the CA1 region of hippocampus counterstained with hematoxylin reveal normal, baseline GSK-3αβ expression levels in untreated and shRNA-scr-treated mice. ShRNA-α treatment significantly reduced GSK-3α expression without altering GSK-3β expression and vice versa for shRNA-β-treated mice. Neurons with reduced GSK-3 expression are highlighted by black circles and neurons expressing normal GSK-3 levels are highlighted by dashed black circles. Scale bar, 100 μm. B, Immunoblot analyses of hippocampal lysates probed with GSK-3 isoform-specific antibodies. C, Corresponding quantification showing that shRNA-α treatment significantly reduced GSK-3α expression without altering GSK-3β expression (F(3,12) = 68.47, p < 0.0001) and (D) vice versa for shRNA-β-treated mice (F(3,12) = 29.05, p < 0.0001). Wt injected and uninjected mice were evaluated at 11 months of age to confirm targeted-gene KD by analyzing GSK-3 activity using a 32P-labeled assay (n = 4–6/group). E, ShRNA-α significantly decreased GSK-3α activity levels (F(3,14) = 38.64, p < 0.0001) and (F) shRNA-β significantly decreased GSK-3β activity (F(3,12) = 12.87, p < 0.0005). Data show mean ± SEM; ***p < 0.001 and **p < 0.01 compared with all other groups. Uninjected, un; shRNA-scr-injected mice, scr; shRNA-α-injected mice, α; and shRNA-β-injected mice, β.
Second, immunoblot analyses showed that GSK-3α protein levels were reduced by ∼58–72% in shRNA-α-injected mice compared with noninjected, shRNA-scr-injected, and shRNA-β-injected mice whereas GSK-3β protein levels were reduced by ∼40–54% in shRNA-β-injected mice (Fig. 2B–D). No statistically significant difference in GSK-3 protein expression levels was observed between noninjected and shRNA-scr-injected mice (t = 1.365, p > 0.05 for GSK-3α and t = 2.055, p > 0.05 for GSK-3β) and no change in the total levels of the nontargeted isoform was observed when compared with uninjected control (t = 1.625, p > 0.05 for GSK-3α levels in shRNA-β mice, and t(6) = 1.180, p > 0.05 for GSK-3β levels in shRNA-α mice) (Fig. 2B–D). These results validate the specificity of the shRNA-α and shRNA-β used in this study, and suggest that silencing one isoform does not induce a compensatory response in the other.
Third, since total protein levels may not necessarily correlate with enzyme activity, GSK-3α and GSK-3β activity levels in hippocampal lysates of shRNA-α-injected, shRNA-β-injected, and shRNA-scr-injected 11-month-old wt mice were compared with noninjected controls for GSK-3 enzyme activity. Importantly, a significant 71% decrease in GSK-3α activity in shRNA-α-injected mice (Fig. 2E), and a comparable 67% reduction in GSK-3β activity in shRNA-β-injected mice (Fig. 2F) were observed. Significantly, GSK-3α KD did not affect GSK-3β activity levels (t = 0.9417, p > 0.05), and GSK-3β KD did not affect GSK-3α activity (t = 1.673, p > 0.05), suggesting that a single isoform KD did not trigger a compensatory effect in the alternate isoform. Further, shRNA-scr had no effect on GSK-3α or GSK-3β activity levels (t = 1.641 and t = 0.6733, respectively, p > 0.05). Last, these results suggest that KD of the targeted genes was persistent over 11 months.
Finally, we sought to confirm whether GSK-3 KD would have deleterious effects on life span. Survival analysis of wt mice injected with shRNA-α, shRNA-β, or -scr rAAV2/1 monitored up to 12 months showed no difference in life span between injected and uninjected controls (χ2 = 2.210, p = 0.53, n = 10 per group) suggesting that GSK-3 KD does not alter survival in wt mice.
GSK-3α or GSK-3β KD reduces NFTs and tau phosphorylation as well as mitigates conformational changes in PDAPP+/−;PS19+/− bigenic mice
To evaluate the effect of GSK-3α and GSK-3β KD on tau phosphorylation and NFT formation, newborn PDAPP+/−;PS19+/− bigenic mice were injected with shRNA-α, shRNA-β, or shRNA-scr rAAV2/1 and were examined at 11 months of age (n = 15 per group). Although shRNA-α-injected and shRNA-β PDAPP+/−;PS19+/−-injected mice had lower reduction of GSK-3α and GSK-β protein expression than previous experiments with wt or PDAPP+/− mice, they were, nonetheless, statistically significant (30%, F(2,57) = 6.271, p = 0.0035, and 29%, F(2,57) = 32.38, p < 0.0001, respectively).
