

Abstract
In the past decade, engineered G-protein-coupled receptors activated solely by synthetic ligands (RASSLs) have been implemented as a new means to study neurotransmission, which is controlled by G-protein-coupled receptors in vitro and in vivo. In this study, we report an engineered dopamine receptor D2L F3906.52W, which is the first identified RASSL for the dopamine receptor family. The mutant receptor is characterized by a disrupted ligand binding and complete loss of efficacy for the endogenous ligand, dopamine, which is putatively due to a sterically induced perturbation of H-bonding with conserved serine residues in TM5. Based on this model, we rationally developed an aminoindane-derived set of agonists. Because these agonists forgo analogous H-bonding functionalities, their binding energy does not depend on the respective interactions. Binding affinity and potency were optimized by ligand modifications bearing molecular appendages that obviously interact with a secondary recognition site provided by four hydrophobic residues in TM2 and TM3. Thus, the ferrocenyl carboxamide 5b (FAUC 185) was identified as a synthetic agonist that is able to stimulate the mutant receptor in a manner similar to that by which endogenous dopamine activates the D2L wild-type receptor. The engineered dopamine receptor D2L F3906.52W in combination with FAUC 185 (5b) provides a new tool to probe GPCR functions selectively in specific cell populations in vitro and in vivo.
Keywords: Dopamine D2L receptor; aminoindane; engineered receptor; G-protein-coupled receptor, GPCR; ferrocenyl carboxamide; receptor activated solely by synthetic ligands, RASSL
As a complement to genetic knockouts, artificial regulatory systems have been discovered as powerful tools for deciphering cellular pathways. Thus, orthogonal ligand−protein pairs have been engineered for nuclear hormone receptors (1−3), and synthetic tetracycline derivatives that selectively induce mutant Tet regulatory systems have been developed in our laboratory (4−6). The largest protein superfamily in the human genome, G-protein-coupled receptors (GPCRs), represents the pharmacological target for about 50% of approved medications (7). Because of the great complexity of GPCR signaling in vivo, engineered GPCRs were suggested as tools to modulate and elucidate neurotransmission of these receptors (8−12). Strader et al. (13) reported the first engineered β2-adrenergic receptor that lost the affinity toward the endogenous neurotransmitter and responded on the synthetic ligand. Since then, further engineered receptors activated solely by synthetic ligands were reported, including the 5-HT414, α2-adrenergic (15), H1-histamine (16), and melacortin-4 (17) receptors (for the detailed review, see Pei et al. (8)). The term RASSL (receptor activated solely by synthetic ligand) was introduced by Conklin and collaborators, who modified κ opioid peptide receptors (18), which were successfully expressed in different tissues of transgenic animals and yielded important information about generation of diverse phenotypes, such as heart-rate control, cardiomyopathy, and taste sensations (19,20). Jacobson and colleagues (11), who modified adenosine receptors based on predictions from molecular modeling, designated newly engineered receptor/ligand complexes as “neoreceptor” and “neoligand”. With the means of molecular evolution, Bryan Roth and colleagues (12) developed muscarinic-1-5 receptors that can be exclusively activated by clozapine-N-oxide (8,12).
Although dopamine receptors are known as key players in GPCR-based neurotransmission, dopamine receptors insensitive to the endogenous neurotransmitter but responding to a synthetic agonist have not yet been reported. Neve and co-workers described the mutant D2S S1945.43A displaying strongly attenuated efficacy but significant antagonist binding of dopamine (21). With the aim to better understand the specificity of receptor−ligand interactions and to find novel lead structures for the treatment of neurological and psychiatric diseases, drug discovery in the field of subtype-selective dopamine D2-like receptor agonists and antagonists is the main focus of our work (22−24). Because of the importance of the D2-like receptors in health (25) and disease (25−27), we envisioned engineering of a receptor−ligand complex that would simulate the functional properties of the D2L receptor. To perturb the molecular recognition of dopamine, we envisaged manipulating the structure of the binding pocket in position F3906.52, which was expected to be highly crucial for both ligand binding and activation. Because functional properties of the engineered receptor should be highly similar to wild-type D2L, a subtle exchange of F3906.52 residue was intended in accord with the concept of “safe” mutations (28). In the second step, we tried to identify a lead dopamine receptor agonist that could take advantage of or at least tolerate the structural modifications on the primary binding site of the receptor. Starting from a suitable lead compound, binding affinity and potency should be optimized by ligand modifications bearing molecular appendages that could interact with secondary recognition sites of the receptor and thus increase binding energy. Finally, a comparison of the functional properties of the newly generated system with those of wild-type D2L activated by dopamine should be performed.
Results and Discussion
Dopamine receptors are important modulators of the nervous system, associated with a variety of neurological and psychiatric disorders, including schizophrenia, Parkinson's disease, and substance abuse (26), and with obesity (27). They are importantly involved in immunomodulation during health and disease (25), for example, in the regulation of human T cell functions (29,30) with the fundamental implications for neurodegeneration. Because of the importance of D2-like receptors in health and disease, we envisioned an artificial regulation system that simulated the functional properties of the D2L receptor. The principle of engineered receptors activated solely by synthetic ligands has been proposed as a tool to study multiple ligand-activation binding sites and different molecular cascades elicited by alternate ligand-activation binding sites (8−10,12,31). These engineered receptors have been achieved by mutation in different receptor domains involved in the endogenous ligand binding: the second cellular loop (18), TM3 (14), TM5 (15), and TM6 (16). In this report, we present a dopamine D2L receptor modified in TM6, which demonstrates the characteristics of a RASSL in our test systems. Additionally, we present the rational design of a ligand that mimics the properties of dopamine on D2L wild-type receptor. Finally, we compare our artificial system consisting of the ferrocenyl carboxamide FAUC 185 (5b) and the mutated D2LF3906.52W receptor with the native system of dopamine and the D2L receptor in experiments measuring [35S]GTPγS incorporation, cAMP production, and ERK1/2 phosphorylation.
Phenotype of the Mutant D2L F3906.52W Receptor
Neve and co-workers reported the D2S S1945.43A mutant displaying strongly attenuated receptor activation but significant antagonist binding of dopamine (21). To approach a functional mutant dopamine D2L receptor that would not bind dopamine, the residue F3606.52 was chosen for mutation. Recent crystal structures of the β2-adrenergic (32,33), β1-adrenergic (34), and A2A adenosine (35) receptors showed a critical role for position 6.52 in ligand binding. This residue was earlier suggested to be involved in a “toggle switch” for receptor activation (36,37). The amino acid phenylalanine at position 6.52 is completely conserved within the catecholamine-binding GPCRs (38). Mutational studies of the D2L receptor indicated that the presence of phenylalanine in the position 6.52 is critical for both binding interaction and receptor activation of the D2L receptor (39−41). On the other hand, only 20% of all rhodopsin-like receptors (42) display this particular conservation. Thus, F6.52 seems to be essential for a specific and effective recognition of the neurotransmitter’s catechol unit. Since we intended to develop a receptor system that should be insensitive to catecholamines but bound the synthetic analogs with bioisosteric moieties different from the catechol structure, the exchange of F6.52 should be a promising approach. With the intention to introduce a “safe” amino acid substitution, which would least likely disturb the protein structure and most likely allow the probing of the structural and functional significance of the substituted site (28), we mutated the highly conserved residue F3906.52 in the dopamine D2L receptor to tryptophan. In our mutant receptor, the introduction of a bulky tryptophan at the position of F3906.52 caused different changes in the affinities of the investigated antagonists and agonists. The binding affinities of [3H]spiperone and haloperidol were comparable to those with the wild-type receptor (0.09 nM for the wild-type vs 0.14 nM for the mutant and 0.41 vs 0.60 nM, respectively; Table 1), whereas the affinity of nemonapride was 11-fold reduced (0.14 vs 1.5 nM). Among the agonists investigated, the endogenous ligand dopamine showed no detectable binding. The affinity of 7-OH-DPAT was reduced 32-fold (from 69 to 2200 nM) and the affinity of quinpirole was reduced 4-fold (from 610 to 2500 nM).
