

Abstract
Most intercellular glutamate signaling in the nervous system occurs at synapses. Some intercellular glutamate signaling occurs outside synapses, however, and even outside the nervous system where high ambient extracellular glutamate might be expected to preclude the effectiveness of glutamate as an intercellular signal. Here, I briefly review the types of intercellular glutamate signaling in the nervous system and beyond, with emphasis on the diversity of signaling mechanisms and fundamental unanswered questions.
Keywords: Glutamate, synaptic, paracrine, receptor
All organisms, and all tissues within organisms, rely on intercellular chemical signals. One of the most important intercellular chemical signals used in nervous systems is glutamate. Brain tissue contains an unusually high concentration of glutamate, approximately 5−15 mmol/kg (1,2). Most of this glutamate is found in neurons. The glutamate concentration in the cytoplasm of glutamatergic neurons is approximately 5−10 mM (1,3−6). This is several times more than any other amino acid, and much more than in other tissue types. Neuronal cytoplasmic glutamate concentrations are highest in axon terminals, where glutamate is 2−3 times that of cell bodies or dendrites (7). This implies that axon terminals somehow restrict the movement of glutamate or (more likely) synthesize and utilize glutamate locally. Local glutamate synthesis in axon terminals may be attributable to localization of glutaminases, which are responsible for synthesis of most neuronal glutamate (8). Consistent with this, immunolocalization studies suggest that glutaminases or glutaminase interacting proteins (GIPs) are associated with mitochondria (9−12), which are particularly abundant in axon terminals (13). In other words, glutamate is likely abundant in axon terminals because glutamate is made in mitochondria and mitochondria are abundant in axon terminals. It is therefore possible that regulation of mitochondrial localization or enzymatic activity could modulate synaptic transmission by controlling glutamate availability. Indeed, Drosophila mutants lacking kinesins critical for mitochondrial transport in axons show activity-dependent loss of synaptic glutamate release (14), though it is not clear whether this is due to glutamate depletion.
Cytoplasmic glutamate in neuronal terminals is further concentrated within synaptic vesicles by proton/glutamate antiport proteins called vesicular glutamate transporters (VGluts) (15,16). The glutamate concentration in synaptic vesicles can reach 100 mM or more (17−19). Synaptic vesicles are small (30−40 nm) proteinaceous organelles that cluster near synapses and fuse with the plasma membrane in response to high cytosolic calcium, which is typically the result of voltage-gated calcium channels opening in response to plasma membrane voltage changes (i.e., action potentials) (20,21). When they fuse with the plasma membrane, synaptic vesicles spill their glutamate into the synaptic cleft. The synaptic cleft is the small (∼20 nm wide) gap between two cells connected by a synapse. Synapses are specialized cell−cell junctions where fast chemical signaling occurs (there are also “electrical synapses”, which are really gap junctions, but we will not discuss them here). Synapses are asymmetric cell junctions. They are formed from one presynaptic cell, which releases the chemical signal, and one postsynaptic cell, which receives and transduces the signal. Any particular neuron could, however, form synapses with multiple other cells (and most do). When synaptic vesicles fuse with the presynaptic neuronal plasma membrane, the synaptic cleft glutamate concentration is thought to reach several millimolar, depending on the shape of the synaptic cleft and activity of nearby glutamate transporters (22−24).
Once secreted into the synaptic cleft, glutamate can diffuse to and interact with any nearby targets. The closest target is postsynaptic cell membrane. Postsynaptic cell plasma membrane associated with the synaptic cleft contains an unusually large amount of membrane-associated protein, so much so that these “postsynaptic densities (PSDs)” are visible by electron microscopy (25). PSDs contain, among other things, many glutamate receptor proteins. Glutamate receptor proteins bind glutamate and trigger changes in the postsynaptic cell, thus completing the synaptic transmission of glutamatergic signals from presynaptic to postsynaptic cells. Glutamate released from the presynaptic cell could also bind to receptors on the presynaptic plasma membrane (“autocrine” signaling) or on other nearby cells (“paracrine” signaling).
There are two general types of glutamate receptor: ionotropic receptors (iGluRs) and metabotropic receptors (mGluRs) (26). Metabotropic glutamate receptors are seven-pass membrane proteins coupled to intracellular heterotrimeric G-proteins. Binding of glutamate to mGluRs triggers activation of G-protein-dependent intracellular signaling cascades (27,28). There are three subtypes of mammalian mGluR, called type I, type II, and type III. Type I mGluRs tend to activate phospholipase C, while type II and type III mGluRs are usually coupled to cAMP pathways (29). Ionotropic glutamate receptors, in contrast, are multimeric glutamate-gated ion channels. When glutamate binds to the extracellular domains of iGluR subunits, the entire protein changes conformation to allow cation flow through the plasma membrane (which usually leads to postsynaptic cell depolarization). Mammalian iGluRs are also subdivided into three types: AMPA (α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate) receptors, NMDA (N-methyl-d-aspartic acid) receptors, and kainate receptors (26,30,31). Fast information flow in the nervous system is thought to occur mostly via AMPA receptors (30,32,33). Kainate receptors, though found postsynaptically in some synapses, are (like mGluRs) also present on presynaptic terminal membranes, where they serve to autoregulate synaptic vesicle function and glutamate secretion (34). NMDA receptors tend to remain open longer than AMPA or kainate receptors and allow significant calcium influx. Thus, NMDA receptor activation can initiate intracellular calcium-dependent signaling cascades that lead to changes in gene expression and synaptic strength (which manifest as, for example, learning and memory) (30,35,36). Glycine and serine function as coligands for NMDA receptors (37). It is possible that aspartate may also be an endogenous ligand for glutamate receptors in vivo (38,39).