To determine whether specific phosphorylation sites of tau were affected by GSK-3α or GSK-3β KD, we conducted IHC using antibodies on a number of different phosphorylation-dependent epitopes, as well as on abnormal conformation epitopes of tau and total tau in the CA3 hippocampus region. Quantification of the IR load showed that total human tau levels measured by T14 were not altered by silencing GSK-3α or GSK-3β (Fig. 3A). In contrast, NFTs and tau hyperphosphorylation associated with pathological tau on Ser202/Thr205 (detected by mAb AT8), Thr231 (mAb AT180), and Ser262 (mAb 12E8) were markedly decreased (Fig. 3A) and quantitation of IHC with both AT180 (Fig. 3B) and 12E8 (Fig. 3C) showed significant reductions after GSK-3α or GSK-3β KD. Interestingly, reduction in tau hyperphosphorylation was accompanied by a decrease in the accumulation of abnormal tau conformers as detected by conformation-specific antibodies Alz50, MC1, and a dramatic reduction in NFTs as monitored by ThS, which recognizes β-pleated conformations in tau amyloid inclusions. Together, these results suggest that both GSK-3α and GSK-3β KD were effective in reducing tau hyperphosphorylation, pathological tau conformational changes, and NFT formation.
Figure 3.
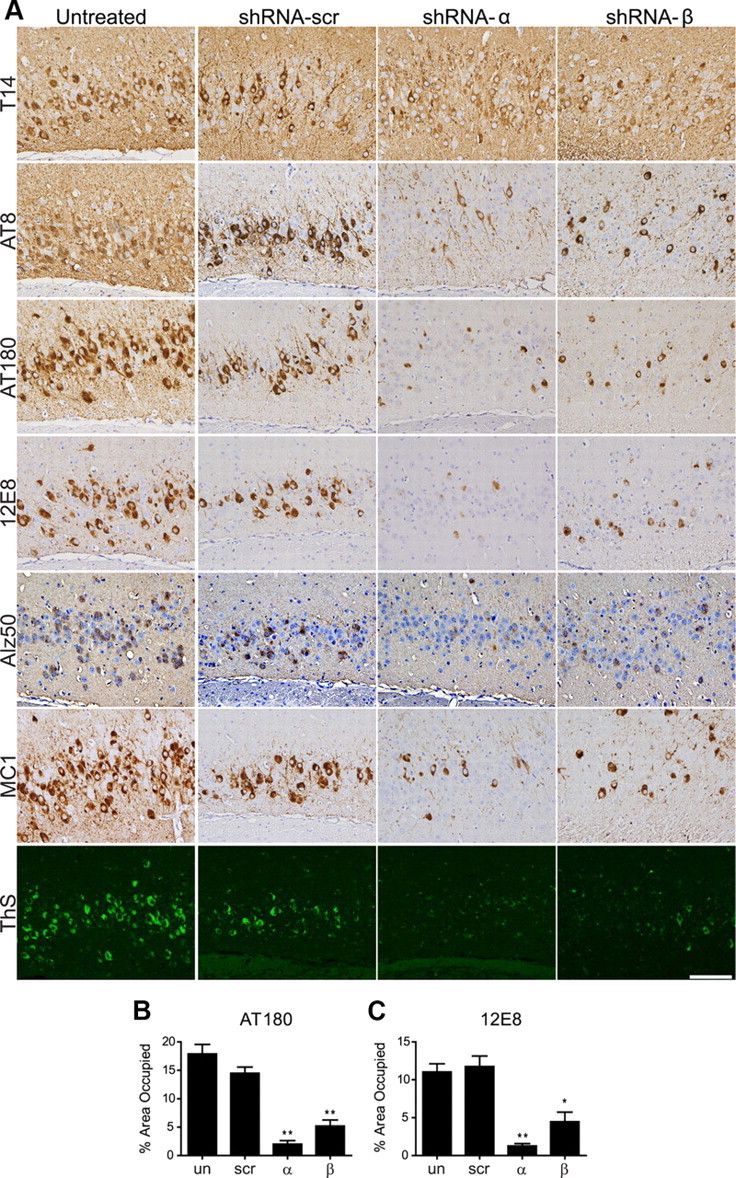
GSK-3α or GSK-3β KD reduces tau phosphorylation and conformational changes in PS19+/−;PDAPP+/− mice. PS19+/−;PDAPP+/− neonates were uninjected (un) or injected with shRNA-α, shRNA-β, or shRNA-scr and evaluated at 11 months of age (n = 15 per group). A, Representative IHC images counterstained with hematoxylin show CA3 regions of posterior hippocampus stained with antibodies to total tau (T14), phospho-tau Ser202/Thr205 (AT8), phospho-tau Thr231 (AT180), phospho-tau Ser262 (12E8), or conformational specific antibodies Alz50, MC1, or ThS as indicated. Scale bar, 100 μm. PS19+/−;PDAPP+/− mice treated with shRNA-α or shRNA-β showed a significant reduction in phospho-tau, Alz50, MC1, and ThS staining, but not in total tau load compared with shRNA-scr and noninjected controls. The phospho-tau burden in the CA3 region was quantified using ImageJ for mAb AT180 (B) and 12E8 (C) in n = 3–4 mice/group, to demonstrate the quantification of >50% reduction with shRNA-α and shRNA-β treatment compared with controls (F(3,8) = 8.283, p = 0.0078 and F(3,9) = 15.28, p = 0.0007, respectively). Data show mean ± SEM; **p < 0.01, *p < 0.05 compared with shRNA-scr controls.
