

Abstract
The finding that acetylcholinesterase (AChE) colocalizes with β-amyloid (Aβ) and promotes and accelerates Aβ aggregation has renewed an intense interest in developing new multifunctional AChE inhibitors as potential disease-modifying drugs for Alzheimer’s therapy. To this end, we have developed a new class of selective AChE inhibitors with site-activated chelating activity. The identified lead, HLA20A, exhibits little affinity for metal (Fe, Cu, and Zn) ions but can be activated following inhibition of AChE to liberate an active chelator, HLA20. HLA20 has been shown to possess neuroprotective and neurorescuing activities in vitro and in vivo with the ability to lower amyloid precursor holoprotein (APP) expression and Aβ generation and inhibit Aβ aggregation induced by metal (Fe, Cu, and Zn) ion. HLA20A inhibited AChE in a time and concentration dependent manner with an HLA20A−AChE complex constant (Ki) of 9.66 × 10−6 M, a carbamylation rate (k+2) of 0.14 min−1, and a second-order rate (ki) of 1.45 × 10 4 M−1 min−1, comparable to those of rivastigmine. HLA20A showed little iron-binding capacity and activity against iron-induced lipid peroxidation (LPO) at concentrations of 1−50 μM, while HLA20 exhibited high potency in iron-binding and in inhibiting iron-induced LPO. At a concentration of 10 μM, HLA20A showed some activity against monoamine oxidase (MAO)-A and -B when tested in rat brain homogenates. Defined restrictively by Lipinski’s rules, both HLA20A and HLA20 satisfied drug-like criteria and possible oral and brain permeability, but HLA20A was more lipophilic and considerably less toxic in human SHSY5Y neuroblastoma cells at high concentrations (25 or 50 μM). Together our data suggest that HLA20A may represent a promising lead for further development for Alzheimer's disease therapy.
Keywords: Acetylcholinesterase inhibitor, β-amyloid aggregation, lipid peroxidation, metal, prochelator, tau hyperphosphorylation
Alzheimer’s disease (AD) is a complex disorder with different but related dysfunctions involved in its progression. The present treatments offer at best moderate symptomatic benefit without arresting the disease progression. Because neurofibrillary tangles (NFTs) and β-amyloid (Aβ) plaques in selectively vulnerable brain regions are the chief hallmarks in AD brains, drugs targeting NFTs, Aβ, or both are considered to have disease-modifying effects (1). Metal (Fe, Cu, and Zn) ions may crucially participate in the pathological processes of AD, because they favor protein misfolding and aggregation (formation of Aβ plaques and NFTs), catalyze the production of reactive oxygen species (ROS), and induce oxidative damage associated with neuronal death. In fact, the metal levels are found to increase 3−5-fold in the brains of AD patient compared with those in age-matched controls, and the metals are significantly accumulated in and around Aβ plaques or NFTs (2). Iron accumulation in the brain is a source of redox-generated free radicals, which contribute to the source of radicals in AD (3). The highest iron levels are found both in the cortex and in the cerebellum from patients with preclinical AD/mild cognitive impairment; glial accumulations of the redox-active iron in the cerebellum are also evident in preclinical AD patients and tend to increase as patients became progressively cognitively impaired (4). The Alzheimer’s amyloid precursor holoprotein (APP) is an iron-regulated protein with an iron-responsive element (IRE) in its 5′ untranslated regions (UTRs). Iron closely regulates the intracellular levels of APP holoprotein by a mechanism that is similar to the translation control of the ferritin L and H mRNAs through an IRE in their 5′ UTRs; it increases the production of APP translation via activation of APP mRNA IRE and consequently increases Aβ formation (5,6). Metal (Fe, Cu, and Zn) dyshomeostasis and the interactions of the metals with Aβ cause Aβ aggregation and deposition. Both zinc and iron can induce aggregation of hyperphosphorylated tau. The interaction between Aβ and copper can promote hyperphosphorylation of tau, and subsequent NFT formation. In addition, iron and copper participate in the Fenton reaction, producing ROS, which aggravates oxidative stress (7). It is now established that oxidative stress is one of the earliest, if not the earliest, change that occurs in AD pathogenesis, because oxidative stress occurs significantly before the development of Aβ plaques and NFTs and serves to increase Aβ and tau hyperphosphorylation (8). The apparently critical involvement of metals, particularly iron, copper, and zinc, in both oxidative stress and the formation of NFTs and Aβ plaques therefore renders chelators as promising disease-modifying drugs for AD (9). Indeed, many chelators have shown inhibition of metal-induced Aβ aggregation and ROS formation and neuroprotection of neuronal cells exposed to AD-relevant reagents or conditions such as synthetic Aβ1−40 or Aβ1−42 fibrils (9,10). Some chelators, including desferrioxamine (DFO) and clioquinol (CQ), have shown promising results in transgenic AD models and achieved limited success in clinical trials, with decreased Aβ aggregate deposits and improved cognition (9−11). DFO is a FDA-approved chelator used for about 3 decades to treat iron overload. It has a high affinity for metal ions with stability constants (log K) for Fe3+, Al3+, Cu2+, and Zn2+ of 30.6, 22.0, 14.1, and 11.1, respectively (12). Studies have shown