Table 1. The Affinity of the Dopamine Receptor Antagonists and Agonists on D2L Wild-Type and D2L F3906.52W Receptora.
| compound | Ki measured | Ki for D2L wild-type (nM) | Ki for D2L F3906.52W (nM) | Kimut/Ki |
|---|---|---|---|---|
| Antagonists | ||||
| spiperone | Ki | 0.09 (0.08−0.12)b (−1.05)c | 0.14 (0.08−0.19)b (−1.16)c | 1.6 |
| haloperidol | Ki | 0.41 (0.24−0.69)b (−0.84)c | 0.60 (0.52−0.69)b (−1.04)c | 1.5 |
| nemonapride | Ki | 0.14 (0.10−0.18)b (−0.84)c | 1.5 (1.2−1.8)b (−0.98)c | 11 |
| Agonists | ||||
| dopamine | K0.5 | 450 (380−660)b (−0.43)c | >20 000 | >44 |
| Khigh | 8.4 (4.6−25)b (31%)d | |||
| Klow | 2000 (1400−2800)b | >20 000 | >10 | |
| 7-OH-DPAT | K0.5 | 69 (49−98)b (−0.40)c | 2200 (1600−3200)b (−0.45)c | 32 |
| Khigh | 1.1 (0.45−2.5)b (39%)d | 21 (11−41)b (28%)d | 19 | |
| Klow | 190 (110−340)b | 5600 (3700−8500)b | 29 | |
| quinpirole | K0.5 | 610 (460−790)b (−0.44)c | 2500 (1700−3700)b (−0.56)c | 4 |
| Khigh | 13 (6.9−25)b (31%)d | 22 (11−41)b (25%)d | 1.7 | |
| Klow | 2100 (1500−3200)b | 5000 (3200−7500)b | 2.3 | |
The affinities of investigated substances were determined on membrane preparations of stably transfected CHO cells expressing either D2L wild-type or D2L F3906.52W receptor using [3H]spiperone displacement study. Data are derived from normalized curves of 3-4 experiments done in triplicate. The factor Kimut/Ki indicates the changes in the affinity.
95% confidence interval.
Hill slope.
Fraction of high-affinity sites.
The complete loss of binding affinity for dopamine and the significant decrease in the binding affinities of the agonists 7-OH-DPAT and quinpirole could be thus interpreted in terms of steric hindrance or minute local conformational changes within the binding pocket caused by the bulky tryptophan. The affinities of the long-chain antagonists spiperone and haloperidol were unaffected by this mutation, indicating that long-chain compounds can efficiently compensate for the induced changes within the mutated binding pocket.
Because of the absence of detectable binding of dopamine and preserved moderate affinity for the synthetic agonist quinpirole, the activation status of the mutant D2L F3906.52W receptor was inspected by functional assays measuring [35S]GTPγS incorporation and the inhibition of cAMP accumulation in CHO cells stably expressing the mutant receptor. To exclude the possibility that dopamine, despite the absence of measurable binding in the low micromolar range, exerts an allosteric effect to the receptor, its efficacy at the mutant D2L F3906.52W receptor was investigated. The endogenous ligand dopamine was not able to induce [35S]GTPγS incorporation or inhibit cAMP production, whereas quinpirole displayed an activity at the D2L F3906.52W receptor at high concentrations (EC50 value 0.5−1.5 μM, Supplemental Figure 1, Supporting Information). Spiperone readily suppressed the D2L F3906.52W receptor activation induced by quinpirole (Supplemental Figure 1, Supporting Information), confirming that the activation of the mutant receptor is reversible. Because it was reported that dopamine can act as an antagonist on dopamine D2S S194A receptor mutant (21), we tested whether dopamine can act as an antagonist at the D2L F3906.52W receptor. In fact, dopamine was not able to antagonize the activation of the D2L F3906.52W receptor by quinpirole (Supplemental Figure 2, Supporting Information).
Development of Synthetic Agonists
In the next step, we identified a dopamine agonist that was able to tolerate the structural modifications on the primary binding site of the receptor. The dopamine binding crevice is expected to be lined by the highly conserved amino acids D3.32, V3.33 S5.42, S5.46, W6.48, F6.51, F6.52, and H6.55. In the wild-type dopamine D2 receptor, the aromatic catecholic ring of dopamine is sandwiched between an aromatic microdomain at TM6 (W6.48, F6.51, F6.52, and H6.55) and the hydrophobic residue V3.33 on TM3 (36−38). The catecholic hydroxyl groups point toward TM5 where they are suggested to interact with S5.42 and S5.46 (21,38,43). Introduction of a bulky tryptophan at position 6.52 could thus interfere with this sandwiching of the catechol ring of dopamine and directly disrupt the hydrogen bonding between the aromatic hydroxyl groups and the serines in TM5, leading to a significant weakening of the binding of dopamine to the mutant D2L F3906.52W receptor (Figure 1).
Figure 1.
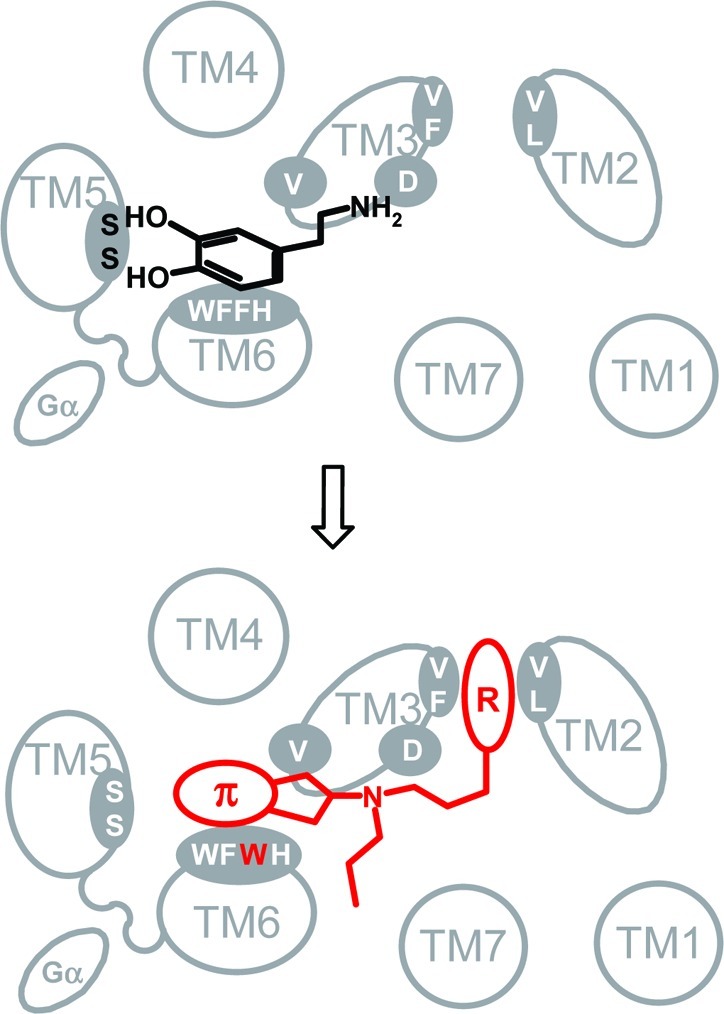
The primary binding site of D2 is expected to be lined by the highly conserved amino acids D3.32, V3.33 S5.42, S5.46, W6.48, F6.51, F6.52, and H6.55. Hydrogen bonds between the aromatic hydroxyl groups of dopamine and S5.42 and S5.46 in TM5 substantially contribute to the binding energy of dopamine. Mutation of phenylalanine to a bulky tryptophan at position 6.52 sterically interferes with this stabilization leading to a significant weakening of the binding of dopamine. An aminoindane moiety, forgoing H-bonding, is more flexible and, thus, can better tolerate local steric modifications. Molecular appendages (R) can be attracted by hydrophobic interactions with a secondary binding site provided by F3.28, V3.29, V2.61, and L2.64.