GSK-3α but not GSK-3β KD decreases Aβ levels and SPs in PDAPP+/− and PDAPP+/−;PS19+/− brains
The effects of GSK-3 KD on Aβ amyloid deposition in SPs and Aβ levels in brain were also evaluated in PDAPP+/− and PDAPP+/−; PS19+/− mice injected with rAAV2/1 containing shRNA-α, shRNA-β, or shRNA-scr and examined at 11 months of age. ShRNA-α and shRNA-β KD resulted in a reduction of GSK-3α (∼70%) and GSK-3β (∼50%) protein expression, respectively, in PDAPP+/− mice (Fig. 4E,G). Aβ levels were measured using IHC and Aβ sandwich ELISA. IHC to monitor Aβ SP burden in hippocampal regions of PDAPP+/− and PDAPP+/−;PS19+/− mice showed a statistically significant reduction in Aβ load after GSK-3α but not GSK-3β KD in PDAPP+/− and PDAPP+/−;PS19+/− mice (Fig. 4A,B). In general, the reduction in Aβ deposition was more pronounced in PDAPP+/−;PS19+/− mice and a modest increase in Aβ accumulation was detected in shRNA-β-treated PDAPP+/− and PDAPP+/−;PS19+/− mice when compared with shRNA-scr-treated and shRNA-scr-untreated mice (Fig. 4B). Moreover, measurements of brain Aβ levels by ELISA corroborated IHC studies of Aβ load by demonstrating a similar reduction in soluble and insoluble Aβ 40/42 levels in hippocampus from both RIPA (Fig. 4C) and FA extracted fractions (Fig. 4D). Overall, Aβ 40 and Aβ 42 levels were statistically different between treatment groups in both fractions and genotypes. Moreover, we observed that GSK-3α KD significantly decreased both soluble and insoluble Aβ 40 and Aβ 42 levels compared with noninjected controls, whereas mice treated with shRNA-scr or shRNA-β had little to no change in Aβ 40/42 levels (Fig. 4C,D). Thus GSK-3α KD but not GSK-3β KD reduces Aβ levels in PDAPP+/− and PDAPP+/−;PS19+/− mice.
Figure 4.
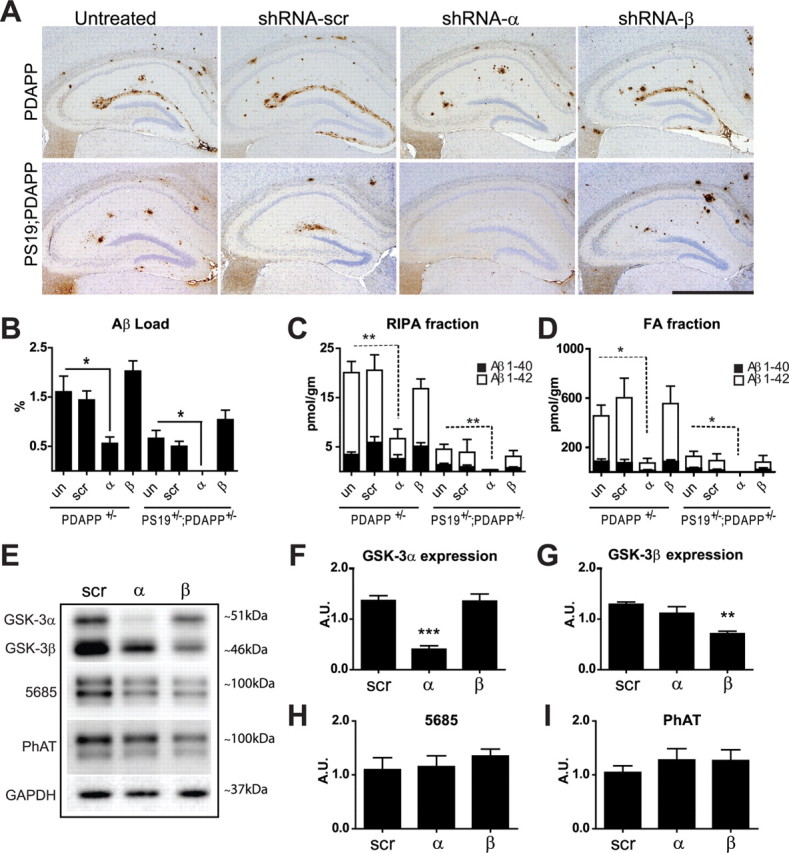
GSK-3α KD decreases Aβ levels and plaque deposition in PDAPP+/− and PDAPP+/−;PS19+/− brains. PDAPP+/− or PDAPP+/−;PS19+/− neonates were uninjected (un) or injected with shRNA-α, shRNA-β, or shRNA-scr and evaluated at 11 months of age. A, Representative images of sections immunostained for Aβ (Nab228) and counterstained with hematoxylin demonstrated a significant reduction in plaque load after GSK-3α KD (n = 15 per group). Scale bar, 1 mm. B, Hippocampal Aβ load was quantified using ImageJ software (n = 7–12 per group). In both PDAPP+/− and PDAPP+/−;PS19+/− models, shRNA-α treatment decreased Aβ burden compared with untreated mice (F(3,124) = 5.269, p = 0.0019 and F(3,91) = 7.701, p = 0.0001, respectively). C, D, In PDAPP+/− and PDAPP+/−;PS19+/− mice, hippocampal regions were analyzed for soluble and insoluble Aβ levels using Aβ sandwich ELISA (n = 5–8/group). In PDAPP+/− mice, shRNA-α decreased soluble Aβ 40 and Aβ 42 levels in the RIPA fraction (C) (Aβ40 H4 = 16.84, p = 0.0008 and Aβ42 H4 = 13.39, p = 0.0039); and FA fraction (D) (Aβ 40 H4 = 14.67, p = 0.0021 and Aβ 42 H4 = 13.07, p = 0.0045). Likewise, shRNA-α decreased soluble Aβ 40 and Aβ 42 levels in PS19+/−;PDAPP+/− mice in the RIPA fraction (C) (Aβ 40 H4 = 10.55, p = 0.0144 and Aβ 42 H4 = 10.74, p = 0.0132), and FA fraction (D) (Aβ 40 H4 = 10.44, p = 0.0152 and Aβ 42 H4 = 9.178, p = 0.0270). Data are shown as mean ± SEM. *p < 0.05, **p < 0.01. E, Western blot and densitometric analysis were performed in triplicate with n = 4–5 mice/group. Representative blots show GSK-3αβ, total APP (5685), phosphorylated APP (PhAT), and a loading control (GAPDH). GSK-3α levels (F) were significantly reduced in mice treated with shRNA-α (F(2,15) = 26.96, p < 0.001), and GSK-3β levels (G) were significantly reduced in mice treated with shRNA-β (F(2,15) = 12.40, p = 0.0007) compared with shRNA-scr controls. However, neither the total APP (H) nor phospho-APP levels (I) were altered across treatment groups (F(2,21) = 0.3396, p = 0.7159, and F(2,21) = 0.4014, p = 0.6744, respectively). Data show mean ± SEM; ***p < 0.001 compared with untreated controls.