There is little or no biochemical conversion of glutamate in the extracellular space (1), meaning that glutamate is capable of continuously interacting with receptors until it diffuses away or is removed from the extracellular fluid by excitatory amino acid transporters (EAATs). There are five types of EAATs, which differ slightly in function and localization: glutamate−aspartate transporter (GLAST/EAAT1), glutamate transporter (GLT/EAAT2), excitatory amino acid carrier (EAAC/EAAT3), and excitatory amino acid transporters 4 and 5 (EAAT 4 and EAAT5) (1). EAATs are clearly important for extracellular glutamate homeostasis; when EAATs are pharmacologically inhibited, extracellular glutamate concentration rises from basal levels of 0.5−5 uM to several hundred micromolar (1,40,41). All EAATs couple the import of l-glutamate (and l- or d-aspartate to some degree) to movement of sodium or potassium (1). Most EAATs are present in astrocytes (a common type of glia) near glutamatergic synapses, suggesting that glutamate released at synapses is mostly recycled by astrocytes rather than neurons. The conventional “textbook” view is that glutamate released from synaptic vesicles is pumped into astrocytes via EAATs, converted to glutamine, released to the extracellular space, taken up by neurons, and converted back to glutamate. However, EAATs are also expressed in nerve axon terminals and dendritic spines, which form the pre- and postsynaptic membranes of glutamatergic synapses, respectively. Because of their proximity to the synaptic cleft, neuronal EAATs are thought to be particularly important for shaping the time course of glutamate in the synaptic cleft, and therefore might regulate synaptic signaling in biologically important ways.
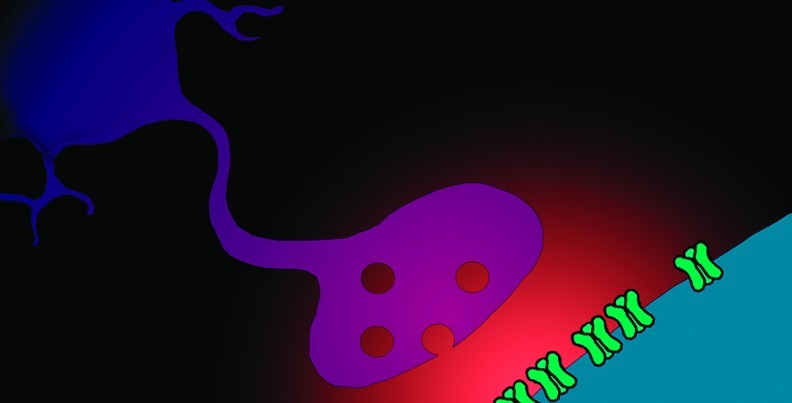
Synaptic glutamate signaling can be summarized as follows (Figure 1): (1) Glutamate synthesized in the cytoplasm of glutamatergic neurons is pumped into synaptic vesicles and then released into the extracellular space when these vesicles fuse with the plasma membrane. (2) This transiently increases the concentration of glutamate in the synaptic cleft. (3) This transient increase in glutamate is detected and transduced by glutamate receptors. (4) Glutamate is removed from the synaptic cleft by EAATs.
Figure 1.

A canonical (but probably atypical) glutamatergic synapse. Cytoplasmic glutamate is put into synaptic vesicles by vesicular glutamate transporter (VGlut) proteins and eventually released into the synaptic cleft when the vesicles fuse with the presynaptic plasma membrane. After being released into the synaptic cleft, glutamate may bind to and activate glutamate receptor proteins. The postsynaptic density (PSD) contains by far the highest concentration of glutamate receptor protein, but receptors are also found in presynaptic terminals, in the neuronal membrane outside the synapse, in astrocytes, and in other cell types. There are two main types of glutamate receptor: ionotropic glutamate receptors (iGluRs) and metabotropic glutamate receptors (mGluRs). Glutamate secreted into the extracellular space is transported back into cells by excitatory amino acid transporters (EAATs). Most EAATs are present in astrocytes, although EAATs also exist in neurons and other cell types. Glutamate taken up by astrocytes is recycled by being converted to glutamine and exported into the extracellular space, where it is then transported into neurons and converted back into glutamate. Glia, neurons, and (to a lesser degree) other cell types also express xCT cystine−glutamate antiport proteins, which comprise the xc-system of amino acid transport. xCT proteins export glutamate from cells and import cystine; they are the largest source of ambient extracellular glutamate in the nervous system. Ambient extracellular glutamate modulates synaptic transmission via constitutive activation of mGluRs or constitutive desensitization of iGluRs (see text).
Interestingly, synaptic transmission does not appear to be the major source of extracellular glutamate in the brain, since extracellular glutamate, measured by microdialysis, is not affected by blockade of synaptic glutamate release (40,42−44). The majority of extracellular glutamate in the brain must therefore be nonvesicular. Nonvesicular glutamate release could be mediated by several different mechanisms, including reversed uptake, swelling-activated anion channels, gap junction hemichannels, purinergic (P2X) receptors, and heteromeric sodium-independent amino acid transporters, including, in particular xCT-based cystine−glutamate antiporters (43,45−50). Of these different mechanisms, cystine−glutamate transporters appear to be responsible for most extracellular glutamate in the brain, because pharmacological inhibition in rats reduces ambient extracellular glutamate to approximately one-half normal (43,51), and genetic elimination of cystine−glutamate transporters in Drosophila and mice also show significantly less extracellular glutamate (ref (52) and unpublished work). Cystine−glutamate proteins, although also expressed in neurons, are most highly expressed in glia (53,54). Thus, most extrasynaptic glutamate in the nervous system likely comes from glia. This is interesting because, as mentioned above, glia also express the majority of EAATs. The continual “give and take” of glutamate into the extracellular space likely allows glial cells to shape the form and intensity of extracellular glutamate pools to a remarkable degree and for various modulatory purposes. It would be interesting to visualize and study the ebb and flow of extracellular glutamate through the brain, which is after all 20% extracellular space by volume. Unfortunately, this is a difficult proposition, because noninvasive glutamate sensors (such as the optical sensors recently developed by Hires et al. (84)) have not been utilized for in vivo studies. Other widely used and generally successful approaches, such as cell culture or slice preparations, disrupt the spatial and chemical milieu under investigation.
Extracellular glutamate in the nervous system may thus be divided into two functionally and spatially distinct pools: (1) “synaptic glutamate”, a transient (few milliseconds) burst of high (0.5−5 mM) glutamate that appears in the synaptic cleft when glutamate-filled synaptic vesicles in neurons fuse with the presynaptic membrane, and (2) “ambient extracellular glutamate”, a relatively steady (but still likely somewhat dynamic) background of glutamate, mostly from glia, that fills most of the extracellular space at lower concentration (1−5 uM).
