

Abstract
The loss of glomerular visceral epithelial cells (podocytes) has been associated with the development of glomerular sclerosis and loss of renal function. Viability of podocytes recovered from urine of subjects with glomerular disease and of healthy controls was investigated by propidium iodide exclusion and TUNEL staining. Podocyte loss was quantified by cytospin. The growth behavior in culture of urinary cells and their expression of specific markers were examined. The majority of urinary podocytes are viable, although apoptosis occurs in about one-half of the cells. Patients with active glomerular disease excreted up to 388 podocytes/mg creatinine, whereas healthy controls and patients with quiescent disease generally excreted <0.5 podocytes/mg creatinine. The identity of cultured cells was confirmed by their morphology, growth behavior, and expression of podocyte-specific markers. The difference in growth behavior between healthy controls and subjects with active glomerular disease suggests that in active disease viable podocytes detach from the glomerular tuft due to local environmental factors rather than defects in the podocytes per se, whereas in healthy individuals mostly senescent podocytes are shed.
Keywords: glomerulus, cell culture, podocalyxin, cytospin
Podocytes are highly specialized epithelial cells lining the urinary surface of the glomerular capillary tuft. They are part of the filtration barrier together with capillary endothelial cells and the glomerular basement membrane (GBM), ensuring the selective permeability of the glomerular capillary wall (2). Podocytes furthermore synthesize components of the GBM and provide structural stability to the glomerular tuft (10). Their foot processes have a well-developed contractile apparatus and adhere to the GBM at focal contacts via integrins and dystroglycan complexes (3, 8).
Recent observations have revealed that the development of glomerular sclerosis in several human and experimental diseases is associated with a decrease in the number or density of podocytes in the glomerular capillary tuft (4, 9, 13, 14, 20). During glomerular injury, podocytes retract and broaden their foot processes and may detach from the GBM. As a consequence of podocyte loss, the remaining podocytes may fail to completely cover the outer surface of the GBM. As a result, parietal epithelial cells of Bowman’s capsule may gain access to bare areas of the GBM, forming adhesions and leading to segmental glomerular sclerosis (11, 12).
Although the above-mentioned clinical studies (13, 14, 20) support the model of podocyte loss, they have not addressed the mechanism by which podocytes disappear from the glomerular tuft. Hara, Nakamura, and colleagues (5, 6, 17, 18) previously described podocalyxin-positive cells in urinary sediments of patients with a variety of kidney diseases, including lupus nephritis. These authors showed that changes in semi-quantitative measures of podocyturia seem to correlate with disease activity as assessed by biopsy and to decline with treatment (17, 18), suggesting that urinary shedding of podocytes may represent a real-time measure of podocyte loss from the glomerulus.
However, it remains unclear whether podocytes are shed because they undergo necrosis or apoptosis in situ, or if they are still viable when they detach from the GBM. It has also not been resolved definitively whether podocytes are lost into the urine of healthy individuals, although this has not been reported to occur in previous studies (5, 6, 16–18). To address these problems, we examined the viability and growth characteristics of cells excreted into the urine of healthy controls and patients with glomerular disease. We also examined the expression of specific markers in viable urinary cells to establish their identity more definitely than with podocalyxin alone.
MATERIALS AND METHODS
Reagents
Rabbit polyclonal anti-WT1 and goat polyclonal anti-CD68 antibodies were both obtained from Santa Cruz Biotechnology, Santa Cruz, CA. Mouse monoclonal antibody to cytokeratin 8 was purchased from Sigma, St. Louis, MO; mouse monoclonal anti-human podocalyxin (PHM5) was from Chemicon (Boronia, Victoria, Australia). Rabbit polyclonal antipodocalyxin was kindly provided by Dr. M. Farquhar, Department of Cellular and Molecular Medicine, University of California San Diego, La Jolla, CA; mouse monoclonal antibody to glomerular epithelial protein 1 (GLEPP1) by Dr. R. Wiggins, University of Michigan, Ann Arbor, MI; and rabbit polyclonal anti-podocin by Dr. C. Antignac, Necker Hospital, Paris, France. Mouse monoclonal anti-syn-aptopodin was purchased from Progen, Heidelberg, Germany; mouse monoclonal anti-P-cadherin from BD Transduction Laboratories, Lexington, KY; and mouse monoclonal anti-α-smooth muscle actin from Sigma. Mouse monoclonal anti-CD 31 was purchased from DAKO Denmark; rabbit polyclonal anti-ZO1 from Zymed, San Francisco, CA; and mouse monoclonal anti-human PGP9.5 from Biomeda, Foster City, CA. In addition, phalloidin-rhodamine conjugate was used to stain for actin (Sigma) and Hoechst nuclear stain (Sigma) was used for nuclei. The rat monoclonal antibody used to detect E-cadherin (DECMA-1) was a gift from Dr. Rolf Kemmler, Max-Planck-Institut, Freiburg, Germany. Positive controls consisted of human podocytes cultured from isolated glomeruli (see below), whereas negative controls were Madin-Darby canine kidney tubule epithelial cells.