To investigate potential mechanisms by which GSK-3α KD reduces Aβ levels and plaque load, we evaluated APP levels in the hippocampus of treated PDAPP+/− mice by immunoblots (Fig. 4E) and showed no group differences in APP expression (Fig. 4E,H, 5685) or phosphorylation at Thr668 (Fig. 4E,I, PhAT) of APP. Together, these results suggest that the reduction in Aβ plaque load by lowering GSK-3α expression is not mediated by alterations in total APP levels or APP phosphorylation.
Conditional knockout of GSK-3α decreases Aβ amyloid deposition and prevents memory deficits in PDAPP+/− mice
To further confirm the effects of GSK-3α on Aβ deposition, we used a genetic approach and the cre-loxP system to generate tg cko mice overexpressing mutant APP in four genotypes: GSK-3α wt, GSK-3α cko, GSK-3α wt;PDAPP+/−, and GSK-3α cko;PDAPP+/−, all of which were analyzed at 17 months of age. IHC studies showed a dramatic reduction in GSK-3α immunostaining in brains of GSK-3α cko mice without altering GSK-3β immunostaining (Fig. 5A). Although complete knockout (KO) of GSK-3α was not achieved, only a small number of GSK-3α-positive neurons were detected in clearly defined regions, and this was consistent throughout our cohort. Specifically, consistent with a previous report (Tsien et al., 1996), the most robust reduction overall was observed in the cortex, limbic system, and thalamus. To a lesser extent, we observed a reduction in the CA2 and CA3 of hippocampus, layers 4–5 of cortex, lateral amygdale nuclei, caudate, putamen, and reticular part of substantia nigra, and there were no changes in the hypothalamus, fornix, septal nuclei, brainstem, and cerebellum (data not shown). Moreover, ∼70% reduction in hippocampal GSK-3α levels was detected by immunoblot analyses (Fig. 5B,C), without any evident compensatory increase in GSK-3β expression (Fig. 5D).
To evaluate the effect of GSK-3α KO on Aβ SP burden in our genetic models, hippocampi from age-matched GSK-3α wt;PDAPP+/− and GSK-3α cko;PDAPP+/− mice were interrogated by IHC with anti-Aβ mAb Nab228 (Fig. 6A), and Aβ IR load was quantified using ImageJ (Fig. 6B). In the CA1, we observed a significant reduction in Aβ IR SP load in GSK-3α cko;PDAPP+/− compared with GSK-3α wt;PDAPP+/− mice.
Figure 6.

GSK-3α cko decreases amyloid deposition and prevents memory deficits in PDAPP+/− mice. At 17 months of age, GSK-3α wt, GSK-3α cko, GSK-3α wt;PDAPP, and GSK-3α cko;PDAPP mice were evaluated for Aβ levels and behavioral phenotype (n = 10–15/group). A, Representative images show hippocampal Aβ using mAb Nab228 in GSK-3α wt;PDAPP and GSK-3α cko;PDAPP mice. Scale bar, 500 μm. GSK-3α cko showed significantly reduced Aβ immunoreactivity (t(12) = 2.253, p = 0.0438) which was quantified as Aβ load using ImageJ (B). Mice were also assessed for locomotor activity on the open field test (C) and spatial memory (D) and search strategy (E) on the Barnes maze. Compared with GSK-3α wt and GSK-3α cko mice, GSK-3α wt;PDAPP displayed hyperactivity on the open field test (F(3,25) = 5.234, p = 0.0061) (C). Compared with GSK-3α wt mice, GSK-3α wt;PDAPP mice performed significantly worse (longer latency to find the target) in the Barnes maze (F(3,43) = 3.589, p = 0.021) while GSK-3α cko;PDAPP mice were statistically indistinguishable from both groups (D). Last, GSK-3α wt;PDAPP mice were more likely to use a poor search strategy (random) on the Barnes maze compared with the other three genotype groups (χ(6)2 = 26.198, p < 0.0001), while GSK-3α cko;PDAPP were more likely to use beneficial search strategies (serial and spatial) (E). Search strategy data are shown as the percentage of mice using each strategy per genotype column. *p < 0.05, **p < 0.01, ***p < 0.001. GSK-3α wt, αwt; GSK-3α cko, αcko; GSK-3α wt;PDAPP, αwt-APP; and GSK-3α cko;PDAPP, αcko-APP.