To address this issue in the ligand design, we envisioned developing a smaller hydrophobic agonist, which would gain binding energy only from its hydrophobic interactions with the aromatic cluster and is thus liberated from the requirements for the formation of the hydrogen bonds with the serines of TM5, which is required for dopamine. A sterically less demanding hydrophobic ligand forgoing H-bonding functionalities should better tolerate steric modifications and local conformational changes within the TM5/TM6 microdomain (Figure 1). Our initial investigations were directed toward our atypical dopaminergics of the type FAUC 73 (44) and FAUC 460 (23) incorporating a small conjugated enyne functionality as a catechol bioisostere. Unfortunately, both agents were unable to activate D2L F3906.52W. Screening of our in-house compound library identified 2-dipropylaminoindane (3) as a promising candidate for the development of a compound that would readily bind and activate the mutant D2L F3906.52W receptor. The aminoindane 3 showed a K0.5 value of 2200 nM at the mutant receptor and a fairly good EC50 value in the inhibition of cAMP accumulation (170 nM, efficacy of 86%, Table 2). Taking advantage of our previously reported SAR studies indicating that biphenyl (23,45), paracyclophanyl (46), and metallocenyl (47) carboxamides are superior molecular appendages for the construction of high-affinity dopamine receptor ligands addressing a secondary binding site provided by F3.28, V3.29, V2.61, and L2.64 (24), we coupled the aminoindane moiety with the respective building blocks resulting in formation of the biphenyl (5a), ferrocene (5b), and paracyclophane (5c) derivatives, as described in Scheme 1.
Table 2. D2L F3906.52W receptor binding data and inhibition of cAMP accumulation as assay for intrinsic activity of test compounds 3 and 5a-c.a.
| |
cAMP inhibition |
|||
|---|---|---|---|---|
| compound | Ki measured | binding affinity (nM) | EC50 (nM) | efficacy (%) |
| 3 | K0.5 | 2200 (1600−3000)b (−0.55)c | 170 | 86 |
| 5a | Ki | 190 (170−220)b (−0.82)c | 1900 | 98 |
| 5b | K0.5 | 64 (54−76)b (−0.56)c | ||
| Khigh | 2.5 (1.3−4.9)b (29%)d | 53 | 105 | |
| Klow | 200 (150−270)b | |||
| (R)-5c | Ki | 61 (43−86)b (−0.77)c | 260 | 80 |
| (S)-5c | Ki | 110 (90−130)b (−0.74)c | 180 | 77 |
Data are derived from normalized curves of three to four experiments done in triplicate.
95% confidence interval.
Hill slope.
Fraction of high-affinity states.
Scheme 1.

Reagents and conditions: (a) (1) propylamine, NaBH(OAc)3, THF, HOAc, RT, 1 h, reflux 18 h; (2) HCl in Et2O, Et2O; (b) 4-bromobutyronitrile, K2CO3, KI, CH3CN, reflux, 24 h; (c) LiAlH4 in Et2O, Et2O, RT, 1 h; (d) 4-biphenylcarboxylic acid chloride, Et3N, CH2Cl2, RT, 21 h; (e) ferrocene carboxylic acid, HATU, DIPEA, DMF, RT, 3 h; (f) [2.2]paracyclophane carboxylic acid, HATU, DIPEA, CH2Cl2, NMP, RT, 2.75−3 h; (g) propionaldehyde, NaBH(OAc)3, THF, HOAc, reflux 21 h.
Because of the chirality of the substituted cyclophane unit, 5c was synthesized in both enantiomeric forms (R)-5c and (S)-5c. The affinities of the new compounds for the mutant receptor increased significantly (K0.5 value 64 nM for 5b, Table 2). These new compounds were tested employing an inhibition of cAMP accumulation in which the feroccene 5b (FAUC 185) was identified as the best substance with an EC50 value of 53 nM and a ligand efficacy of 105% (relative to the reference agent quinpirole, Table 2, Figure 2).
Figure 2.

FAUC 185 (5b) exerts full agonist effect on the D2LF3906.52W receptor. The compounds 3 and 5a−c were tested for their ability to inhibit cAMP accumulation in stably transfected CHO cells expressing the D2L F3906.52W receptor. Normalized curves with error bars representing the SEM are shown. Efficacy was determined relative to that of the reference substance quinpirole. The EC50 values and the efficacies are summarized in Table 2.
Overall, the ferrocene derivative 5b improved the binding affinity for the D2L F3906.52W receptor by over 310-fold compared with dopamine, a 39-fold increase compared with the synthetic agonists quinpirole and a 34-fold improvement of binding compared with the lead compound 3. Because of the relatively shallow binding curve of 5b with the Hill slope value −0.56, detailed biphasic analysis of the binding profile was performed. Derivative 5b demonstrated binding character on the D2L F3906.52W receptor indistinguishable from that on the D2L wild-type receptor. The fraction of high-affinity sites was 30% in D2L wild-type and 29% in D2L F3906.52W receptor. Ki values of 1.1 nM (Khigh) and 180 nM (Klow) for D2L wild-type and 2.5 nM and 200 nM for D2L F3906.52W mutant receptor were determined for the high-affinity binding sites representing the active ternary complex (Figure 3). Interestingly, whereas the F3906.52W mutation caused a substantial decrease in binding for reference agonists and antagonists, ligand affinity could be maintained for the ferrocene derivative 5b.
Figure 3.

FAUC 185 (5b) displays binding character on the D2LF3906.52W receptor undistinguishable from that on the D2L wild-type receptor. Data obtained from [3H]spiperone displacement studies with FAUC 185 (5b) on membrane preparations from stably transfected CHO cells that expressed either wild-type D2L or D2L F3906.52W receptor yielded biphasic curves. The fraction of high-affinity sites was 30% in D2L wild-type and 29% in D2L F3906.52W receptor. The affinities were 1.1 nM (Khigh) and 180 nM (Klow) for D2L wild-type and 2.5 nM and 200 nM for D2L F3906.52W receptor. Normalized curves with error bars representing the SEM are shown.
To inspect the specificity of the newly synthesized compounds, the binding profiles of the active agents 3 and 5a−c were determined by radioligand displacement studies using the human dopamine receptor subtypes D2L, D2S, D3, and D4.4, as well as porcine D1, serotonin 5-HT1A and 5-HT2, and adrenergic α1 receptors. Detailed binding profiles displayed subnanomolar D3/D4 receptor affinities for the newly synthesized compounds 5a−c and comparable binding affinities for the wild-type D2L and the mutant D2L F3906.52W receptor (Supplemental Table 1, Supporting Information). Because of the high degree of homology between the binding sites of D2-like receptors and the less-abundant subtypes D3 and D4, the synthesis of selective D2 agonists poses a continuous challenge (48−50). This issue, which is continuously investigated in our laboratory, is still poorly resolved despite great efforts in medicinal chemistry.
Comparison of the Dopamine/D2L Wild-Type Receptor System with the Ligand−Receptor Pair FAUC 185/D2L F3906.52W
To test whether our artificial system incorporating the mutant GPCR D2L F3906.52W and the synthetic agonist FAUC 185 (5b) mimics the properties of natural D2L receptor with its endogenous ligand dopamine, both the wild-type and the mutant receptor were tested in experiments measuring [35S]GTPγS incorporation, inhibition of cAMP accumulation, and increase in ERK1/2 phosphorylation. CHO cells stably expressing D2L or D2L F3906.52W receptor were used in all assays.
FAUC 185 (5b) activated the D2L F3906.52W receptor in a manner comparable to that of dopamine in the wild-type D2L receptor when the [35S]GTPγS incorporation and inhibition of cAMP accumulation assays were used. In the [35S]GTPγS incorporation assay indicating the agonist-induced nucleotide exchange, dopamine exerted full agonist effect on the wild-type D2L receptor with an EC50 value of 330 nM (Figure 4A). FAUC 185 was a full agonist on the D2L F3906.52W receptor with an EC50 value 260 nM (Figure 4A), thus demonstrating an activity very similar to the effect of dopamine on the wild-type receptor. In the assay investigating inhibition of cAMP accumulation, cAMP production was stimulated by 20 μM forskolin and the inhibition of the cAMP production mediated by the activation of the Gi/o-coupled D2L receptors was subsequently measured. Dopamine, as expected, displayed a full agonist effect at the wild-type receptor with EC50 value 26 nM (Figure 4B). FAUC 185 was a full agonist on the D2L F3906.52W receptor with an EC50 value 55 nM (Figure 4B), again demonstrating a very similar profile to that of dopamine on the wild-type receptor.