Quantitation of podocytes in cytospin of urine sediments using anti-podocalyxin antibody
Urine specimens were divided for use in cell counting by cytospin and for cell culture. Podocytes in urine sediments were quantified according to the method of Hara and colleagues (5) with some modifications. Freshly voided urine (20–100 ml) was centrifuged at 700 g for 5 min. The supernatant was carefully aspirated and the sediment pellet was washed two times with human diploid fibroblast (HDF) solution (distilled water, 137 mM NaCl, 5 mM KCl, 5.5 mM glucose, 4 mM NaHCO3, and 0.2% EDTA). The pellet was then resuspended in 1 ml of HDF. Aliquots of 100 μl of the resuspended sediment were then centrifuged onto polylysine-coated slides using a Wescor Cytopro 7620 cytocentrifuge (Wescor, Logan, UT). The slides were air-dried and then fixed with 3% paraformaldehyde at room temperature for 15 min and permeabilized using Triton X-100. After washing with PBS, the slides were incubated with blocking buffer (PBS containing 0.2% BSA, 50 mM NH4Cl, and 1% goat serum) overnight at 4°C.
The slides were then incubated with anti-human podocalyxin monoclonal antibody (PHM5) at a dilution of 1:100 for 60 min. After being washed with PBS, secondary antibody (FITC-labeled goat anti-mouse IgG) was added at a dilution of 1:100 for 30 min. The sediments were counterstained with Hoechst nuclear stain to distinguish whole cells from cell fragments. Coverslips were mounted with Vectashield (Vector Labs, Burlington, CA) and the slides were viewed with an Axioplan fluorescence microscope (Zeiss, Oberkochen, Germany) at 488 nm for podocalyxin (FITC) and 460 nm for the Hoechst nuclear stain. Nucleated, podocalyxin-positive cells were considered to be podocytes (6). Two to four cytospin slides were examined per patient sample.
Staining with propidium iodide to determine viability and staining with TdT-mediated dUTP nick-end labeling to determine apoptotic cells
Staining for cell viability was performed on specimens prepared for cytospin by adding pro-pidium iodide (1 μM/ml) to urine aliquots before they were cytospun onto slides for podocalyxin antibody staining. TdT-mediated dUTP nick-end-labeling (TUNEL) staining for apoptosis was also performed on specimens prepared by cytospin. The TUNEL assay was performed using a standard rhodamine incorporation kit according to package instructions (Intergen, Purchase, NY). Briefly, urinary sediment cytospun onto a polylysine slide was fixed, blocked, and stained with podocalyxin as described above. After equilibration with buffer for 10 s at room temperature, ice-cold working-strength TdT enzyme was added. The preparation was incubated at 37°C for 60 min in a dark chamber. The reaction was terminated by addition of stop-and-wash buffer for 10 min at room temperature, followed by incubation with rhodamine solution for 30 min at room temperature. The slide was washed three times with PBS and then counterstained with Hoechst stain for 2 min. After being washed with water, coverslips were mounted with Vectashield and the slide was viewed in the fluorescence microscope at 590 nm for rhodamine and 460 nm for Hoechst. Apoptosis was characterized by positive TUNEL staining.
Culture of urine sediments
Cells from urine were harvested as follows: freshly voided urine was centrifuged at 700 g for 5 min. The supernatant was carefully aspirated and the pellets were resuspended in sterile HDF. The suspension was gently mixed and centrifuged two more times. The pellet was taken up in DMEM F-12 medium with 10% FBS supplemented with antibiotics (0.5 U/l penicillin, 0.5 mg/dl streptomycin, 1 mg/ml kanamycin) and cultured in Falcon tissue flasks at 37°C in 5% CO2. Medium was changed three times per week. The number of colonies in the tissue flasks was counted every 2–3 days over the first 4 wk. The maximum number of colonies was recorded.
Culture of glomerular epithelial cells
To generate human glomerular epithelial cells from a nonurinary source, glomeruli were isolated by sieving from unaffected poles of a tumor-bearing human nephrectomy specimen as previously described (15) and were cultured for 7 days. The outgrowing epithelial cells were trypsinized and passed through sieves with a 100-μm pore size. These cells were then cultured under conditions identical to those described above for cells isolated from urine sediments.
Immunofluorescence studies
Immunofluorescence studies were performed on cultured cells that had been trypsinized and grown on collagen-coated coverslips for 1–5 wk. Cells were fixed in 3% paraformaldehyde at room temperature for 15 min and permeabilized for 3 min with cytoskeleton extraction buffer (CSK; 50 mM NaCl; 300 mM sucrose; 10 mM PIPES, pH 6.8; 3 mM MgCl2; 0.5% Triton X-100). After fixation, samples were washed in PBS and incubated overnight at 4°C in blocking solution. Samples were incubated with primary antibodies at dilutions from 1:20 to 1:200 in PBS and 0.2% BSA for 1 h at room temperature. After incubation and washing with PBS and 0.2% BSA, they were incubated for 1 h with rhodamine- or FITC-conjugated goat anti-rabbit, goat anti-mouse, or donkey anti-goat secondary antibodies at 1:50 to 1:100 dilutions. The samples were washed in PBS and mounted in Vectashield before being analyzed by immunofluorescence microscopy.