Last, to determine whether the abovementioned changes in Aβ IR load corresponded to alterations in motor activity or spatial memory, GSK-3α wt, GSK-3α cko, GSK-3α wt;PDAPP+/−, and GSK-3α cko;PDAPP+/− mice were tested in the open field and Barnes maze. GSK-3α wt;PDAPP+/− mice manifested an increased level of locomotor activity in the open field compared with control groups (Fig. 6C), displayed significant deficits in latency to find the target on the Barnes maze (Fig. 6D), and used a poor maze strategy compared with all three other groups (Fig. 6E). However, GSK-3α cko;PDAPP+/− performed at a level indistinguishable from GSK-3α wt on all three behavioral measures (Fig. 6C–E), suggesting that GSK-3α KO can prevent the locomotor hyperactivity and memory deficits demonstrated in PDAPP mice. Importantly, these results are not confounded by GSK-3α KO as we did not detect any differences between GSK-3α wt and GSK-3α cko mice on locomotor activity or spatial memory. Together, these data suggest that GSK-3α KO can prevent behavioral deficits and reduce Aβ plaque burden in PDAPP+/− mice.
Discussion
To identify the respective roles of both GSK-3α and GSK-3β in modulating formation of AD SPs and NFTs in vivo, we used a novel viral approach and a genetic approach to manipulate the levels of these two GSK-3 isoforms. Importantly, both GSK-3α and GSK-3β underwent successful KD using isoform-specific shRNA rAAV2/1. Moreover, we successfully generated cko of GSK-3α in GSK-3α cko and GSK-3α cko;PDAPP+/− mice as a second approach to further confirm our findings using the viral approach. Based on both approaches, we report that GSK-3α, but not GSK-3β, KD reduced Aβ levels and formation of SPs and that both GSK-3α and GSK-3β KD reduced tau phosphorylation, tau misfolding, and NFT numbers in these tg mice. These results suggest that GSK-3α contributes to both SP and NFT pathogenesis while GSK-3β only modulates NFT formation.
Our study corroborates previous reports demonstrating that GSK-3 may regulate APP metabolism (Sun et al., 2002; Li et al., 2003; Phiel et al., 2003; Ryder et al., 2003; Su et al., 2004; Serenó et al., 2009). However, this is the first study to analyze the impact of manipulating individual GSK-3 isoforms on the formation of SPs and NFTs in aged tg mouse models of these two signature lesions of AD. In accordance with a previous cell-based study (Phiel et al., 2003), our data suggest that inhibition of GSK-3α but not GSK-3β is sufficient to reduce Aβ levels and plaque load, suggesting differential roles of GSK-3α and GSK-3β in Aβ production, accumulation, and SP formation. In contrast, a recent study failed to report any changes in Aβ 40 and Aβ 42 levels after 3 weeks of AAV-APP injection in GSK-3α cko mouse brains (Jaworski et al., 2011). However, this difference is most likely due to technical issues such as the different levels of APP expression or the drastically shorter treatment term (3 weeks) compared with 11 months in our study. Further studies are necessary to elucidate the mechanism of GSK-3α in Aβ and APP metabolism.
Currently, it is unclear if inhibition of GSK-3 directly regulates APP proteolysis or indirectly through interaction with an unidentified protein. Our results are consistent with previous reports demonstrating no change in APP (Ryder et al., 2003; Jaworski et al., 2011) or phospho-APP levels (Plattner et al., 2006; Jaworski et al., 2011) after GSK-3 reduction. Furthermore, GSK-3 inhibition could lead to the accumulation and increased activity of various GSK-3 phosphorylation substrates including β-catenin, CREB, c-Myc, NFAT, and Notch (Jope and Johnson, 2004; Rayasam et al., 2009), that may activate an array of genes and modulate APP metabolism. Importantly, this effect may have the greatest impact in brain regions specifically relevant to AD such as the hippocampus where GSK-3 is highly expressed and active in resting neuronal and glia cells (Yao et al., 2002). Our study and others highlight the importance of further effort aimed at uncovering the mechanism behind GSK-3 regulation of APP and Aβ processing.