Changes in nonsynaptic extracellular glutamate are associated with developmental, physiological, or behavioral effects (44,55−58), consistent with the idea that ambient extracellular glutamate, released nonvesicularly and modulated dynamically, has important paracrine signaling functions. In other words, ambient extracellular glutamate probably acts like a paracrine modulator. For example, nonvesicular paracrine glutamate released by cystine−glutamate transporters has been linked to drug withdrawal behavior (59,60). Chronic treatment with cocaine reduces cystine−glutamate transporter expression in the nucleus accumbens of rats. This leads to a decrease in ambient extracellular glutamate, measured by microdialysis, and increased drug-seeking behavior (60). Importantly, these behavioral alterations are mimicked by cystine−glutamate antagonists (61).
Glutamate released by cystine−glutamate transporters presumably affects behavior by modifying neuronal function and does this via either synaptic or extrasynaptic glutamate receptors. Extrasynaptic glutamate receptors, not being subject to the dramatic changes in synaptic glutamate experienced by synaptic receptors, are obviously in an excellent position to mediate paracrine glutamate signaling. Although glutamate receptors in the nervous system are present in highest density in postsynaptic membrane, many glutamate receptors are also present in extrasynaptic membrane (62). Extrasynaptic glutamate receptors seem to play unique roles in development and disease (62−66), but they are very understudied compared with synaptic receptors, which might also be somewhat “protected” from ambient extracellular glutamate due to high EAAT activity near the synaptic cleft.
In part, the presumed protection of synaptic receptors by EAATs is why changes in drug-seeking behavior regulated by cystine−glutamate transporters are thought to be due to altered activation of extrasynaptic mGluRs. Specifically, loss of cystine−glutamate transporters and reduced ambient extracellular glutamate is thought to reduce tonic activation of extrasynaptic group 2 and group 3 mGluRs, leading to increased synaptic transmission and altered drug-seeking behavior (60). Consistent with this, drug-seeking behavior is altered in expected ways by mGluR antagonists and agonists (59−61,67,68). mGluR antagonists simulate loss of “ambient extracellular glutamate tone” and mimic addiction, while mGluR agonists promote constitutive mGluR activity and ameliorate the effects of chronic dug administration. Ambient extracellular glutamate and cystine−glutamate transporters might modulate many other behaviors in a similar manner, since mGluR activity regulates many aspects of cell biology (29,69). mGluR activation in a variety of synapse types, for example, leads to reductions in synaptic transmission or “long-term depression (LTD)” (70). In other synapses or under other conditions, mGluR activation leads to synaptic strengthening (LTP) (71). LTP and LTD both underlie various forms of learning.
Ambient extracellular glutamate might also regulate brain function by altering the number of functional ionotropic glutamate receptors in the synapse. Specifically, it has been proposed that ambient extracellular glutamate causes constitutive desensitization of ionotropic glutamate receptors, and subsequent receptor protein loss from the synapse (44). Ionotropic receptors are desensitized at glutamate concentrations of a few micromolar or less, well within the ambient range measured in vivo (44). Desensitization itself effectively removes receptors from the functional pool, but because desensitization affects receptor trafficking and localization, high ambient extracellular glutamate can also remove receptors from the synapse (44,52,56,57). This idea was tested explicitly in Drosophila, which showed dramatic changes in the number of synaptic receptors and behavior after genetic reductions in cystine−glutamate transporter function (52,56,72). These changes were desensitization dependent and appeared to involve alterations in the number of both ionotropic and metabotropic receptors (56). The mechanism by which receptors are lost from the synapse after desensitization remains unknown.
In a way, glutamate in the nervous system might be thought of as noise at a party. Synaptic glutamate signaling can be compared with individual conversations between people, spatially isolated, fast, and information-rich. Ambient extracellular glutamate, on the other hand, is like background music; it can drown out (and thus modulate the effectiveness of) conversation between individuals, but it also sets the mood. Like music at a party, ambient extracellular glutamate varies spatially and temporally (73−77). For example, changes in extracellular glutamate in the nervous system are associated with circadian rhythms and sleep/wake states (78,79). Melatonin alters glial glutamate uptake and thereby ambient extracellular glutamate (80). It is not clear whether synaptic suppression by ambient extracellular glutamate might contribute to the particular quiescence of sleep but is easy to imagine that modulation of mGluRs or synaptic receptor availability by ambient extracellular glutamate might contribute to various behavioral states, including hunger, anxiety, depression, or other moods (62,81,82). Alterations in cystine−glutamate transporter expression are also linked to schizophrenia (68).
Despite increasing evidence that ambient extracellular glutamate profoundly affects brain function, the idea is still quite controversial. In part, this is due to a pervasive belief that nonpathological ambient extracellular glutamate levels are extremely low, too low to have any significant biological effect. This belief is based mainly on the idea that the specificity of synaptic glutamate signaling requires virtually nonexistent background levels of glutamate. Specificity of glutamate signaling does not, however, require extraordinarily low extracellular glutamate. Indeed, the idea that ambient extracellular glutamate secreted by cystine−glutamate transporters alters behavior by constitutive desensitization of glutamate receptors relies on the assumption that ambient extracellular glutamate does in fact interfere with synaptic transmission. The situation is not unlike that found in voltage-gated ion channels, which are to a large degree constitutively inactivated at resting membrane potential (83). There is intense interest in the voltage dependence of voltage-gated ion channel inactivation precisely because the steepness of inactivation curves means that the functional number of ion channels can be efficiently modulated by slight changes in resting membrane potential or channel voltage sensitivity. The same principles apply to ligand-gated receptors.