Experimental subjects
We examined the urine of a group of 41 individuals composed of 14 patients with glomerular disease and 27 healthy subjects, who served as controls. The control subjects ranged in age from 17 to 87 yr (median 38 yr). There were 13 men and 14 women among the controls. The patients were five children with focal segmental glomerulosclerosis (FSGS) and nine children with lupus nephritis. Their ages ranged from 7 to 19 yr (median 12 yr). We distinguished active disease from quiescent disease in these patients by the presence of an albumin/creatinine ratio >300 μg/mg, which corresponds to dipstick-positive proteinuria, in patients with active disease.
These studies were approved by the local human subjects board, and written informed consent was obtained from all study participants and (if minors) their parents.
Laboratory analysis
An aliquot of the urine was saved for determination of albumin and creatinine concentrations. Concentrations were determined in duplicate (albumin: Beckman Array 360; creatinine: Beckman creatinine analyzer 2, Fullerton, CA). The coefficients of variation of the assays are 3.0 and 4.2%, respectively.
To more accurately quantify the rate of urinary podocyte excretion on these untimed urine samples, the number of podocytes in the sample was expressed as a ratio to the urinary creatinine content of that sample. Urine pH was determined by pH indicator strips (colorpHast, EM Science, Gobbstown, NJ).
Data analysis
Each determination of quantitative urinary podocyte excretion from a single specimen was based on the mean of values of the two to four cytospin slides. Results are reported as median (range) or mean. Values of podocyturia are plotted as box plots, due to the nonnormal distribution of the values. Differences between two groups were analyzed by two-sample Kolmogorov-Smirnov and Mann-Whitney tests. Differences among more than two groups were analyzed by Dunnett’s T3 test. P values <0.05 were considered significant.
RESULTS
Quantitation, identification, and viability of podocytes in cytospin urine sediments
Podocytes in cytospin urine sediments were recognized as either nucleated or multinucleated cells expressing podocalyxin by immunofluorescence microscopy. Eighty-eight percent of urine samples from patients with glomerular disease and 42% of samples from healthy controls had podocalyxin-positive cells present on cytospin. The median value of podocyturia per sample among the patients was 10.9 podocytes/mg creatinine (n = 51, range 0–388 podocytes/mg creatinine, mean 53.4) and 0 podocytes/mg creatinine for healthy controls (n = 40, range 0–5.3 podocytes/mg creatinine, mean 0.32). All but one of the control samples had a podocyte content of <0.5 podocytes/mg creatinine. Among the control subjects, podocyturia did not differ between the older and younger subjects (<38 yr, median 0.025 vs. 38 yr, median 0; P = 0.98 by 2-sample Kolmogorov-Smirnov test). Ninety-three percent of patients with glomerular disease and 44% of healthy controls had podocalyxin-positive cells in at least one urine sample.
When the patient samples were divided into those from patients with active disease and those with inactive disease, it was clear that both inactive lupus nephritis and inactive FSGS were indistinguishable from control values of podocyturia (medians 0.17, 0.47, and 0 podocytes/mg creatinine, respectively, P > 0.94). There was only a trend toward a higher level of podocyturia in active lupus nephritis than in active FSGS (74.8 vs. 19 podocytes/mg creatinine, P = 0.14; Fig. 1).
Fig. 1.

Podocyte excretion expressed as podocytes/mg creatinine. Comparison between urine specimens from healthy individuals and patients with glomerular disease is shown. Five groups are expressed as box plots: A, control subjects; B, inactive focal segmental glomerulosclerosis (FSGS); C, active FSGS; D, inactive lupus nephritis; E, active lupus nephritis. Median values (podocytes/mg creatinine) for the groups (horizontal line within box): A, 0; B, 0.47; C, 19; D, 0.17; E, 74.8.
Fewer than 1% of the nucleated cells in the urinary sediment showed positive podocalyxin staining (Fig. 2A). Cytospin sediments were also stained for other podocyte-specific markers including synaptopodin, GLEPP1, podocin, and WT1. Approximately 30–40% of samples positive for podocalyxin also stained for synaptopodin, GLEPP1, or podocin. Staining for WT1 was not found in any of the sediments. Many cells in the sediments stained for the epithelial cell marker cytokeratin 8; none of these cells stained for podocalyxin. The most inclusive marker for podocytes was podocalyxin. Virtually no cells that stained for another podocyte marker failed to stain for podocalyxin. Binucleate podocytes were fairly common in patients (10–50% of podocalyxin-positive, nucleated cells) but were not seen among controls.