Figure 4.

The D2L F3906.52W receptor does not respond to dopamine in different functional assays but demonstrates full activation upon stimulation with synthetic agonist. Normalized curves with error bars representing the SEM are shown. (A) The [35S]GTPγS binding assay was performed on membrane preparations of stably transfected CHO cells that expressed either wild-type D2L or D2L F3906.52W receptor. The EC50 value for dopamine at the D2L wild-type receptor was 330 nM. The EC50 value for 5b at the D2L F3906.52W receptor was 260 nM. (B) The determination of inhibition of cAMP accumulation was performed on stably transfected CHO cells that expressed either wild-type D2L or mutant D2L F3906.52W receptor. The EC50 value for dopamine at the D2L wild-type receptor was 26 nM. The EC50 value for 5b at the D2L F3906.52W receptor was 47 nM. (C) The phosphoERK1/2 ELISA assay was performed on stably transfected CHO cells that expressed either wild-type D2L or mutant D2L F3906.52W receptor. The EC50 value for dopamine at the D2L wild-type receptor was 16 nM. The EC50 value for 5b at the D2L F3906.52W receptor was 220 nM with an efficacy of 91%.
In the phosphoERK1/2 ELISA assay, noticeable differences could be determined in the effects of the compounds at the receptor types. FAUC 185 displayed a significantly reduced EC50 value on the D2L F3906.52W receptor at 220 nM with an efficacy of 91% (of quinpirole-mediated effect), thus displaying a profile that differs from the activation profile of dopamine on the wild-type D2L receptor showing full agonistic effect and EC50 value 16 nM (Figure 4C). This could indicate that already the subtle modification of one amino acid in the region critical for the ligand binding and receptor activation can elicit minor changes in the signaling properties, as already proposed by Pauwels (31).
In conclusion, we describe the rational design and development of a dopamine-insensitive artificial D2L receptor system as the first RASSL for the family of dopamine receptors, the D2L F3906.52W receptor being specifically stimulated by the new synthetic ligand FAUC 185 (5b). The system mimics the endogenous receptor−ligand pair dopamine/D2L wild-type receptor, thus, offering a powerful tool to probe GPCR functions selectively in specific cellular populations. To further approach to a fully orthogonal system, additional modifications at the mutant receptor and the synthetic agonists will be performed when particular attention will be given to the development of a synthetic ligand with high selectivity over D2 wild-type and related GPCRs.
Methods
Materials
Dulbecco's modified Eagle's medium/F-12, l-glutamine, fetal bovine serum (FBS), penicillin−streptomycin, zeocin, and hygromycin-B were purchased from Invitrogen. [3H]Spiperone (97 Ci/mmol) and [35S]GTPγS (1250 Ci/mmol) were purchased from Amersham and PerkinElmer Biosciences, respectively. cAMP-Glo assay was purchased from Promega. Dopamine (3,4-dihydroxyphenethylamine), quinpirole ((−)-quinpirole hydrochloride), spiperone, haloperidol, 7-OH-DPAT (R-(+)-7-hydroxy-2-(N,N-di-n-propylamino)tetralin hydrobromide) and other substances were purchased from Sigma, unless otherwise stated. Nemonapride was purchased from BioTred.
Site-Directed Mutagenesis and Cloning
The cDNA of the human dopamine D2long (D2L) receptor was purchased at the Missouri S&T cDNA Resource Center and subcloned into a pcDNA3.1 vector (Invitrogen) using NheI/XbaI restriction sites. The mutation was induced with overlap PCR that used flanking primers TAATACGACTCACTATAGGG and ACTAGAAGGCACAGTCGAGG, and the mutation F3906.52W inducing primers CTGCTGGTTACCCTTCTGGATCACAC and TGTGATCCAGAAGGGTAACCAGCAG. The final product of D2L receptor mutagenesis was digested with BstEII/XbaI (New England Biolabs) and cloned into a D2L wild-type pcDNA3.1 vector. The D2L wild-type and D2L F3906.52W receptors were additionally subcloned into pcDNA5/FRT (Invitrogen) using NheI/XhoI restriction sites. The entire coding region of the D2L receptor clones was sequenced to ensure that the correct mutation was introduced and to confirm the absence of unwanted mutations.
Cell Lines and Transfection
The Flp-in CHO cell line (Invitrogen) was maintained in DMEM/F-12 supplemented with 10% FBS, 2 mM l-glutamine, 1% pen−strep, and 0.25 μg/mL zeocin and kept in a humid atmosphere at 37 °C with 5% CO2.
The Flp-in CHO cells were transfected with the pOG44 vector encoding Flp recombinase and the pcDNA5/FRT vector encoding the specific dopamine receptor in the ratio 9:1 using TransIT-LT transfection reagent (Mirus Bio Corporation). Forty-eight hours after transfection, cells were subcultured and the medium was supplemented with 750 μg/mL hygromycin-B to obtain colonies stably expressing dopamine receptors.
Cell Harvest and Membrane Preparation
Cells were washed with phosphate-buffered saline (PBS), briefly treated with Tris−EDTA buffer (10 mM Tris, 0.5 mM EDTA, 5 mM KCl, 140 mM NaCl, pH 7.4), and dissociated using a cell scraper. Cells were pelleted at 1000g for 6 min at 4 °C, resuspended in Tris−EDTA−MgCl2 buffer (50 mM Tris, 5 mM EDTA, 1.5 mM CaCl2, 5 mM MgCl2, 5 mM KCl, 120 mM NaCl, pH 7.4) and subsequently lysed with an Ultraturrax. After additional centrifugation at 50 000g, the membranes were resuspended in the binding buffer (50 mM Tris, 1 mM EDTA, 5 mM MgCl2, 100 μg/mL bacitracin, 5 μg/mL soybean trypsin inhibitor) and homogenized 10 times with a glass−Teflon homogenizer at 4 °C. The homogenized membranes were shock-frozen in liquid nitrogen and stored at −80 °C. The protein concentration was determined with the Lowry method (51) using bovine serum albumin as a standard.
Receptor Binding Studies
Receptor binding studies were carried out as described by Hübner et al. (44). In brief, the dopamine D1 receptor assay was done with porcine striatal membranes at a final protein concentration of 40 μg/well and the radioligand [3H]SCH 23390 at 0.3 nM (Kd = 0.42−0.67 nM). Competition experiments with human D2L and D2L F3906.52W, D2S, D3, and D4.4 receptors were run with preparations of membranes from CHO cells stably expressing the corresponding receptor and [3H]spiperone at a final concentration of 0.1−0.2 nM. The assays were carried out with a protein concentration of 1.5−22 μg/well and Kd values of 0.10−0.15, 0.03−0.08, 0.06−0.20, and 0.13−0.23 nM for the D2L, D2S, D3, and D4.4 receptors, respectively. Protein concentration was determined by the method of Lowry et al. (51) using bovine serum albumin as a standard. 5-HT and α1 receptor binding experiments were performed with homogenates prepared from porcine cerebral cortex and the selective radiologands [3H]WAY 100635 (5-HT1A), [3H]ketanserin (5-HT2), and [3H]prazosin (α1) at a concentration of 0.1 nM for [3H]WAY 100635 and [3H]prazosin and 0.5 nM for [3H]ketanserin, as described by Heindl et al. (52). Assays were run with membranes at a protein concentration of 80 μg/mL for 5-HT1A and 5-HT2 and 55−60 μg/mL for α1 receptor with Kd values of 0.03−0.06 nM for 5-HT1A, 0.55−1.5 nM for 5-HT2, and 0.04−0.16 nM for the α1 receptor. All assays were performed in 96-well plates at a final volume of 200 μL. After incubation for 1 h at 37 °C, we stopped the assay by filtration through Whatman GF/B filters presoaked with 0.3% polyethylenimine. The filters were rinsed 5 times with ice-cold Tris−NaCl buffer. After 3 h of drying at 60 °C, filters were sealed with melt-on scintillator sheets (MeltiLex B/HS, Perkin-Elmer), and the filter-bound radioactivity was measured in a MicroBeta TriLux liquid scintillation counter (Perkin-Elmer). Two to ten experiments per compound were performed with each concentration in triplicate.