There are other more interesting arguments against paracrine glutamate signaling. For example, recent experiments using baseline NMDA receptor activity as an indicator of ambient glutamate have argued that baseline synaptic cleft glutamate is approximately 25 nM (41). This is much less than the 1−5 μM typically measured by microdialysis. Why the discrepancy? First, there is no doubt that microdialysis probes damage cells as they penetrate brain tissue. As described above, glutamatergic neurons contain very high levels of cytoplasmic glutamate. Therefore, it is easy to imagine that probe-based glutamate measurements might be artifactually high due to glutamate leakage from cells, though it is more difficult to imagine how such an “artifact” would persist several hours (or days) after probe insertion, which is when measurements are typically made. It is also possible that EAATs in close proximity to the synaptic cleft keep synaptic glutamate levels extremely low relative to glutamate levels outside the synaptic cleft. However, this would preclude synaptic modulation by constitutive desensitization of glutamate receptors and leave certain observations unexplained (44). It will be difficult to know for sure what the relative extracellular glutamate concentrations are in vivo until optical glutamate sensors are applied to the problem and studied with high-resolution microscopy. A daunting task to be sure, but one of fundamental importance.
What about glutamate signaling outside the synapse? Astrocytes (the most abundant glial subtype) release vesicular glutamate in response to high intracellular calcium, just like neurons do (85). They also contain glutamate receptors and participate in local glutamate signaling (86−88), like neurons. For example, glutamate release from neurons activates NMDA receptors in astrocytes, triggering calcium-dependent release of glutamate which in turn activates neuronal NMDA receptors and modulates synaptic transmission (89). This glutamate signaling between neurons and glia is thought to modulate drug behavior, and glutamate secretion by astrocytes in hippocampal slices also potentiates GABAergic transmission between interneurons and pyramidal cells (90), to presumably affect memory or cognition.
Finally, what about glutamate signaling outside the brain, where plasma glutamate levels are 30−50 μM and can vary wildly depending on diet, metabolism, and protein turnover (91−93)? Surely intercellular glutamate signaling cannot exist in such an environment, even allowing for substantial receptor desensitization and perhaps some local glutamate sequestration. But it apparently does. Bone, skin, pancreas, heart, intestine, lung, and liver also all contain proteins associated with interneuronal glutamate signaling, including vesicular glutamate transporters, AMPA receptors, kainate receptors, NMDA receptors, and EAATs (94−98). Furthermore, these components seem to play active roles in intercellular glutamate signaling.
For example, the islets of Langerhans are pancreatic micro-organs containing several types of endocrine cells, which include alpha, beta, delta, PP, and epsilon cells. Glutamate secreted from alpha cells activates AMPA receptors on beta and delta cells, which stimulates them to secrete GABA and somatostatin, which in turn inhibits secretion of glucagon and glutamate from alpha cells (99). Glutamate also autoregulates alpha cell secretion directly via mGluRs (99).
Glutamate signaling outside the nervous system most often seems to regulate cell differentiation. Several studies, for example, strongly suggest that intercellular glutamate signaling regulates bone formation in vivo (94,100). Rat osteoblasts in culture contain mGluRs that modulate functional NMDA receptors whose activation leads to biologically significant increases in intracellular calcium (101). These NMDA receptors have electrophysiological properties similar to NMDA receptors in neurons (102), and NMDA receptor blockers inhibit bone formation (103,104). AMPA receptor blockade, meanwhile, alters osteoblast precursor cell fate (105). Mutant mice without the primary vesicular glutamate transporter (VGLUT1) develop osteoporosis (99).
Similarly, NMDA receptor blockade in human keratinocytes induces signs of differentiation (106,107). Glutamate in this case probably comes from the direction of the basal lamina, since glutamate receptors are expressed basally. This form of glutamate signaling may play an important role in wound healing (94).
Plants also apparently contain glutamate receptors and rely on intercellular glutamate signaling for a variety of purposes, including light signal transduction and growth (108−110). Thus, intercellular glutamate signaling is not even specific to animals.
Until recently, synaptic glutamate signaling was generally assumed to comprise the entirety of intercellular glutamate signaling. However, it is important to remember that glutamate was only reluctantly accepted as a neurotransmitter, in part because of its ubiquity. When one considers the diversity of intercellular glutamate signaling and abundance of apparent glutamate signaling under circumstances previously thought unlikely (high extracellular glutamate), synaptic glutamate signaling does not seem quite so special anymore. For those of us interested in glutamate signaling without being hung up about neurons, this is good news. It means there are vast unexplored worlds ripe for exploration.
References
- Danbolt N. C. (2001) Glutamate uptake. Prog. Neurobiol. 65, 1–105. [DOI] [PubMed] [Google Scholar]
- Basic Neurochemistry: Molecualar, Cellular, and Medical Aspects (2006) 7th ed., Elsevier Academic Press, Burlington, MA. [Google Scholar]