Fig. 2.

Urine sediment cytospun onto polylysine-coated slide and stained with antipodocalyxin antibody, Hoechst nuclear stain, and propidium iodide. A: podocalyxin-positive cell (binucleate) with podocalyxin in green and nuclear stain in blue. B: podocalyxin-positive cell (green), surrounded by podocalyxin-negative cells (visible by Hoechst nuclear stain in blue). C: propidium iodide (red) stains nuclei of the podocalyxin-negative cells only, indicating lack of viability. D: merged image with anti-podocalyxin staining. Scale bar 10 μm.
Viability of podocalyxin-positive cells on cytospin was determined by propidium iodide exclusion. Results from cytospin showed that most of the podocytes in the urine of patients and healthy controls were viable (Fig. 2, B–D). Seventy-five percent of samples from patients with glomerular disease (n = 20) and 100% of controls had viable podocytes in their urine sediment (n = 9). The viability of urinary podocytes did not seem to be influenced by the pH of the urine (Fig. 3). In some cytospin samples, the viability of cytokeratin 8-positive cells was also assessed, but the vast majority of these cells were not viable by propidium iodide exclusion.
Fig. 3.

Podocyte viability [percentage of podocalyxin-positive cells that were propidium iodide (PI) negative] as a function of urinary pH in healthy controls (△) and patients with glomerular disease (■). Viability is not influenced by the pH of the urine.
TUNEL staining of podocalyxin-positive cells in the sediments confirmed the presence of apoptotic podocytes in the urine of both patients and controls. Figure 4 shows three podocytes identified on cytospin with anti-podocalyxin antibody. Two of them are viable and one is apoptotic. Occasional fragmented podocyte nuclei were also observed with the Hoechst stain. We identified apoptotic podocytes in urine sediments of many patients with kidney disease as well as in healthy controls, although about one-half of urinary podocytes did not show evidence of apoptosis.
Fig. 4.
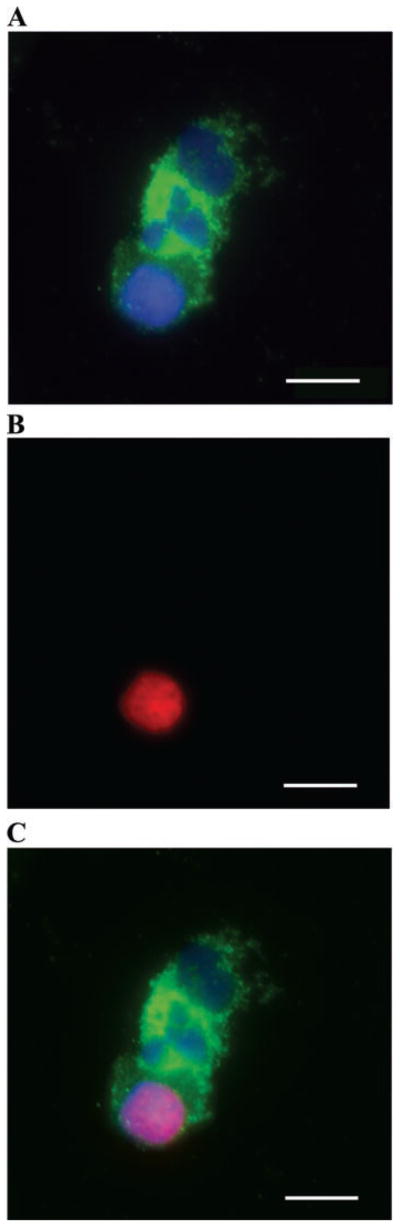
TdT-mediated dUTP-nick-end labeling (TUNEL) staining of podocalyxin-positive cells in sediment confirmed the presence of apoptotic podocytes in urine of both patients and controls. Three podocytes, identified on cytospin with anti-podocalyxin antibody. Two of them are nonapoptotic and one is apoptotic. Antipodocalyxin staining is in green, nuclear stain is in blue, and TUNEL staining is in red. A: urine sediment stained with anti-podocalyxin antibody and Hoechst nuclear stain. B: TUNEL staining with 1 TUNEL-positive cell. C: merged image. Scale bar 10 μm.
Relationship of podocyturia to albuminuria
Because albumin-to-creatinine ratios were also measured in the patient urine samples, we were able to investigate the relationship between podocyturia and albuminuria. There was no simple relationship between these indexes. In fact, among the patients with active disease, rather than being positively correlated, podocyturia tended to be greatest in the patients with the lowest levels of albuminuria (Fig. 5).
Fig. 5.

Relationship of podocyturia to albuminuria in patients with active FSGS (■) and lupus nephritis (□). Active disease is defined as presence of an albumin-to-creatinine ratio >300 μg/mg. Notice the absence of a simple positive relationship of podocyturia to albuminuria.