Our data on tau phosphorylation by GSK-3α and GSK-3β corroborate previous findings that both isoforms contribute to normal and pathological tau phosphorylation (Ishiguro et al., 1988, 1993; Pérez et al., 2003; Noble et al., 2005; Serenó et al., 2009). Other isoform-specific studies in vivo have shown that overexpression of GSK-3β increases phosphorylation of tau (Spittaels et al., 2000; Lucas et al., 2001) while overexpression of a dominant-negative form of GSK-3β (Gómez-Sintes et al., 2007) or KO of GSK-3α (Alon et al., 2011) decreases endogenous tau phosphorylation. Our results strengthen these conclusions by revealing that targeting either GSK-3 isoform is sufficient to reduce levels of pathological phospho-tau species linked to NFT formation and conformational changes associated with formation of NFTs in a double tg mice model of AD. While our results are largely consistent with previous studies, it is important to note that this study is the first to separately assess the impact of individual GSK-3α and GSK-3β isoforms in NFT pathology.
The use of a viral injection model and a conditional KO approach in AD tg mice in the present study offers significant strengths and advantages over previous studies. For example, we avoided the use of GSK-3 inhibitors which act indiscriminately on both GSK-3 isoforms and other kinases or cellular processes and therefore may confound data interpretation. By individually targeting GSK-3α and GSK-β isoforms, we circumvented the broad and poorly understood targets of kinase inhibitors and instead focused on one kinase and one isoform at a time. This allowed us to confirm and extend our previous conclusion that selectively silencing GSK-3α reduces Aβ plaque load as well as the tau tangle burden. Moreover, in addition to demonstrating that silencing GSK-3α with shRNA reduced soluble and insoluble levels of both Aβ 40 and Aβ 42 and plaque load in multiple AD tg lines, we were able to further demonstrate a significant amelioration of cognitive deficits in our GSK-3α cko;PDAPP+/− mice. Thus, the utilization of two distinct experimental approaches strengthens our conclusion that selectively silencing GSK-3α reduces the Aβ plaque load.
However, one significant limitation of this study was the variability of KD efficiency between GSK-3 α and GSK-3β isoforms. Specifically, GSK-3β protein levels were only reduced by ∼50% compared with∼70% for GSK-3α. In an attempt to achieve higher GSK-3β KD, we attempted to generate a triple tg mouse (GSK-3βflox/flox;α CaMKII-Cre+/−;PDAPP+/−). However, this cross failed to generate viable offspring that survived beyond 6 months of age (9 of 11 mice died by 6 months) similar to a previous report (Jaworski et al., 2011). Therefore, we were limited to achieving 50% GSK-3β KD using our shRNA approach. Although this GSK-3β KD efficiency was sufficient to confirm the effects of this in the behavioral bioassay (forced swim test, data not shown) and to quantify changes in tau phosphorylation, it was not sufficient to observe any effects on Aβ. Therefore, we cannot rule out the possibility that full GSK-3β KD could influence APP proteolysis in vivo and future studies using conditional, inducible, brain-specific KO mice is required to address this issue.
Similarly, this study was complicated by the variability of GSK-3 KD efficiency between tg mouse genotypes. Using the viral approach, GSK-3 was sufficiently silenced in wt and PDAPP+/− mice; however, the KD in PS19+/−;PDAPP+/− mice was ∼40% lower. All genotypes were injected at the same time and under the same conditions by one of two researchers. However, reducing protein levels by injection of rAAV2 containing shRNA is fraught with several technical problems such as the reproducibility of the injections and virus stability. Alternatively, this difference may have been caused by differential tolerability of the virus and mortality as PS19+/−;PDAPP+/− mice displayed a higher mortality rate compared with other lines (χ(3)2 = 34.24, p < 0.0001) regardless of the shRNA injection. Therefore it is possible that mice with lower GSK-3 KD preferentially survived. Despite this issue, bigenic PS19+/−;PDAPP+/− mice displayed a more profound reduction in Aβ plaques after GSK-3 KD compared with monogenic PDAPP+/− with greater KD efficiency, suggesting that other factors contribute to reducing Aβ pathology in these mice.
In terms of mortality, we did observe an unexpected shortened life span of PS19+/−;PDAPP+/− and PDAPP+/− mice treated with shRNA-α (data not shown). This effect has not been observed in other GSK-3α KD models (MacAulay et al., 2007; Jaworski et al., 2011), our GSK-3α cko mice (χ(31,26)2 = 0.8387, p = 0.3598), or our GSK-3α cko;PDAPP mice (χ(20,31)2 = 2.317, p = 0.1279), suggesting that it might be an artifact or technical issue of our viral injection paradigm. We speculate that abruptly reducing GSK-3α levels at the 12 h postnatal developmental stage when critical neocortical regions are still under development (Rice et al., 1985) may contribute to additional deleterious brain abnormalities separate from transgene expression of APP and tau, as supported by recent studies (Kaidanovich-Beilin et al., 2009; Kim et al., 2009).