- Ottersen O. P.; Zhang N.; Walberg F. (1992) Metabolic compartmentation of glutamate and glutamine: morphological evidence obtained by quantitative immunocytochemistry in rat cerebellum. Neuroscience 46, 519–534. [DOI] [PubMed] [Google Scholar]
- Osen K. K.; Storm-Mathisen J.; Ottersen O. P.; Dihle B. (1995) Glutamate is concentrated in and released from parallel fiber terminals in the dorsal cochlear nucleus: a quantitative immunocytochemical analysis in guinea pig. J. Comp. Neurol. 357, 482–500. [DOI] [PubMed] [Google Scholar]
- Bramham C. R.; Torp R.; Zhang N.; Storm-Mathisen J.; Ottersen O. P. (1990) Distribution of glutamate-like immunoreactivity in excitatory hippocampal pathways: a semiquantitative electron microscopic study in rats. Neuroscience 39, 405–417. [DOI] [PubMed] [Google Scholar]
- Ottersen O. P.; Storm-Mathisen J.; Bramham C.; Torp R.; Laake J.; Gundersen V. (1990) A quantitative electron microscopic immunocytochemical study of the distribution and synaptic handling of glutamate in rat hippocampus. Prog. Brain Res. 83, 99–114. [DOI] [PubMed] [Google Scholar]
- Ottersen O. P. (1989) Quantitative electron microscopic immunocytochemistry of neuroactive amino acids. Anat. Embryol. (Berlin) 180, 1–15. [DOI] [PubMed] [Google Scholar]
- Marquez J.; Tosina M.; de la Rosa V.; Segura J. A.; Alonso F. J.; Mates J. M.; Campos-Sandoval J. A. (2009) New insights into brain glutaminases: beyond their role on glutamatergic transmission. Neurochem. Int. 55, 64–70. [DOI] [PubMed] [Google Scholar]
- Fisher R. S. (2007) Co-localization of glutamic acid decarboxylase and phosphate-activated glutaminase in neurons of lateral reticular nucleus in feline thalamus. Neurochem. Res. 32, 177–186. [DOI] [PubMed] [Google Scholar]
- Olalla L.; Gutierrez A.; Jimenez A. J.; Lopez-Tellez J. F.; Khan Z. U.; Perez J.; Alonso F. J.; de la Rosa V.; Campos-Sandoval J. A.; Segura J. A.; Aledo J. C.; Marquez J. (2008) Expression of the scaffolding PDZ protein glutaminase-interacting protein in mammalian brain. J. Neurosci. Res. 86, 281–292. [DOI] [PubMed] [Google Scholar]
- Laake J. H.; Takumi Y.; Eidet J.; Torgner I. A.; Roberg B.; Kvamme E.; Ottersen O. P. (1999) Postembedding immunogold labelling reveals subcellular localization and pathway-specific enrichment of phosphate activated glutaminase in rat cerebellum. Neuroscience 88, 1137–1151. [DOI] [PubMed] [Google Scholar]
- Aoki C.; Kaneko T.; Starr A.; Pickel V. M. (1991) Identification of mitochondrial and non-mitochondrial glutaminase within select neurons and glia of rat forebrain by electron microscopic immunocytochemistry. J. Neurosci. Res. 28, 531–548. [DOI] [PubMed] [Google Scholar]
- Ly C. V.; Verstreken P. (2006) Mitochondria at the synapse. Neuroscientist 12, 291–299. [DOI] [PubMed] [Google Scholar]
- Guo X.; Macleod G. T.; Wellington A.; Hu F.; Panchumarthi S.; Schoenfield M.; Marin L.; Charlton M. P.; Atwood H. L.; Zinsmaier K. E. (2005) The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379–393. [DOI] [PubMed] [Google Scholar]
- Takamori S.; Rhee J. S.; Rosenmund C.; Jahn R. (2000) Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407, 189–194. [DOI] [PubMed] [Google Scholar]
- Takamori S. (2006) VGLUTs: 'exciting' times for glutamatergic research?. Neurosci Res 55, 343–351. [DOI] [PubMed] [Google Scholar]
- Riveros N.; Fiedler J.; Lagos N.; Munoz C.; Orrego F. (1986) Glutamate in rat brain cortex synaptic vesicles: influence of the vesicle isolation procedure. Brain Res. 386, 405–408. [DOI] [PubMed] [Google Scholar]
- Burger P. M.; Mehl E.; Cameron P. L.; Maycox P. R.; Baumert M.; Lottspeich F.; De Camilli P.; Jahn R. (1989) Synaptic vesicles immunoisolated from rat cerebral cortex contain high levels of glutamate. Neuron 3, 715–720. [DOI] [PubMed] [Google Scholar]
- Shupliakov O.; Brodin L.; Cullheim S.; Ottersen O. P.; Storm-Mathisen J. (1992) Immunogold quantification of glutamate in two types of excitatory synapse with different firing patterns. J. Neurosci. 12, 3789–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Chacon R.; Sudhof T. C. (1999) Genetics of synaptic vesicle function: toward the complete functional anatomy of an organelle. Annu. Rev. Physiol. 61, 753–776. [DOI] [PubMed] [Google Scholar]
- Takamori S.; Holt M.; Stenius K.; Lemke E. A.; Gronborg M.; Riedel D.; Urlaub H.; Schenck S.; Brugger B.; Ringler P.; Muller S. A.; Rammner B.; Grater F.; Hub J. S.; De Groot B. L.; Mieskes G.; Moriyama Y.; Klingauf J.; Grubmuller H.; Heuser J.; Wieland F.; Jahn R. (2006) Molecular anatomy of a trafficking organelle. Cell 127, 831–846. [DOI] [PubMed] [Google Scholar]
- Clements J. D.; Lester R. A.; Tong G.; Jahr C. E.; Westbrook G. L. (1992) The time course of glutamate in the synaptic cleft. Science 258, 1498–1501. [DOI] [PubMed] [Google Scholar]
- Clements J. D. (1996) Transmitter timecourse in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 19, 163–171. [DOI] [PubMed] [Google Scholar]
- Choi S.; Klingauf J.; Tsien R. W. (2000) Postfusional regulation of cleft glutamate concentration during LTP at 'silent synapses'. Nat. Neurosci. 3, 330–336. [DOI] [PubMed] [Google Scholar]
- Kennedy M. B. (1997) The postsynaptic density at glutamatergic synapses. Trends Neurosci. 20, 264–268. [DOI] [PubMed] [Google Scholar]
- Kew J. N.; Kemp J. A. (2005) Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology (Berlin) 179, 4–29. [DOI] [PubMed] [Google Scholar]
- Pin J. P.; Duvoisin R. (1995) The metabotropic glutamate receptors: structure and functions. Neuropharmacology 34, 1–26. [DOI] [PubMed] [Google Scholar]