Establishment and characterization of primary cultures of podocytes from urine
Viable cells were cultured from the sediments of clean-catch urine specimens. On plating of urine sediments of the patients, cultured urinary cells often started to grow rapidly into large colonies with a densely packed, cobblestone morphology similar to that of podocytes recovered from explanted glomeruli. Figure 6A shows a colony in early stages of growth from a patient with glomerular disease. Figure 6B shows cells grown out from isolated glomeruli after 14 days in culture.
Fig. 6.

A, C, E: cells cultured from sediments of patient urine. B, D, F: podocytes grown out of explanted glomeruli isolated from healthy parts of a kidney removed for localized tumor. A and B: early dense, cobblestone-like pattern. C and D: cultured cells undergoing crisis. E and F: differentiated podocytes. Comparison between cultured sediments and outgrowths shows substantial similarity. G and H: urinary podocytes with primary and secondary processes. Scale bar 100 μm.
After growing for 1–2 wk, podocytes grown from urine usually underwent “crisis” (Fig. 6, C and D), during which many of the cells underwent apoptosis. The remaining cells developed primary and secondary cell processes (Fig. 6E) similar to those in glomerular epithelial cell cultures (Fig. 6F). Sometimes, the urinary cells that remained after crisis grew as small clusters of very large, often multinucleated cells. Some of these cells also developed long secondary or even tertiary processes (Fig. 6, G and H).
To test the influence of serum on cell growth and morphology, growth medium was supplemented with 5.0% FBS. Neither overt phenotype nor growth rate of cells was altered and, therefore, subsequent experiments included 10% FBS in the growth medium. Cells were routinely plated on plastic, because plating on collagen (types I and IV), fibronectin, or laminin had no apparent influence on cell growth. To address the influence of plating density on growth behavior, subcultured cells were grown at low (20,000 cells/ml) or high density (200,000 cells/ml), but again no influence on cell growth characteristics was evident.
Cultured cells grown on coverslips were stained with antibodies to the podocyte markers podocalyxin, synaptopodin, podocin, WT1, P-cadherin, and GLEPP1. To identify cells of other lineages, cultured urinary cells were also stained with antibodies to markers of tubule epithelial cells (E-cadherin and cytokeratin 8), mesangial cells (α-smooth muscle actin), endothelial cells (CD31), macrophages (CD68), and glomerular parietal epithelial cells (PGP9.5). Cultured cells did not stain for the tubule cell marker E-cadherin either during the period of rapid proliferation or in the confluent cobblestone phase. Some cells in culture expressed cytokeratin 8, but the same cells also expressed WT1 or podocin. A small number of cells (<1/200) in some older cultures stained for α-smooth muscle actin, but these cells also expressed WT1. Cultured cells did not stain for CD31, CD68, or PGP 9.5.
Cultured cells showed expression of all podocyte marker proteins examined except GLEPP1. A nuclear staining pattern of WT1 was observed. The majority of cells expressed WT1 at some point in culture (Fig. 7D), but WT1 staining tended to disappear within a few weeks. The synaptopodin staining pattern was similar to that of actin-containing stress fibers and colocalized with actin as stained by phalloidin (Fig. 7, A–C). In most cases, the majority of cells expressed synaptopodin after they had grown in culture for 21–28 days and had undergone crisis. Staining for P-cadherin, which colocalized with ZO-1 (Fig. 7, E–G), was present in cells in early phases of growth, when the cells had a cobblestone-like morphology. In more differentiated cells, ZO-1 was concentrated at sites of cell-cell contact (Fig. 7H). Cells also expressed podocin, which showed either cell-cell contact or netlike filamentous distributions (Fig. 7, I and J), as has been described before in cultures of immortalized human podocytes (21). Podocalyxin stained the apical membrane or filopodia of cultured podocytes (Fig. 7, K and L), but levels of expression declined as cells continued to grow in culture.
Fig. 7.

Immunofluorescence of cultured urinary cells grown on collagen-coated coverslips. A: anti-synaptopodin staining (green). B: phalloidin staining (red). C: merged image shows colocalization of synaptopodin and actin. D: podocyte expressing WT1 (red). E: cultured cells expressing P-cadherin at cell-cell contact sites (green). F: cultured, cobblestone-like cells expressing ZO-1 at cell-cell contact sites (red). G: merged image. H: more differentiated podocytes expressing ZO-1 at rudimentary slit diaphragms (red). I: podocin staining in less differentiated cells at cell-cell contact sites (red). J: podocin staining in a filamentous pattern (red). K: podocalyxin staining the apical membrane of cells early in culture (green). L: antipodocalyxin staining of filopodia later in culture (green). Scale bar 10 μm.
Growth behavior in culture of urinary podocytes from healthy individuals and patients with glomerular disease
Primary glomerular epithelial cells were grown in culture from glomeruli isolated from uninvolved parts of a kidney removed for localized tumor. As shown above, these cells had the same morphology as the cells cultured from urine of patients with active glomerular disease and stained for synaptopodin with the same intracellular pattern and for WT1; none stained for CD31 or CD68.