In conclusion, our study highlights the importance of GSK-3α in the formation of Aβ plaques as well as the role both GSK-3α and GSK-3β play in the pathological development of NFTs formed by pathological tau. Our observations demonstrate that reducing GSK-3α expression ameliorates the key pathological signatures of AD, i.e., SPs and NFTs, and improves AD-like cognitive impairment in our mouse models. These results have important implications for future design of therapeutics that may preferentially target GSK-3α over GSK-3β. However, before therapeutic development can proceed in a fully informed manner, the differential roles of GSK-3 isoforms in various cellular mechanisms need to be delineated to fully appreciate such specific targeting. The broad functions of both GSK-3 isoforms allow them to be potential drug targets in a number of diseases including cancer, bipolar mood disorder, diabetes, and AD (Jope and Johnson, 2004). Through the use of cko models and RNAi techniques, we may begin to understand the critical functions of both isoforms in the pathogenesis of a variety of diseases.
Footnotes
This study is supported by National Institutes of Health grants AG11542, AG17586, T32-GM07229, and T32-AG000255, the Marian S. Ware Alzheimer Program, the Fundación Caja Madrid, and Instituto de Salud Carlos III. We thank Jon Toledo, Amanda Piarulli, Brigid Jensen, Erin Coffey, and Stylianos Monos for technical and statistical assistance.
The author(s) declare(s) no competing financial interests.
References
- Akiyama H, Shin RW, Uchida C, Kitamoto T, Uchida T. Pin1 promotes production of Alzheimer's amyloid beta from beta-cleaved amyloid precursor protein. Biochem Biophys Res Commun. 2005;336:521–529. doi: 10.1016/j.bbrc.2005.08.130. [DOI] [PubMed] [Google Scholar]
- Alon LT, Pietrokovski S, Barkan S, Avrahami L, Kaidanovich-Beilin O, Woodgett JR, Barnea A, Eldar-Finkelman H. Selective loss of glycogen synthase kinase-3alpha in birds reveals distinct roles for GSK-3 isozymes in tau phosphorylation. FEBS Lett. 2011;585:1158–1162. doi: 10.1016/j.febslet.2011.03.025. [DOI] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Holmes A. Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic- and antidepressant-like effects in mice. Neuropsychopharmacology. 2006;31:2405–2414. doi: 10.1038/sj.npp.1301039. [DOI] [PubMed] [Google Scholar]
- Brunden KR, Zhang B, Carroll J, Yao Y, Potuzak JS, Hogan AM, Iba M, James MJ, Xie SX, Ballatore C, Smith AB, 3rd, Lee VM, Trojanowski JQ. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci. 2010;30:13861–13866. doi: 10.1523/JNEUROSCI.3059-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JC, Boyce-Rustay JM, Millstein R, Yang R, Wiedholz LM, Murphy DL, Holmes A. Effects of mild early life stress on abnormal emotion-related behaviors in 5-HTT knockout mice. Behav Genet. 2007;37:214–222. doi: 10.1007/s10519-006-9129-9. [DOI] [PubMed] [Google Scholar]
- Carroll JC, Iba M, Bangasser DA, Valentino RJ, James MJ, Brunden KR, Lee VM, Trojanowski JQ. Chronic stress exacerbates tau pathology, neurodegeneration, and cognitive performance through a corticotropin-releasing factor receptor-dependent mechanism in a transgenic mouse model of tauopathy. J Neurosci. 2011;31:14436–14449. doi: 10.1523/JNEUROSCI.3836-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Gao G, Lu Y, Calcedo R, Grant RL, Bell P, Wang L, Figueredo J, Lock M, Wilson JM. Biology of AAV serotype vectors in liver-directed gene transfer to nonhuman primates. Mol Ther. 2006;13:77–87. doi: 10.1016/j.ymthe.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Giese KP. GSK-3: a key player in neurodegeneration and memory. IUBMB Life. 2009;61:516–521. doi: 10.1002/iub.187. [DOI] [PubMed] [Google Scholar]
- Gómez-Sintes R, Hernández F, Bortolozzi A, Artigas F, Avila J, Zaratin P, Gotteland JP, Lucas JJ. Neuronal apoptosis and reversible motor deficit in dominant-negative GSK-3 conditional transgenic mice. EMBO J. 2007;26:2743–2754. doi: 10.1038/sj.emboj.7601725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Hurtado DE, Molina-Porcel L, Iba M, Aboagye AK, Paul SM, Trojanowski JQ, Lee VM. A{beta} accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am J Pathol. 2010;177:1977–1988. doi: 10.2353/ajpath.2010.100346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro K, Ihara Y, Uchida T, Imahori K. A novel tubulin-dependent protein kinase forming a paired helical filament epitope on tau. J Biochem. 1988;104:319–321. doi: 10.1093/oxfordjournals.jbchem.a122465. [DOI] [PubMed] [Google Scholar]
- Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993;325:167–172. doi: 10.1016/0014-5793(93)81066-9. [DOI] [PubMed] [Google Scholar]
- Jaworski T, Dewachter I, Lechat B, Gees M, Kremer A, Demedts D, Borghgraef P, Devijver H, Kügler S, Patel S, Woodgett JR, Van Leuven F. GSK-3alpha/beta kinases and amyloid production in vivo. Nature. 2011;480:E4–E5. doi: 10.1038/nature10615. discussion E6. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Lipina TV, Takao K, van Eede M, Hattori S, Laliberté C, Khan M, Okamoto K, Chambers JW, Fletcher PJ, MacAulay K, Doble BW, Henkelman M, Miyakawa T, Roder J, Woodgett JR. Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol Brain. 2009;2:35. doi: 10.1186/1756-6606-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Lee JE, Kim MJ, Cho EG, Cho SG, Choi EJ. Glycogen synthase kinase 3 beta is a natural activator of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase 1 (MEKK1) J Biol Chem. 2003;278:13995–14001. doi: 10.1074/jbc.M300253200. [DOI] [PubMed] [Google Scholar]