- Ferraguti F.; Shigemoto R. (2006) Metabotropic glutamate receptors. Cell Tissue Res. 326, 483–504. [DOI] [PubMed] [Google Scholar]
- Conn P. J.; Pin J.-P. (1997) Pharamcology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 37, 205–237. [DOI] [PubMed] [Google Scholar]
- Monaghan D. T., and Wenthold R. J., Eds. (1997) The Ionotropic Glutamate Receptors, Humana Press, Totowa, NJ. [Google Scholar]
- Ozawa S.; Kamiya H.; Tsuzuki K. (1998) Glutamate receptors in the mammalian central nervous system. Prog. Neurobiol. 54, 581–618. [DOI] [PubMed] [Google Scholar]
- Derkach V. A.; Oh M. C.; Guire E. S.; Soderling T. R. (2007) Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci. 8, 101–113. [DOI] [PubMed] [Google Scholar]
- Kessels H. W.; Malinow R. (2009) Synaptic AMPA receptor plasticity and behavior. Neuron 61, 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro P.; Mulle C. (2006) Kainate receptors. Cell Tissue Res. 326, 457–482. [DOI] [PubMed] [Google Scholar]
- Monaghan D. T.; Bridges R. J.; Cotman C. W. (1989) The excitatory amino acid receptors: their classes, pharmacology, and distinct properties in the function of the central nervous system. Annu. Rev. Pharmacol. Toxicol. 29, 365–402. [DOI] [PubMed] [Google Scholar]
- Whitlock J. R.; Heynen A. J.; Shuler M. G.; Bear M. F. (2006) Learning induces long-term potentiation in the hippocampus. Science 313, 1093–1097. [DOI] [PubMed] [Google Scholar]
- Johnson J. W.; Ascher P. (1992) Equilibrium and kinetic study of glycine action on the N-methyl-D-aspartate receptor in cultured mouse brain neurons. J. Physiol. 455, 339–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen V.; Chaudhry F. A.; Bjaalie J. G.; Fonnum F.; Ottersen O. P.; Storm-Mathisen J. (1998) Synaptic vesicular localization and exocytosis of L-aspartate in excitatory nerve terminals: a quantitative immunogold analysis in rat hippocampus. J. Neurosci. 18, 6059–6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallero A.; Marte A.; Fedele E. (2009) L-Aspartate as an amino acid neurotransmitter: mechanisms of the depolarization-induced release from cerebrocortical synaptosomes. J. Neurochem. 110, 924–934. [DOI] [PubMed] [Google Scholar]
- Jabaudon D.; Shimamoto K.; Yasuda-Kamatani Y.; Scanziani M.; Gahwiler B. H.; Gerber U. (1999) Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc. Natl. Acad. Sci. U.S.A. 96, 8733–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman M. A.; Jahr C. E. (2007) Extracellular glutamate concentration in hippocampal slice. J. Neurosci. 27, 9736–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman W.; Westerink B. H. (1997) Brain microdialysis of GABA and glutamate: what does it signify?. Synapse 27, 242–261. [DOI] [PubMed] [Google Scholar]
- Baker D. A.; Xi Z. X.; Shen H.; Swanson C. J.; Kalivas P. W. (2002) The origin and neuronal function of in vivo nonsynaptic glutamate. J. Neurosci. 22, 9134–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone D. E.; Shippy S. A. (2008) Regulation of synaptic transmission by ambient extracellular glutamate. Neuroscientist 14, 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatkowski M.; Barbour B.; Attwell D. (1990) Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature 348, 443–446. [DOI] [PubMed] [Google Scholar]
- Kimelberg H. K.; Goderie S. K.; Higman S.; Pang S.; Waniewski R. A. (1990) Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J. Neurosci. 10, 1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S.; Anderson C. M.; Keung E. C.; Chen Y.; Swanson R. A. (2003) P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J. Neurosci. 23, 1320–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z. C.; Wyeth M. S.; Baltan-Tekkok S.; Ransom B. R. (2003) Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J. Neurosci. 23, 3588–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperlagh B.; Kofalvi A.; Deuchars J.; Atkinson L.; Milligan C. J.; Buckley N. J.; Vizi E. S. (2002) Involvement of P2X7 receptors in the regulation of neurotransmitter release in the rat hippocampus. J. Neurochem. 81, 1196–1211. [DOI] [PubMed] [Google Scholar]
- Parpura V.; Scemes E.; Spray D. C. (2004) Mechanisms of glutamate release from astrocytes: gap junction “hemichannels”, purinergic receptors and exocytotic release. Neurochem. Int. 45, 259–264. [DOI] [PubMed] [Google Scholar]
- Piyankarage S. C.; Augustin H.; Grosjean Y.; Featherstone D. E.; Shippy S. A. (2008) Hemolymph amino acid analysis of individual Drosophila larvae. Anal. Chem. 80, 1201–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin H.; Grosjean Y.; Chen K.; Sheng Q.; Featherstone D. E. (2007) Nonvesicular release of glutamate by glial xCT transporters suppresses glutamate receptor clustering in vivo. J. Neurosci. 27, 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W. J.; Lyons S. A.; Nelson G. M.; Hamza H.; Gladson C. L.; Gillespie G. Y.; Sontheimer H. (2005) Inhibition of cystine uptake disrupts the growth of primary brain tumors. J. Neurosci. 25, 7101–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Bella V.; Valentino F.; Piccoli T.; Piccoli F. (2007) Expression and developmental regulation of the cystine/glutamate exchanger (xc-) in the rat. Neurochem. Res. 32, 1081–1090. [DOI] [PubMed] [Google Scholar]
- Liu R. R.; Brown C. E.; Murphy T. H. (2007) Differential regulation of cell proliferation in neurogenic zones in mice lacking cystine transport by xCT. Biochem. Biophys. Res. Commun. 364, 528–533. [DOI] [PubMed] [Google Scholar]