Urinary podocyte viability was confirmed by their ability to grow in culture. Cultured urinary cells often grew rapidly into large colonies with a densely packed, cobblestone morphology that later underwent crisis, resulting in fewer but more differentiated cells (as reflected in their morphology and marker protein expression). This growth pattern resembles that of colonies that grow out of glomerular explants (15) and of an immortalized human podocyte cell line (22). In the early phase of culture, cells stained for podocalyxin, podocin, and P-cadherin. In later phases, in addition to podocin, synaptopodin was a consistent marker, whereas WT1 was variably expressed. These results confirm the identity of the cultured urinary cells as podocytes.
Urine sediments of ~90% of patients and 60% of healthy controls grew podocytes in culture. As described above, cells cultured from urine sediments with active glomerular disease grew rapidly as densely packed colonies (Fig. 6A) and proliferated until they underwent crisis and developed primary and secondary processes. Such cells exhibited a very similar morphology and growth pattern to colonies of podocytes grown from glomerular explants. Podocytes of patients grew to confluent colonies of >50,000 cells. When these cells were trypsinized and replated, they again grew to confluence for up to five passages.
Among the densely growing cobblestone cells, it was common to observe cell processes crossing neighboring cells (Fig. 6, C and D). To investigate the possibility that malignant transformation had occurred in these rapidly proliferating cells, we grew cultured urinary cells suspended in 0.8% agar. The suspended cells did not proliferate when grown in agar, showing that the lack of strict contact inhibition in culture was not a sign of malignant transformation.
Cultures from urine of healthy persons grew smaller colonies (typically 10–100 cells, but always <1,000). The time of active proliferation was shorter and the cells died sooner in culture. Among the small number of control urine cultures that were large enough to attempt subculture, almost none grew after trypsinization and replating. In addition, the urinary sediments of both control subjects and subjects with inactive disease produced significantly smaller numbers of colonies in culture than those of subjects with active disease (median maximal colony values: active disease, 4.34 colonies/mg creatinine; inactive disease, 0.15 colonies/mg creatinine; control, 0 colonies/mg creatinine; active vs. inactive, P = 0.011; active vs. control, P = 0.003).
DISCUSSION
Loss of podocytes appears to play an important role in the progression of glomerular disease and the development of glomerular sclerosis. However, it remains unclear whether podocytes are lost because they undergo necrosis or apoptosis in situ, or if they detach from the GBM when they are still viable. It is also not clear whether podocytes are lost into the urine of healthy individuals.
We found podocytes not only in the urine of patients with glomerular disease but also in the urine of healthy subjects. Podocytes were identified by expression of several specific markers. The most inclusive marker on cytospin was podocalyxin. Cells expressing other podocyte markers (WT1, synaptopodin, podocin) almost invariably expressed podocalyxin, although the converse was not true. With the use of a cytospin method to quantitate podocyte excretion (5), and normalizing podocyte number by the urinary creatinine content, we found that the number of podocytes shed by healthy controls and patients with inactive disease was significantly lower than in patients with active glomerular disease.
The rate of podocyte excretion appears to reflect both disease activity and disease type. Quantitative podocyturia tended to be greater in patients with active lupus nephritis than it was in those with FSGS. Podocyturia was significantly greater in active disease than it was in inactive disease. Among patients with active disease (as defined by a urinary albumin/creatinine ratio >300 μg/mg), urinary albumin excretion and podocyturia did not show a simple positive correlation (Fig. 5). This phenomenon will need to be confirmed in a greater number of patients, including patients with different glomerular diseases. We submit that the quantitative rate of urinary podocyte excretion observed in the present study accords well with the rate at which kidney function can be lost in the corresponding renal diseases. If there are 1.2 million glomeruli in the two kidneys (19) with 300 podocytes/glomerulus (14) and 1,000 mg of urinary creatinine excretion/day, then at an excretion rate of 100 podocytes/mg creatinine, one-half of the initial complement of podocytes would be lost in just 5 yr. This degree of podocyte loss would probably lead to global sclerosis in most of the glomeruli. A comparable time course of development of renal failure can be seen in severe cases of lupus nephritis or FSGS.
We showed that most of the podocytes in urine sediments of both healthy controls and patients with glomerular disease were viable, as judged by propidium iodide exclusion. Negative TUNEL staining confirmed that podocytes in urine sediment are not all undergoing apoptosis, although apoptosis was present in about one-half of the urinary podocytes examined. Apoptosis of glomerular podocytes in situ has been described in animal models of progressive glomerular sclerosis (23). Although the majority of urinary podocytes seemed to be viable, our findings suggest that apoptosis and non-apoptotic cell death also accompany shedding of podocytes from the glomerulus into the urine. We cannot rule out that in our subjects with glomerular disease podocytes detach from the glomerular tuft intact and viable and that it is subsequent exposure to extreme conditions in the final urine that first induces them to undergo apoptosis. Interestingly, podocyte viability seems not to be influenced by urinary pH, suggesting that other factors may be involved.