[35S]GTPγS Assay
The [35S]GTPγS binding assay was performed on membrane preparations of stably transfected CHO cells that expressed either the wild-type D2L or D2L F3906.52W receptor. The cells expressed comparable amounts of the receptor as determined by saturation experiments (2100 ± 200 fmol/mg and 2700 ± 300 fmol/mg of protein, respectively). The assay was carried out in 96-well plates at the final volume of 200 μL. The incubation buffer contained 20 mM Hepes, 10 mM MgCl2·6H2O, and 100 mM NaCl (pH 7.4). Membranes (10−20 μg/mL of membrane protein), compounds, and 10 μM GDP were preincubated in the absence of [35S]GTPγS for 30 min at 37 °C. After the addition of 0.1 nM [35S]GTPγS, membranes were incubated for additional 30 min at 37 °C. Reactions were terminated by filtration through Whatman GF/B filters soaked with ice-cold PBS. The filter-bound radioactivity was measured as described above. Four to twelve experiments per compound were performed with each concentration in quadruplicate.
Inhibition of cAMP Accumulation Assay
Bioluminescence-based cAMP-Glo assay (Promega) was performed according to the manufacturer's instructions. Briefly, CHO cells expressing D2L wild-type and D2L F3906.52W receptor were seeded into a white 96-well plate (10 000 cells/well) 24 h prior to the assay. The cells expressed comparable amounts of the receptor as determined by saturation experiments (2100 ± 200 fmol/mg and 2700 ± 300 fmol/mg of protein, respectively). Cells were first briefly washed with Krebs−Ringer buffer (pH 7.4) to remove traces of serum and were incubated with various concentrations of substances in the presence of 20 μM forskolin in Krebs−Ringer buffer that contained 500 μM IBMX and 100 μM Ro 20-1724, pH 7.4. After 15 min of incubation at room temperature, the cells were lysed with cAMP-Glo lysis buffer. After lysis, the kinase reaction was performed with a reaction buffer containing PKA. At the end of the kinase reaction, an equal volume of Kinase-Glo reagent was added. The plates were read with a luminescence protocol on a microplate reader (Victor3V, Perkin-Elmer). The experiments were performed three to nine times per compound with each concentration in duplicate.
PhosphoERK1/2 ELISA Assay
The PathScan phospho-p42/44 MAPK (Thr202/Tyr204) sandwich ELISA (Cell Signaling) was performed according to manufacturer's instructions. Briefly, 6 × 106 CHO cells that expressed D2L or D2L F6.526.52W receptor were seeded in a 100 mm plate. The next day, cells were washed once with serum-free media and incubated in the presence of serum-free media for an additional 24 h. On the day of the experiment, the medium was removed and replaced with serum-free media containing various concentrations of the test substances as indicated and incubated for 5 min at 37 °C. The wash with ice-cold PBS and the addition of the lysis buffer stopped the reaction. The plates were kept on ice, and cells were scraped, briefly sonicated (UP50H, Hielscher Ultrasound Technologies), and centrifuged at 15 000g for 10 min. The supernatant was promptly diluted with the sample dilutent and incubated overnight at 4 °C in the well. After intensive washing steps, the detection of the phosphorylated ERK1/2 followed. The absorbance was read at 450 nm within 2 min after addition of STOP solution on a Victor3V (Perkin-Elmer) microplate reader. The experiment was performed four times per compound.
Data Analysis
The resulting competition curves of the receptor binding experiments and activity assays were analyzed by nonlinear regression using the algorithms in PRISM 3.0 (GraphPad Software, San Diego, CA). Competition curves were fitted to the sigmoid curve by nonlinear regression analysis in which the log EC50 value and the Hill coefficient were free parameters. EC50 values were transformed to K0.5 values according to the equation of Cheng and Prusoff (53).
Chemistry
2-Propylaminoindane hydrochloride 2(54), which was readily prepared by reductive amination of indan-2-one 1, was substituted in a nucleophilic manner with 4-bromobutyronitrile to afford the (N-indan-2-yl-N-propyl)-4-aminobutyronitrile 4a. Reduction of the nitrile 4a resulted in the primary amine 4b, which could be transformed into the carboxamides 5a−c by taking HATU-promoted coupling with the respective carboxylic acids. The enantiomers of [2.2]paracyclophane carboxylic acid were prepared according to the literature (46,55,56). 2-Dipropylaminoindane 3(54) was synthesized via reductive amination of the monopropylamine-substituted precursor 2.
(N-Indan-2-yl-N-propyl)-4-aminobutyronitrile (4a)
To a suspension of 2-propylaminoindane 2 (2.89 g, 13.7 mmol), KI (2.06 g, 12.4 mmol), and K2CO3 (10.6 g, 76.8 mmol) in CH3CN (70 mL), 4-bromobutyronitrile (3.27 mL, 32.7 mmol) was added dropwise. After being refluxed for 24 h, the mixture was allowed to cool to room temperature, and the solvent was evaporated. The residue was dissolved in H2O, basified with 2 N NaOH, and extracted with CH2Cl2. The combined organic layers were dried (Na2SO4) and evaporated. The residue was purified by flash chromatography (hexane−EtOAc 9:1 + 0.5% NEtMe2) to give 4a as a yellow liquid (2.57 g, 78% yield): IR 2958 s, 2245 m (Nitril), 1462 m, 1084 m, 746 s cm−1; 1H NMR (360 MHz, CDCl3) δ 0.88 (t, J = 7.4 Hz, 3 H), 1.48 (m, 2 H), 1.79 (m, 2 H), 2.39−2.49 (m, 4 H), 2.61 (m, 2 H), 2.85 (dd, J = 15.4, 8.4 Hz, 2 H), 3.01 (dd, J = 15.4, 7.8 Hz, 2 H), 3.67 (m, 1 H), 7.09−7.19 (m, 4 H); 13C NMR (150 MHz, CDCl3) δ 11.84, 14.69, 20.40, 23.84, 36.11, 49.27, 53.21, 62.50, 119.94, 124.42, 126.31, 141.68; EI-MS m/z 242; Anal. (C16H22N2·0.4H2O) C, H, N.
(N-Indan-2-yl-N-propyl)butane-1,4-diamine (4b)
To a cooled solution of 4a (1.21 g, 4.96 mmol) in Et2O (35 mL), a solution of LiAlH4 (1 M) in Et2O (12.4 mL, 12.4 mmol) was added dropwise. After being stirred at room temperature for 1 h, the mixture was cooled to 0 °C, quenched with aqueous NaHCO3, filtered over Celite/Mg2SO4/Celite, and washed several times with CH2Cl2 and EtOAc. The solvent was evaporated to give 4b as a red liquid (1.14 g, 93% yield): IR 3363 m, 2935 s, 2864 m, 1577 m, 1464 m, 1309 m, 1076 m, 742 s cm−1; 1H NMR (360 MHz, CDCl3) δ 0.88 (t, J = 7.4 Hz, 3 H), 1.37−1.57 (m, 6 H), 2.49 (m, 2 H), 2.54 (m, 2 H), 2.71 (m, 2 H), 2.87 (dd, J = 15.3, 8.9 Hz, 2 H), 3.01 (dd, J = 15.3, 7.7 Hz, 2 H), 3.65 (m, 1 H), 7.08−7.20 (m, 4 H); 13C NMR (150 MHz, CDCl3) δ 11.97, 20.22, 24.58, 31.79, 36.58, 42.17, 51.24, 53.35, 63.11, 124.39, 126.30, 141.90; EI-MS m/z 246; Anal. (C16H26N2·0.5H2O) C, H, N.