- Grosjean Y.; Grillet M.; Augustin H.; Ferveur J. F.; Featherstone D. E. (2008) A glial amino-acid transporter controls synapse strength and courtship in Drosophila. Nat. Neurosci. 11, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.; Augustin H.; Featherstone D. E. (2009) Effect of ambient extracellular glutamate on Drosophila glutamate receptor trafficking and function. J. Comp. Physiol., A 195, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt L. A.; LaRowe S.; Mardikian P.; Malcolm R.; Upadhyaya H.; Hedden S.; Markou A.; Kalivas P. W. (2009) The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol. Psychiatry 65, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D. A.; McFarland K.; Lake R. W.; Shen H.; Tang X. C.; Toda S.; Kalivas P. W. (2003) Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat. Neurosci. 6, 743–749. [DOI] [PubMed] [Google Scholar]
- Kalivas P. W. (2009) The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 10, 561–572. [DOI] [PubMed] [Google Scholar]
- Madayag A.; Lobner D.; Kau K. S.; Mantsch J. R.; Abdulhameed O.; Hearing M.; Grier M. D.; Baker D. A. (2007) Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J. Neurosci. 27, 13968–13976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopanitsa M. V. (1997) Extrasynaptic receptors of neurotransmitters: distribution, mechanisms of activation, and physiological role. Neurophysiology 29, 357–365. [Google Scholar]
- Nguyen L.; Rigo J. M.; Rocher V.; Belachew S.; Malgrange B.; Rogister B.; Leprince P.; Moonen G. (2001) Neurotransmitters as early signals for central nervous system development. Cell Tissue Res. 305, 187–202. [DOI] [PubMed] [Google Scholar]
- Vanhoutte P.; Bading H. (2003) Opposing roles of synaptic and extrasynaptic NMDA receptors in neuronal calcium signalling and BDNF gene regulation. Curr. Opin. Neurobiol. 13, 366–371. [DOI] [PubMed] [Google Scholar]
- Manent J. B.; Represa A. (2007) Neurotransmitters and brain maturation: early paracrine actions of GABA and glutamate modulate neuronal migration. Neuroscientist 13, 268–279. [DOI] [PubMed] [Google Scholar]
- Sierra-Paredes G.; Sierra-Marcuno G. (2007) Extrasynaptic GABA and glutamate receptors in epilepsy. CNS Neurol. Disord.: Drug Targets 6, 288–300. [DOI] [PubMed] [Google Scholar]
- Moran M. M.; McFarland K.; Melendez R. I.; Kalivas P. W.; Seamans J. K. (2005) Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J. Neurosci. 25, 6389–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D. A.; Madayag A.; Kristiansen L. V.; Meador-Woodruff J. H.; Haroutunian V.; Raju I. (2008) Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine. Neuropsychopharmacology 33, 1760–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho V.; Knopfel T. (2002) Metabotropic glutamate receptors: electrical and chemical signaling properties. Neuroscientist 8, 551–561. [DOI] [PubMed] [Google Scholar]
- Bellone C.; Luscher C.; Mameli M. (2008) Mechanisms of synaptic depression triggered by metabotropic glutamate receptors. Cell. Mol. Life Sci. 65, 2913–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwyl R. (2009) Metabotropic glutamate receptor-dependent long-term potentiation. Neuropharmacology 56, 735–740. [DOI] [PubMed] [Google Scholar]
- Featherstone D. E.; Yanoga F.; Grosjean Y. (2008) Accelerated bang recovery in Drosophila genderblind mutants. Commun. Integr. Biol. 1, 14–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhasz G.; Kekesi K.; Emri Z.; Soltesz I.; Crunelli V. (1990) Sleep-promoting action of excitatory amino acid antagonists: a different role for thalamic NMDA and non-NMDA receptors. Neurosci. Lett. 114, 333–338. [DOI] [PubMed] [Google Scholar]
- Juhasz G.; Kekesi K. A.; Emri Z.; Ujszaszi J.; Krogsgaard-Larsen P.; Schousboe A. (1991) Sleep promoting effect of a putative glial gamma-aminobutyric acid uptake blocker applied in the thalamus of cats. Eur. J. Pharmacol. 209, 131–133. [DOI] [PubMed] [Google Scholar]
- Rada P.; Tucci S.; Murzi E.; Hernandez L. (1997) Extracellular glutamate increases in the lateral hypothalamus and decreases in the nucleus accumbens during feeding. Brain Res. 768, 338–340. [DOI] [PubMed] [Google Scholar]
- Lena I.; Parrot S.; Deschaux O.; Muffat-Joly S.; Sauvinet V.; Renaud B.; Suaud-Chagny M. F.; Gottesmann C. (2005) Variations in extracellular levels of dopamine, noradrenaline, glutamate, and aspartate across the sleep−wake cycle in the medial prefrontal cortex and nucleus accumbens of freely moving rats. J. Neurosci. Res. 81, 891–899. [DOI] [PubMed] [Google Scholar]
- Castaneda T. R.; de Prado B. M.; Prieto D.; Mora F. (2004) Circadian rhythms of dopamine, glutamate and GABA in the striatum and nucleus accumbens of the awake rat: modulation by light. J. Pineal Res. 36, 177–185. [DOI] [PubMed] [Google Scholar]
- Honma S.; Katsuno Y.; Shinohara K.; Abe H.; Honma K. (1996) Circadian rhythm and response to light of extracellular glutamate and aspartate in rat suprachiasmatic nucleus. Am. J. Physiol. 271, R579–585. [DOI] [PubMed] [Google Scholar]
- Lopez-Rodriguez F.; Medina-Ceja L.; Wilson C. L.; Jhung D.; Morales-Villagran A. (2007) Changes in extracellular glutamate levels in rat orbitofrontal cortex during sleep and wakefulness. Arch. Med. Res. 38, 52–55. [DOI] [PubMed] [Google Scholar]
- Adachi A.; Natesan A. K.; Whitfield-Rucker M. G.; Weigum S. E.; Cassone V. M. (2002) Functional melatonin receptors and metabolic coupling in cultured chick astrocytes. Glia 39, 268–278. [DOI] [PubMed] [Google Scholar]
- Cortese B. M.; Phan K. L. (2005) The role of glutamate in anxiety and related disorders. CNS Spectr. 10, 820–830. [DOI] [PubMed] [Google Scholar]
- Lee Y.; Gaskins D.; Anand A.; Shekhar A. (2007) Glia mechanisms in mood regulation: a novel model of mood disorders. Psychopharmacology (Berlin) 191, 55–65. [DOI] [PubMed] [Google Scholar]