Urinary podocyte viability was also confirmed by their ability to grow in culture. Their growth pattern resembled that of colonies derived from glomerular explants (15) or immortalized human podocyte cell lines (22). Specific staining with markers such as podocalyxin, podocin, P-cadherin, synaptopodin, and WT1 confirmed the identity of the cultured urinary cells as podocytes.
It has been reported previously that viable tubule epithelial cells can be cultured from the urine of patients with acute tubular necrosis or diabetes (1, 21). Identification of these cells was based on their morphology as epithelial cells and expression of γ-glutamyl transferase. Given the plasticity that cells may show in culture, we wished to establish a definite identity of our urinary cells by the use of multiple specific cell markers in addition to cell morphology. Cells grown in culture in our study, for example, were not found to express the tubule cell marker E-cadherin. Although some cells in culture expressed the tubule cell marker cytokeratin 8, coexpression of this marker with WT1 or podocin suggests that podocytes in culture may acquire protein expression profiles of less differentiated cells rather than being evidence of the presence of viable tubule epithelial cells. Similar considerations may apply to coexpression of α-smooth muscle actin and WT1. Within the limits imposed by the effects of partial dedifferentiation and plasticity attendant to in vitro culture conditions, our results provide strong evidence only for the presence of viable podocytes and not of other renal cell types in the urinary sediment of patients with active glomerular disease.
We found that podocytes were present in urine of healthy subjects, as well as individuals with glomerular disease, when assessed either by cytospin or by culture. The growth behavior of urinary podocytes in culture may shed light on the mechanism by which they detach from the glomerular tuft. Cells cultured from urine samples from patients with active glomerular disease showed the same morphology and growth pattern as outgrowths from glomeruli isolated from whole kidney, whereas colonies cultured from healthy control subjects had significantly less growth capability and died much sooner. These results suggest that podocytes are shed from the glomerulus in disease states not as a result of a defect in the podocytes themselves, but rather as a response to some damaging but sublethal stimulus in the tuft environment. For example, activated complement in lupus nephritis might act to disrupt podocyte focal adhesions (24), whereas exposure to a circulating factor in FSGS might disrupt the actin cytoskeleton (7). Podocytes from normal healthy individuals, on the other hand, may be shed principally when they are senescent, resulting in the limited potential for replication in culture that they showed in the majority of cases. Further research is needed into the specific mechanisms of loss of viable podocytes from the glomerular tuft.
Acknowledgments
S. U. Vogelmann was supported by a grant from Satellite Dialysis and by a research fellowship from the National Kidney Foundation of Northern California. This work was also supported in part by the National Institutes of Health (DK-54600, DK-49372), by a Faculty Scholar Award (Satellite Dialysis), and a grant from the Lucile Packard Foundation for Children’s Health to K. V. Lemley.
Footnotes
The authors express gratitude to the members of the laboratory of Dr. W. J. Nelson, as well as to K. Blouch, R. Xu, J. Bialek, and Dr. Z. Ge, for valuable assistance and advice.
Parts of this work have been presented in abstract form at the 3rd International Symposium on Podocyte Biology, Heidelberg, Germany, September 2001, and the American Society of Nephrology, San Francisco, CA, October 2001.
References
- 1.Detrisac CJ, Mayfield RK, Cowell JA, Garvin AJ, Sens DA. In vitro culture of cells exfoliated in the urine by patients with diabetes mellitus. J Clin Invest. 1983;71:170–173. doi: 10.1172/JCI110747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drumond MC, Deen WM. Structural determinants of glomerular hydraulic permeability. Am J Physiol Renal Fluid Electrolyte Physiol. 1994;266:F1–F12. doi: 10.1152/ajprenal.1994.266.1.F1. [DOI] [PubMed] [Google Scholar]
- 3.Endlich K, Kriz W, Witzgall R. Update in podocyte biology. Curr Opin Nephrol Hypertens. 2001;10:331–340. doi: 10.1097/00041552-200105000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Fries JW, Sandstrom DJ, Meyer TW, Rennke HG. Glomerular hypertrophy and epithelial cell injury modulate progressive glomerulosclerosis in the rat. Lab Invest. 1989;60:205–218. [PubMed] [Google Scholar]