N-[(N′-Indan-2-yl-N′-propyl)-4-aminobutyl]-4-biphenyl carboxamide (5a)
Compound 4b (104.6 g, 0.42 mmol) was dissolved in 5 mL of dry CH2Cl2 and 0.18 mL Et3N (1.30 mmol) was added. The mixture was cooled to 0 °C. Then, a solution of 4-biphenylcarboxylic acid chloride (92.0 g, 0.42 mmol) in dry CH2Cl2 (5 mL) was added dropwise. The mixture was stirred for 21 h at room temperature before aqueous NaHCO3 was added. The aqueous layer was extracted with CH2Cl2, and the combined organic layers were dried (Mg2SO4) and evaporated. The residue was purified by flash chromatography (CH2Cl2−MeOH 95:5) to give 5a as a gray-white solid (101.4 mg, 56%): mp 78 °C; IR 3321 s, 2933 s, 2866 m, 1637 s, 1545 s, 1070 s, 1043 s, 746 s cm−1; 1H NMR (600 MHz, CDCl3) δ 0.87 (t, J = 7.3 Hz, 3 H), 1.54 (m, 2 H), 1.75−1.86, 1.67 (m, 4 H), 2.58 (m, 2 H), 2.66 (m, 2 H), 2.92−3.09 (m, 4 H), 3.48 (m, 2 H), 3.71 (m, 1 H), 7.05−7.19 (m, 4 H), 7.35 (m, 1 H), 7.42 (m, 2 H), 7.56 (m, 4 H), 7.88 (m, 2 H); 13C NMR (150 MHz, CDCl3) δ 11.73, 19.10, 24.14, 27.39, 36.02, 39.65, 50.81, 52.92, 63.01, 124.31, 126.45, 126.93, 127.02, 127.48, 127.78, 128.74, 133.34, 139.90, 140.97, 143.85, 167.29; EI-MS m/z 426.
N-[(N′-Indan-2-yl-N′-propyl)-4-aminobutyl]ferrocenyl carboxamide (5b)
To a solution of ferrocene carboxylic acid (76.2 mg, 0.33 mmol) in DMF (8 mL), DIPEA (109 μL, 0.66 mmol) was added. The mixture was cooled to 0 °C before a solution of HATU (161 mg, 0.42 mmol) in DMF (2 mL) was added. Then, a solution of 4b (122 mg, 0.50 mmol) in DMF (5 mL) was added dropwise. The mixture was stirred for 3 h at room temperature before aqueous NaHCO3 was added. The aqueous layer was extracted with CH2Cl2, and the combined organic layers were dried (Mg2SO4) and evaporated. The residue was purified by flash chromatography (CHCl3−MeOH 98:2) to give 5b as an orange solid (162 mg, >99%): mp 96 °C; IR 3643 w, 3431 w, 2974 w, 1618 m, 1541 m, 1460 m, 1298 m, 1026 m, 841 s, 752 m cm−1; 1H NMR (600 MHz, CDCl3) δ 0.99 (t, J = 7.3 Hz, 3 H), 1.72 (m, 2 H), 1.75−1.86 (m, 4 H), 3.03 (m, 2 H), 3.22 (m, 2 H), 3.27 (dd, J = 16.1, 7.5 Hz, 2 H), 3.35−3.42 (m, 4 H), 4.13−4.19 (m, 6 H), 4.33 (t, J = 1.9 Hz, 2 H), 4.70 (t, J = 1.9 Hz, 2 H), 6.61 (m, 1 H), 7.20−7.24 (m, 4 H); 13C NMR (90 MHz, CDCl3) δ 11.05, 17.69, 21.35, 26.81, 34.89, 38.25, 52.02, 53.27, 63.98, 68.22, 69.83, 70.89, 74.96, 124.58, 127.71, 138.42, 172.34; EI-MS m/z 460; purity >99% (HPLC).
N-[(N′-Indan-2-yl-N′-propyl)-4-aminobutyl]-4-(R)-[2.2]paracyclophane carboxamide ((R)-5c)
To a solution of 4-(R)-[2.2]paracyclophane carboxylic acid (38.9 mg, 0.15 mmol) in CH2Cl2 (5 mL), DIPEA (0.05 mL, 0.30 mmol) was added. The mixture was cooled to 0 °C before a solution of HATU (63.2 mg, 0.17 mmol) in NMP (1 mL) was added. Then, a solution of 4b (49.9 mg, 0.20 mmol) in CH2Cl2 (5 mL) was added dropwise. The mixture was stirred for 165 min at 0 °C before the solvent was evaporated. The residue was dissolved in aqueous NaHCO3 and extracted several times with Et2O. The combined organic layers were washed with aqueous NaCl, dried (Mg2SO4), and evaporated. The residue was purified by flash chromatography (CH2Cl2−MeOH 98:2) to give (R)-5c as a white solid (65.9 mg, 89%): [α]D25.5 +68.2°; IR 3348 s, 2927 s, 2854 m, 1639 s, 1523 m, 1435 m, 1072 s, 1045 s cm−1; 1H NMR (360 MHz, CDCl3) δ 0.87 (t, J = 7.4 Hz, 3 H), 1.49 (m, 2 H), 1.62 (m, 4 H), 2.50 (m, 2 H), 2.60 (m, 2 H), 2.80−3.70 (m, 15 H), 5.95 (m, 1 H), 6.40 (m, 1 H), 6.44 (m, 1 H), 6.52−6.65 (m, 4 H), 6.81 (m, 1 H), 7.09−7.17 (m, 4 H); 13C NMR (90 MHz, CDCl3) δ 11.95, 19.75, 24.89, 27.89, 34.77, 35.11, 35.28, 35.46, 36.38, 39.75, 51.03, 53.25, 63.10, 124.43, 126.35, 131.57, 132.01, 132.41, 132.55, 132.58, 134.84, 135.20, 135.84, 138.97, 139.13, 139.84, 140.13, 141.63, 169.36; EI-MS m/z 480.
N-[(N′-Indan-2-yl-N′-propyl)-4-aminobutyl]-4-(S)-[2.2]paracyclophane carboxamide ((S)-5c)
To a cooled solution of 4-(S)-[2.2]paracyclophane carboxylic acid (30.3 mg, 0.12 mmol) in CH2Cl2 (5 mL), DIPEA (0.04 mL, 0.24 mmol) was added. A solution of HATU (48.0 mg, 0.13 mmol) in NMP (1 mL) was added. Then, a solution of 4b (34.9 mg, 0.14 mmol) in CH2Cl2 (5 mL) was added dropwise. Stirring for 180 min at 0 °C, workup, and purification was done according to the protocol for (R)-5c to give (S)-5c as a white solid (37.9 mg, 66%): [α]D27 −67.9°.
Abbreviations
RASSL, receptor activated solely by synthetic ligands; HEK, human embryonic kidney cells; CHO, Chinese hamster ovarian cells; EDTA, ethylenediaminetetraacetic acid; 7-OH-DPAT, 7-hydroxy-2-(N,N-di-n-propylamino)tetralin; IBMX, 3-isobutyl-1-methylxanthine; Ro 20-1724, [4-(3-butoxy-4-methoxybenzyl)imidazoline; K0.5, the concentration of the compound producing 50% inhibition of the specific binding of the radioactive ligand (Ki value independent of the HILL slope, nH); EC50, half-maximal effective concentration; IC50, half-maximal inhibitory concentration; PKA, protein kinase A; THF, tetrahydrofuran; HOAc, acetic acid; HATU, 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; DIPEA, N,N-diisopropylethylamine; DMF, N,N-dimethylformamide; NMP, N-methylpyrrolidone; ELISA, enzyme-linked immunosorbent assay; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; pen−strep, penicillin and streptomycin; Tris, tris(hydroxymethyl)aminomethane; PBS, phosphate-buffered saline; 5-HT, 5-hydroxytryptamine; Hepes, N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid.