- Featherstone D. E.; Richmond J. E.; Ruben P. C. (1996) Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys. J. 71, 3098–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hires S. A.; Zhu Y.; Tsien R. Y. (2008) Optical measurement of synaptic glutamate spillover and reuptake by linker optimized glutamate-sensitive fluorescent reporters. Proc. Natl. Acad. Sci. U.S.A. 105, 4411–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkey E. B.; Parpura V. (2008) Mechanisms of glutamate release from astrocytes. Neurochem. Int. 52, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichberg V. I. (1991) Glial glutamate receptors: likely actors in brain signaling. FASEB J. 5, 3086–3091. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A.; Kirchhoff F. (2007) NMDA receptors in glia. Neuroscientist 13, 28–37. [DOI] [PubMed] [Google Scholar]
- Volterra A.; Meldolesi J. (2005) Astrocytes, from brain glue to communication elements: the revolution continues. Nat. Rev. Neurosci. 6, 626–640. [DOI] [PubMed] [Google Scholar]
- Fellin T.; D'Ascenzo M.; Haydon P. G. (2007) Astrocytes control neuronal excitability in the nucleus accumbens. ScientificWorldJournal 7, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J.; Jiang L.; Goldman S. A.; Nedergaard M. (1998) Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1, 683–692. [DOI] [PubMed] [Google Scholar]
- Andreadou E.; Kapaki E.; Kokotis P.; Paraskevas G. P.; Katsaros N.; Libitaki G.; Zis V.; Sfagos C.; Vassilopoulos D. (2008) Plasma glutamate and glycine levels in patients with amyotrophic lateral sclerosis: the effect of riluzole treatment. Clin. Neurol. Neurosurg. 110, 222–226. [DOI] [PubMed] [Google Scholar]
- Wesseldijk F.; Fekkes D.; Huygen F. J.; van de Heide-Mulder M.; Zijlstra F. J. (2008) Increased plasma glutamate, glycine, and arginine levels in complex regional pain syndrome type 1. Acta Anaesthesiol. Scand. 52, 688–694. [DOI] [PubMed] [Google Scholar]
- Smith Q. R. (2000) Transport of glutamate and other amino acids at the blood-brain barrier. J. Nutr. 130, 1016S–1022S. [DOI] [PubMed] [Google Scholar]
- Skerry T. M.; Genever P. G. (2001) Glutamate signalling in non-neuronal tissues. Trends Pharmacol. Sci. 22, 174–181. [DOI] [PubMed] [Google Scholar]
- Nedergaard M.; Takano T.; Hansen A. J. (2002) Beyond the role of glutamate as a neurotransmitter. Nat. Rev. Neurosci. 3, 748–755. [DOI] [PubMed] [Google Scholar]
- Gill S., and Pulido O., Eds. (2005) Glutamate Receptors in Peripheal Tissue: Excitatory Transmission Outside the CNS, 1 ed., Kluwer Academic/Plenum Publishers, New York. [Google Scholar]
- Gill S. S.; Pulido O. M. (2001) Glutamate receptors in peripheral tissues: current knowledge, future research, and implications for toxicology. Toxicol. Pathol. 29, 208–223. [DOI] [PubMed] [Google Scholar]
- Hinoi E.; Takarada T.; Ueshima T.; Tsuchihashi Y.; Yoneda Y. (2004) Glutamate signaling in peripheral tissues. Eur. J. Biochem. 271, 1–13. [DOI] [PubMed] [Google Scholar]
- Moriyama Y.; Omote H. (2008) Vesicular glutamate transporter acts as a metabolic regulator. Biol. Pharm. Bull. 31, 1844–1846. [DOI] [PubMed] [Google Scholar]
- Spencer G. J.; McGrath C. J.; Genever P. G. (2007) Current perspectives on NMDA-type glutamate signalling in bone. Int. J. Biochem. Cell Biol. 39, 1089–1104. [DOI] [PubMed] [Google Scholar]
- Gu Y.; Publicover S. J. (2000) Expression of functional metabotropic glutamate receptors in primary cultured rat osteoblasts. Cross-talk with N-methyl-D-aspartate receptors. J. Biol. Chem. 275, 34252–34259. [DOI] [PubMed] [Google Scholar]
- Laketic-Ljubojevic I.; Suva L. J.; Maathuis F. J.; Sanders D.; Skerry T. M. (1999) Functional characterization of N-methyl-D-aspartic acid-gated channels in bone cells. Bone 25, 631–637. [DOI] [PubMed] [Google Scholar]
- Dobson K. R.; Skerry T. M. (2000) The NMDA-type glutamate receptor antagonist MK801 regulates differentiation of rat bone marrow osteoprogenitors and influences adipogenesis. J. Bone Miner. Res. 15, S272–S272. [Google Scholar]
- Dobson K. R.; Skerry T. M. (2001) The NMDA-type glutamate receptor antagonist MK801 regulates differentiation of non-adherent rat bone marrow osteoprogenitors. Bone 28, S75–S75. [Google Scholar]
- Taylor A. F.; Brabbs A. C.; Peet N. M.; Laketic-Ljubojevic I.; Dobson K. R.; Skerry T. M. (2000) Bone formation/resorption and osteoblast/adipocyte plasticity mediated by AMPA/kainate glutamate receptors in vitro and in vivo. J. Bone Miner. Res. 15, S275–S275. [Google Scholar]
- Genever P. G.; Maxfield S. J.; Kennovin G. D.; Maltman J.; Bowgen C. J.; Raxworthy M. J.; Skerry T. M. (1999) Evidence for a novel glutamate-mediated signaling pathway in keratinocytes. J. Invest. Dermatol. 112, 337–342. [DOI] [PubMed] [Google Scholar]
- Morhenn V. B.; Waleh N. S.; Mansbridge J. N.; Unson D.; Zolotorev A.; Cline P.; Toll L. (1994) Evidence for an Nmda receptor subunit in human keratinocytes and rat cardiocytes. Eur. J. Pharmacol.: Mol. Pharmacol. Sect. 268, 409–414. [DOI] [PubMed] [Google Scholar]
- Lam H. M.; Chiu J.; Hsieh M. H.; Meisel L.; Oliveira I. C.; Shin M.; Coruzzi G. (1998) Glutamate-receptor genes in plants. Nature 396, 125–126. [DOI] [PubMed] [Google Scholar]
- Dubos C.; Willment J.; Huggins D.; Grant G. H.; Campbell M. M. (2005) Kanamycin reveals the role played by glutamate receptors in shaping plant resource allocation. Plant J. 43, 348–355. [DOI] [PubMed] [Google Scholar]
- Li J.; Zhu S.; Song X.; Shen Y.; Chen H.; Yu J.; Yi K.; Liu Y.; Karplus V. J.; Wu P.; Deng X. W. (2006) A rice glutamate receptor-like gene is critical for the division and survival of individual cells in the root apical meristem. Plant Cell 18, 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]