- 5.Hara M, Yamamoto T, Yanagihara T, Takada T, Itoh M, Adachi Y, Yoshizumi A, Kawasaki K, Kihara I. Urinary excretion of podocalyxin indicates glomerular epithelial cell injuries in glomerulonephritis. Nephron. 1995;69:397–403. doi: 10.1159/000188509. [DOI] [PubMed] [Google Scholar]
- 6.Hara M, Yanagihara T, Itoh M, Matsuno M, Kihara I. Immunohistochemical and urinary markers of podocyte injury. Pediatr Nephrol. 1998;12:43–48. doi: 10.1007/s004670050401. [DOI] [PubMed] [Google Scholar]
- 7.Kemeny E, Mihatsch MJ, Durmuller U, Gudat F. Podocytes loose their adhesive phenotype in focal segmental glomerulosclerosis. Clin Nephrol. 1995;43:71–83. [PubMed] [Google Scholar]
- 8.Kerjaschki D. Caught flat-footed: podocytes damage, and the molecular bases of focal glomerulosclerosis. J Clin Invest. 2001;108:1583–1587. doi: 10.1172/JCI14629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim YH, Goyal M, Kurnit D, Wharram B, Wiggins J, Holzman L, Kershaw D, Wiggins R. Podocyte depletion and glomerulosclerosis have direct relationship in the PAN-treated rat. Kidney Int. 2001;60:957–968. doi: 10.1046/j.1523-1755.2001.060003957.x. [DOI] [PubMed] [Google Scholar]
- 10.Kriz W, Elger M, Mundel P, Lemley KV. Structure-stabilizing forces in the glomerular tuft. J Am Soc Nephrol. 1995;5:1731–1739. doi: 10.1681/ASN.V5101731. [DOI] [PubMed] [Google Scholar]
- 11.Kriz W, Gretz N, Lemley KV. Progression of glomerular disease: is the podocyte the culprit? Kidney Int. 1998;54:687–697. doi: 10.1046/j.1523-1755.1998.00044.x. [DOI] [PubMed] [Google Scholar]
- 12.Kriz W, Lemley KV. The role of the podocyte in glomerulosclerosis. Curr Opin Nephrol Hypertens. 1999;8:489–497. doi: 10.1097/00041552-199907000-00014. [DOI] [PubMed] [Google Scholar]
- 13.Lemley KV, Abdullah I, Myers BD, Meyer TW, Blouch KS, William E, Bennett PH, Nelson RG. Evolution of incipient nephropathy in type 2 diabetes mellitus. Kidney Int. 2000;58:1228–1237. doi: 10.1046/j.1523-1755.2000.00223.x. [DOI] [PubMed] [Google Scholar]
- 14.Lemley KV, Lafayette RA, Safai M, Derby G, Blouch K, Squarer A, Myers BD. Podocytopenia and disease activity in IgA nephropathy. Kidney Int. 2002;61:1475–1485. doi: 10.1046/j.1523-1755.2002.00269.x. [DOI] [PubMed] [Google Scholar]
- 15.Mundel P, Reiser J, Kriz W. Induction of differentiation in cultured rat and human podocytes. J Am Soc Nephrol. 1997;8:697–705. doi: 10.1681/ASN.V85697. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura T, Ushiyama C, Suzuki S. Effect of angioten-sin-converting enzyme inhibitor, angiotensin II receptor antagonist and calcium antagonist on urinary podocytes in patients with IgA nephropathy. Am J Nephrol. 2000;20:373–379. doi: 10.1159/000013619. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura T, Ushiyama C, Suzuki S, Hara M, Shimada N, Ebihara I, Koide H. Urinary excretion of podocytes in patients with diabetic nephropathy. Nephrol Dial Transplant. 2000;15:1379–1383. doi: 10.1093/ndt/15.9.1379. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura T, Ushiyama C, Suzuki S, Hara M, Shimada N, Sekizuka K, Ebihara I, Koide H. Urinary podocytes for the assessment of disease activity in lupus nephritis. Am J Med Sci. 2000;320:112–116. doi: 10.1097/00000441-200008000-00009. [DOI] [PubMed] [Google Scholar]
- 19.Nyengaard JR, Bendtsen TF. Glomerular number and size in relation to age, kidney weight, and body surface in normal man. Anat Rec. 1992;232:194–201. doi: 10.1002/ar.1092320205. [DOI] [PubMed] [Google Scholar]
- 20.Pagtalunan EP, Miller PL, Jumping-Eagle S, Nelson RG, Meyers BD, Rennke HG, Coplon NS, Sun L, Meyer TW. Podocytes loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Racusen LC, Fivush BA, Li YL, Slatnik I, Solez K. Dissociation of tubular cell detachment and tubular cell death in clinical and experimental “acute tubular necrosis”. Lab Invest. 1991;64:546–556. [PubMed] [Google Scholar]
- 22.Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13:630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- 23.Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, Böttinger EP. Apoptosis in podocytes induced by TGF-β and Smad7. J Clin Invest. 2001;108:807–816. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Topham PS, Haydar SA, Kuphal R, Lightfoot JD, Salant DJ. Complement-mediated injury reversibly disrupts glomerular epithelial cell actin microfilaments and focal adhesions. Kidney Int. 1999;55:1763–1775. doi: 10.1046/j.1523-1755.1999.00407.x. [DOI] [PubMed] [Google Scholar]