Supporting Information Available
Receptor binding data for dopamine and test compounds at various receptors, comparison of dopamine and quinpirole for activation of the mutant receptor, and evaluation of antagonistic effects of dopamine on the mutant receptor. This information is available free of charge via the Internet at http://pubs.acs.org.
GlaxoSmithKline Consumer Healthcare GmbH & Co. KG, Bussmatten 1, D-77815 Bühl, Germany.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG).
Supplementary Material
References
- Koh J. T.; Putnam M.; Tomic-Canic M.; McDaniel C. M. (1999) Selective regulation of gene expression using rationally-modified retinoic acid receptors. J. Am. Chem. Soc. 121, 1984–1985. [Google Scholar]
- Tedesco R.; Thomas J. A.; Katzenellenbogen B. S.; Katzenellenbogen J. A. (2001) The estrogen receptor: A structure-based approach to the design of new specific hormone-receptor combinations. Chem. Biol. 8, 277–287. [DOI] [PubMed] [Google Scholar]
- Doyle D. F.; Braasch D. A.; Jackson L. K.; Weiss H. E.; Boehm M. F.; Mangelsdorf D. J.; Corey D. R. (2001) Engineering orthogonal ligand−receptor pairs from “near drugs”. J. Am. Chem. Soc. 123, 11367–11371. [DOI] [PubMed] [Google Scholar]
- Henssler E. M.; Scholz O.; Lochner S.; Gmeiner P.; Hillen W. (2004) Structure-based design of Tet repressor to optimize a new inducer specificity. Biochemistry 43, 9512–9518. [DOI] [PubMed] [Google Scholar]
- Krueger C.; Danke C.; Pfleiderer K.; Schuh W.; Jäck H.-M.; Lochner S.; Gmeiner P.; Hiller W.; Berens C. (2006) A gene regulation system with four distinct expression levels. J. Gene Med. 8, 1037–1047. [DOI] [PubMed] [Google Scholar]
- Berens C.; Lochner S.; Löber S.; Usai I.; Schmidt A.; Drueppel L.; Hillen W.; Gmeiner P. (2006) Subtype selective tetracycline agonists and their application for a two-stage regulatory system. ChemBioChem 7, 1320–1324. [DOI] [PubMed] [Google Scholar]
- Kroeze W. K.; Sheffler D. J.; Roth B. L. (2003) G-protein-coupled receptors at a glance. J. Cell Sci. 116, 4867–4869. [DOI] [PubMed] [Google Scholar]
- Pei Y.; Rogan S. C.; Yan F.; Roth B. L. (2008) Engineered GPCRs as tools to modulate signal transduction. Physiology 23, 313–321. [DOI] [PubMed] [Google Scholar]
- Conklin B. R. (2007) New tools to build synthetic hormonal pathways. Proc. Natl. Acad. Sci. U.S.A. 104, 4777–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin B. R.; Hsiao E. C.; Claeysen S.; Dumuis A.; Srinivasan S.; Forsayeth J. R.; Guettier J. M.; Chang W. C.; Pei Y.; McCarthy K. D.; Nissenson R. A.; Wess J.; Bockaert J.; Roth B. L. (2008) Engineering GPCR signaling pathways with RASSLs. Nat. Methods 5, 673–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A.; Gao Z. G.; Chen A.; Barak D.; Kim S. A.; Lee K.; Link A.; Rompaey P. V.; van Calenbergh S.; Liang B. T. (2001) Neoceptor concept based on molecular complementarity in GPCRs: A mutant adenosine A(3) receptor with selectively enhanced affinity for amine-modified nucleosides. J. Med. Chem. 44, 4125–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster B. N.; Li X.; Pausch M. H.; Herlitze S.; Roth B. L. (2007) From the Cover: Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U.S.A. 104, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strader C. D.; Gaffney T.; Sugg E. E.; Candelore M. R.; Keys R.; Patchett A. A.; Dixon R. A. (1991) Allele-specific activation of genetically engineered receptors. J. Biol. Chem. 266, 5–8. [PubMed] [Google Scholar]
- Claeysen S.; Joubert L.; Sebben M.; Bockaert J.; Dumuis A. (2003) A single mutation in the 5-HT4 receptor (5-HT4-R D100(3.32)A) generates a Gs-coupled receptor activated exclusively by synthetic ligands (RASSL). J. Biol. Chem. 278, 699–702. [DOI] [PubMed] [Google Scholar]
- Pauwels P. J.; Colpaert F. C. (2000) Disparate ligand-mediated Ca(2+) responses by wild-type, mutant Ser(200)Ala and Ser(204)Ala alpha(2A)-adrenoceptor: G(alpha15) fusion proteins: Evidence for multiple ligand-activation binding sites. Br. J. Pharmacol. 130, 1505–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruysters M.; Jongejan A.; Akdemir A.; Bakker R. A.; Leurs R. (2005) A Gq/11-coupled mutant histamine H1 receptor F435A activated solely by synthetic ligands (RASSL). J. Biol. Chem. 280, 34741–34746. [DOI] [PubMed] [Google Scholar]
- Srinivasan S.; Vaisse C.; Conklin B. R. (2003) Engineering the melanocortin-4 receptor to control Gs signaling in vivo. Ann. N.Y. Acad. Sci. 994, 225–232. [DOI] [PubMed] [Google Scholar]
- Coward P.; Wada H. G.; Falk M. S.; Chan S. D. H.; Meng F.; Akil H.; Conklin B. R. (1998) Controlling signaling with a specifically designed Gi-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 95, 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scearce-Levie K.; Coward P.; Redfern C. H.; Conklin B. R. (2001) Engineering receptors activated solely by synthetic ligands (RASSLs). Trends Pharmacol. Sci. 22, 414–420. [DOI] [PubMed] [Google Scholar]
- Mueller K. L.; Hoon M. A.; Erlenbach I.; Chandrashekar J.; Zuker C. S.; Ryba N. J. (2005) The receptors and coding logic for bitter taste. Nature 434, 225–229. [DOI] [PubMed] [Google Scholar]
- Wiens B. L.; Nelson C. S.; Neve K. A. (1998) Contribution of serine residues to constitutive and agonist-induced signaling via the D2S dopamine receptor: Evidence for multiple, agonist-specific active conformations. Mol. Pharmacol. 54, 435–444. [DOI] [PubMed] [Google Scholar]
- Elsner J.; Boeckler F.; Heinemann F. W.; Hübner H.; Gmeiner P. (2005) Pharmacophore-guided drug discovery investigations leading to bioactive 5-aminotetrahydropyrazolopyridines. Implications for the binding mode of heterocyclic dopamine D3 receptor agonists. J. Med. Chem. 48, 5771–5779. [DOI] [PubMed] [Google Scholar]
- Dörfler M.; Tschammer N.; Hamperl K.; Hübner H.; Gmeiner P. (2008) Novel D3 selective dopaminergics incorporating enyne units as nonaromatic catechol bioisosteres: Synthesis, bioactivity, and mutagenesis studies. J. Med. Chem. 51, 6829–6838. [DOI] [PubMed] [Google Scholar]
- Ehrlich K.; Götz A.; Bollinger S.; Tschammer N.; Bettinetti L.; Härterich S.; Hübner H.; Lanig H.; Gmeiner P. (2009) Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific receptor binding. J. Med. Chem. 52, 4923–4935. [DOI] [PubMed] [Google Scholar]
- Flierl M. A.; Rittirsch D.; Huber-Lang M.; Sarma J. V.; Ward P. A. (2008) Catecholamines-crafty weapons in the inflammatory arsenal of immune/inflammatory cells or opening Pandora's box?. Mol. Med. 14, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault J.-A.; Greengard P. (2004) The neurobiology of dopamine signaling. Arch. Neurol. 61, 641–644. [DOI] [PubMed] [Google Scholar]
- Wang G. J.; Volkow N. D.; Logan J.; Pappas N. R.; Wong C. T.; Zhu W.; Netusil N.; Fowler J. S. (2001) Brain dopamine and obesity. Lancet 357, 354–357. [DOI] [PubMed] [Google Scholar]