- Kim WY, Wang X, Wu Y, Doble BW, Patel S, Woodgett JR, Snider WD. GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci. 2009;12:1390–1397. doi: 10.1038/nn.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Skovronsky DM, Abtahian F, Doms RW, Lee VM. Secretion and intracellular generation of truncated Abeta in beta-site amyloid-beta precursor protein-cleaving enzyme expressing human neurons. J Biol Chem. 2003;278:4458–4466. doi: 10.1074/jbc.M210105200. [DOI] [PubMed] [Google Scholar]
- Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. 2006;281:4292–4299. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, Lee VM. BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J Cell Biol. 2005;168:291–302. doi: 10.1083/jcb.200407070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Ryder J, Su Y, Zhou Y, Liu F, Ni B. FRAT1 peptide decreases Abeta production in swAPP(751) cells. FEBS Lett. 2003;553:347–350. doi: 10.1016/s0014-5793(03)01042-1. [DOI] [PubMed] [Google Scholar]
- Li J, Daly TM. Adeno-associated virus-mediated gene transfer to the neonatal brain. Methods. 2002;28:203–207. doi: 10.1016/s1046-2023(02)00224-4. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernández F, Gómez-Ramos P, Morán MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007;6:329–337. doi: 10.1016/j.cmet.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary TP, Savoie V, Brown RE. Learning, memory and search strategies of inbred mouse strains with different visual abilities in the Barnes maze. Behav Brain Res. 2011;216:531–542. doi: 10.1016/j.bbr.2010.08.030. [DOI] [PubMed] [Google Scholar]
- Passini MA, Wolfe JH. Widespread gene delivery and structure-specific patterns of expression in the brain after intraventricular injections of neonatal mice with an adeno-associated virus vector. J Virol. 2001;75:12382–12392. doi: 10.1128/JVI.75.24.12382-12392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez M, Hernández F, Lim F, Díaz-Nido J, Avila J. Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J Alzheimers Dis. 2003;5:301–308. doi: 10.3233/jad-2003-5405. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Plattner F, Angelo M, Giese KP. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem. 2006;281:25457–25465. doi: 10.1074/jbc.M603469200. [DOI] [PubMed] [Google Scholar]
- Rayasam GV, Tulasi VK, Sodhi R, Davis JA, Ray A. Glycogen synthase kinase 3: more than a namesake. Br J Pharmacol. 2009;156:885–898. doi: 10.1111/j.1476-5381.2008.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice FL, Gomez C, Barstow C, Burnet A, Sands P. A comparative analysis of the development of the primary somatosensory cortex: interspecies similarities during barrel and laminar development. J Comp Neurol. 1985;236:477–495. doi: 10.1002/cne.902360405. [DOI] [PubMed] [Google Scholar]
- Ryan KA, Pimplikar SW. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J Cell Biol. 2005;171:327–335. doi: 10.1083/jcb.200505078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder J, Su Y, Liu F, Li B, Zhou Y, Ni B. Divergent roles of GSK3 and CDK5 in APP processing. Biochem Biophys Res Commun. 2003;312:922–929. doi: 10.1016/j.bbrc.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Serenó L, Coma M, Rodríguez M, Sánchez-Ferrer P, Sánchez MB, Gich I, Agulló JM, Pérez M, Avila J, Guardia-Laguarta C, Clarimón J, Lleó A, Gómez-Isla T. A novel GSK-3beta inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359–367. doi: 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- Sledz CA, Williams BR. RNA interference in biology and disease. Blood. 2005;106:787–794. doi: 10.1182/blood-2004-12-4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR. Activation of the interferon system by short-interfering RNAs. Nat Cell Biol. 2003;5:834–839. doi: 10.1038/ncb1038. [DOI] [PubMed] [Google Scholar]
- Spittaels K, Van den Haute C, Van Dorpe J, Geerts H, Mercken M, Bruynseels K, Lasrado R, Vandezande K, Laenen I, Boon T, Van Lint J, Vandenheede J, Moechars D, Loos R, Van Leuven F. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. 2000;275:41340–41349. doi: 10.1074/jbc.M006219200. [DOI] [PubMed] [Google Scholar]
- Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P, Brune K, Paul S, Zhou Y, Liu F, Ni B. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry. 2004;43:6899–6908. doi: 10.1021/bi035627j. [DOI] [PubMed] [Google Scholar]
- Sun X, Sato S, Murayama O, Murayama M, Park JM, Yamaguchi H, Takashima A. Lithium inhibits amyloid secretion in COS7 cells transfected with amyloid precursor protein C100. Neurosci Lett. 2002;321:61–64. doi: 10.1016/s0304-3940(01)02583-6. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao HB, Shaw PC, Wong CC, Wan DC. Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J Chem Neuroanat. 2002;23:291–297. doi: 10.1016/s0891-0618(02)00014-5. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]