- Bordo D.; Argos P. (1991) Suggestions for “safe” residue substitutions in site-directed mutagenesis. J. Mol. Biol. 217, 721–729. [DOI] [PubMed] [Google Scholar]
- Saha B.; Mondal A. C.; Majumder J.; Basu S.; Dasgupta P. S. (2001) Physiological concentrations of dopamine inhibit the proliferation and cytotoxicity of human CD4+ and CD8+ T cells in vitro: A receptor-mediated mechanism. Neuroimmunomodulation 9, 23–33. [DOI] [PubMed] [Google Scholar]
- Kipnis J.; Cardon M.; Avidan H.; Lewitus G. M.; Mordechay S.; Rolls A.; Shani Y.; Schwartz M. (2004) Dopamine, through the extracellular signal-regulated kinase pathway, downregulates CD4+CD25+ regulatory T-cell activity: Implications for neurodegeneration. J. Neurosci. 24, 6133–6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels P. J. (2003) Unravelling multiple ligand-activation binding sites using RASSL receptors. Trends Pharmacol. Sci. 24, 504–507. [DOI] [PubMed] [Google Scholar]
- Cherezov V.; Rosenbaum D. M.; Hanson M. A.; Rasmussen S. G. F.; Thian F. S.; Kobilka T. S.; Choi H.-J.; Kuhn P.; Weis W. I.; Kobilka B. K.; Stevens R. C. (2007) High-resolution crystal structure of an engineered human 2-adrenergic G protein coupled receptor. Science 318, 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D. M.; Cherezov V.; Hanson M. A.; Rasmussen S. G. F.; Thian F. S.; Kobilka T. S.; Choi H.-J.; Yao X.-J.; Weis W. I.; Stevens R. C.; Kobilka B. K. (2007) GPCR engineering yields high-resolution structural insights into 2-adrenergic receptor function. Science 318, 1266–1273. [DOI] [PubMed] [Google Scholar]
- Warne T.; Serrano-Vega M. J.; Baker J. G.; Moukhametzianov R.; Edwards P. C.; Henderson R.; Leslie A. G.; Tate C. G.; Schertler G. F. (2008) Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454, 486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola V.-P.; Griffith M. T.; Hanson M. A.; Cherezov V.; Chien E. Y. T.; Lane J. R.; Ijzerman A. P.; Stevens R. C. (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz T. W.; Frimurer T. M.; Holst B.; Rosenkilde M. M.; Elling C. E. (2006) Molecular mechanism of 7TM receptor activation--a global toggle switch model. Annu. Rev. Pharmacol. Toxicol. 46, 481–519. [DOI] [PubMed] [Google Scholar]
- Visiers I.; Ballesteros J. A.; Weinstein H. (2002) Three-dimensional representations of G protein-coupled receptor structures and mechanisms. Methods Enzymol. 343, 329–371. [DOI] [PubMed] [Google Scholar]
- Xhaard H.; Rantanen V. V.; Nyronen T.; Johnson M. S. (2006) Molecular evolution of adrenoceptors and dopamine receptors: Implications for the binding of catecholamines. J. Med. Chem. 49, 1706–1719. [DOI] [PubMed] [Google Scholar]
- Floresca C. Z.; Schetz J. A. (2004) Dopamine receptor microdomains involved in molecular recognition and the regulation of drug affinity and function. J. Recept. Signal Transduction Res. 24, 207–239. [DOI] [PubMed] [Google Scholar]
- Cho W.; Taylor L. P.; Mansour A.; Akil H. (1995) Hydrophobic residues of the D2 dopamine receptor are important for binding and signal transduction. J. Neurochem. 65, 2105–2115. [DOI] [PubMed] [Google Scholar]
- Javitch J. A.; Ballesteros J. A.; Weinstein H.; Chen J. (1998) A cluster of aromatic residues in the sixth membrane-spanning segment of the dopamine D2 receptor is accessible in the binding-site crevice. Biochemistry 37, 998–1006. [DOI] [PubMed] [Google Scholar]
- Mirzadegan T.; Benko G.; Filipek S.; Palczewski K. (2003) Sequence analyses of G-protein-coupled receptors: Similarities to rhodopsin. Biochemistry 42, 2759–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley C.; Woodward R.; Johansson A. M.; Strange P. G.; Naylor L. H. (2000) Effect of multiple serine/alanine mutations in the transmembrane spanning region V of the D2 dopamine receptor on ligand binding. J. Neurochem. 74, 358–366. [DOI] [PubMed] [Google Scholar]
- Hübner H.; Haubmann C.; Utz W.; Gmeiner P. (2000) Conjugated enynes as nonaromatic catechol bioisosteres: Synthesis, binding experiments, and computational studies of novel dopamine receptor agonists recognizing preferentially the D(3) subtype. J. Med. Chem. 43, 756–762. [DOI] [PubMed] [Google Scholar]
- Bettinetti L.; Löber S.; Hübner H.; Gmeiner P. (2005) Parallel synthesis and biological screening of dopamine receptor ligands taking advantage of a click chemistry based BAL linker. J. Comb. Chem. 7, 309–316. [DOI] [PubMed] [Google Scholar]
- Schlotter K.; Boeckler F.; Hübner H.; Gmeiner P. (2006) Fancy bioisosteres: Novel paracyclophane derivatives as super-affinity dopamine D3 receptor antagonists. J. Med. Chem. 49, 3628–3635. [DOI] [PubMed] [Google Scholar]
- Schlotter K.; Boeckler F.; Hübner H.; Gmeiner P. (2005) Fancy bioisosteres: Metallocene-derived G-protein-coupled receptor ligands with subnanomolar binding affinity and novel selectivity profiles. J. Med. Chem. 48, 3696–3699. [DOI] [PubMed] [Google Scholar]
- Kumar R.; Riddle L. R.; Griffin S. A.; Chu W.; Vangveravong S.; Neisewander J.; Mach R. H.; Luedtke R. R. (2009) Evaluation of D2 and D3 dopamine receptor selective compounds on l-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology 56, 956–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A.; Neumeyer J. L.; Baldessarini R. J. (2007) Recent progress in development of dopamine receptor subtype-selective agents: Potential therapeutics for neurological and psychiatric disorders. Chem. Rev. 107, 274–302. [DOI] [PubMed] [Google Scholar]
- Boeckler F.; Lanig H.; Gmeiner P. (2005) Modeling the similarity and divergence of dopamine D2-like receptors and identification of validated ligand-receptor complexes. J. Med. Chem. 48, 694–709. [DOI] [PubMed] [Google Scholar]
- Lowry O. H.; Rosebrough N. J.; Farr A. L.; Randall R. J. (1951) Protein measurement with the folin phenol reagent. J. Biol. Chem. 193, 265–275. [PubMed] [Google Scholar]
- Heindl C.; Hübner H.; Gmeiner P. (2003) Ex-chiral pool synthesis and receptor binding studies of 4-substituted prolinol derivatives. Tetrahedron: Asymmetry 14, 3141–3152. [Google Scholar]
- Cheng Y.; Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Cannon J. G.; Perez J. A.; Pease J. P.; Long J. P.; Flynn J. R.; Rusterholz D. B.; Dryer S. E. (1980) Comparison of biological effects of N-alkylated congeners of beta-phenethylamine derived from 2-aminotetralin, 2-aminoindan, and 6-aminobenzocycloheptene. J. Med. Chem. 23, 745–749. [DOI] [PubMed] [Google Scholar]
- Cram D. J.; Day A. C. (1966) Macro Rings, XXXI. Quinone derived from [2.2]paracyclophane, an intramolecular-molecular complex. J. Org. Chem. 31, 1227–1232. [Google Scholar]
- Rozenberg V.; Dunrovina N.; Sergeeva E.; A. D.; Belokon Y. (1998) An improved synthesis of (S)-(+)- and (R)-(-)-[2.2]paracyclophane-4-carbocyclic acid. Tetrahedron: Asymmetry 9, 653–656. [Google Scholar]
- Stemp G., and Johnson C. N. (1999) Bicyclic amine derivatives and their use as anti-psychotic agents: SmithKline Beech p.I.c.: U.S. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.