that DFO can protect neurons against metal ion-mediated toxicity of synthetic Aβ (13), inhibit Aβ-mediated redox activity (14), and remove Fe3+ from hyperphosphorylated tau (15). When used in combination with another chelator, Feralex-G, DFO can efficiently remove Al3+ from neurons (16). In studies with the tissues from brains of AD patients, DFO can completely strip the lesion-associated iron from Aβ deposits and NFTs, which prevents iron from catalyzing a H2O2-dependent oxidation (3). A 2-year blinded phase II study with 48 AD patents showed that intramuscular injection of DFO (125 mg, twice a day for 5 days a week) significantly slowed the rate of cognitive decline of AD patents compared with that in placebo-treated and nontreated controls (17). However, the clinical trials with DFO were halted due to difficulty in administration, because DFO is not orally active and does not efficiently cross the blood−brain barrier (BBB) (9). Clioquinol (CQ) is another chelator that has recently been widely investigated as a potential anti-AD drug. It is an old antibiotic drug with a moderate affinity for Fe3+, Al3+, Cu2+, and Zn2+, similar to other 8-hydroxyquinoline chelators (18). Studies revealed that CQ can capture metal ions from metal−Aβ species, induce the disassembly of Aβ fibrils, and partly dissolve Aβ aggregates (19). It is know that Aβ neurotoxicity in vitro is related to metal-mediated redox-generated free radicals (20). CQ and other chelators were shown to block these redox reactions and correspondingly inhibit Aβ toxicity in cell culture (21). In addition, diverse chelators including CQ were also found to reduce secreted Aβ levels in cell culture by inducing metal-dependent activation of PI3K and JNK, resulting in JNK-mediated up-regulation of metalloprotease activity (22). In Tg2576 APP transgenic mice, treatment with CQ (20 mg/kg daily for 12 weeks) significantly decreased the insoluble Aβ levels in brain extracts and the amyloid deposits in the brain sections (23). In a pilot phase II double-blind, placebo-controlled trial, CQ given orally (125−375 mg, twice daily over 36 weeks) significantly decreased the rate of cognitive decline in moderately severe AD patients (24). Despite these promising results, however, clinical trials with CQ were discontinued because of toxic concerns, because CQ was suspected to be involved in subacute myelooptico-neuropathy (SMON) and was withdrawn from the market in 1970 (25).
In view of the promising results and the limitations of traditional chelators as potential anti-AD drugs, new chelators with low side effects and better BBB permeability and efficacy are greatly needed for Alzheimer’s therapy. Indeed, numerous new chelators with potentially better profiles have been developed, including chelators with specific transporters or nanoparticles as chelator carriers, chelators with amyloid-binding activity, and site-activated chelators with antioxidant or anti-acetylcholinesterase (AChE)/monoamine oxidase (MAO) activity (26−32).
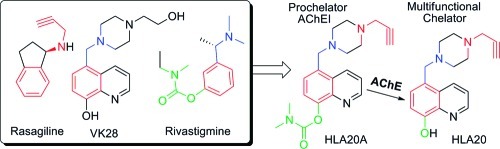
Recently, we have developed a number of new chelators, including 5-(4-propargyl-piperazin-1-ylmethyl)-8-hydroxyquinoline (HLA20, Chart 1) (33,34). HLA20 was rationally designed by incorporating the neuroprotective and neurorestorative propargyl-amine moiety from the anti-Parkinson drug rasageline (Azilect, Teva, Kansas City, MO, USA) into our prototype chelator (5-[4-(2-hydroxyethyl) piperazine-1-ylmethyl]-8-hydroxyquinoline (VK28, Chart 1). HLA20 has relatively poor selectivity for MAO-B, with an IC50 value of 110 μM versus >200 μM for MAO-A. It acts as a neuroprotective chelator with a wide range of pharmacological activities, including pro-survival neurorescue effects, induction of neuronal differentiation and regulation of APP and Aβ levels, and inhibition of oxidative stress and Aβ aggregation induced by metal ions (Cu and Zn) (34−36). Like other nonspecific chelators, HLA20 has the potential to chelate metal ions both in the brain and in the body. To improve the targeting specificity, reduce the potential side effects, and enhance the efficacy of chelators for treating AD, we have recently introduced a new strategy for designing site-activated multifunctional chelators with AChE and neuroprotective−neurorestorative moieties (26). Our design principle involves merging the three pharmacophores of rivastigmine, a marketed AChE inhibitor for AD, into HLA20 without major structural modifications (Chart 1), but it endows the new compounds with anti-AChE activity and improved targeting and permeability to the brain.
Chart 1. Strategy Leading to Site-Activated Chelator−AChE Inhibitor HLA20A.
In recent studies, we have shown that HLA20A has little affinity for metal (Fe, Cu, and Zn) ions and exhibits considerably lower cytotoxicity compared with HLA20. However HLA20A can be activated following inhibition of AChE with a concomitant release of an active chelator HLA20 (26).
In this study, HLA20A is further evaluated for its various properties including inhibition of lipid peroxidation (LPO) and MAO-A/B activity, iron-chelating potency, and drug-like properties, as well as the kinetic analysis of AChE inhibition.
Results
Kinetic Analysis of AChE Inhibition
The inhibition of AChE by carbamates involves a reversible complex (EI) formation between enzyme (E) and inhibitor (I), followed by carbamylation of the enzyme and production of a covalent adduct (E*). The carbamylated adduct hydrolyzes slowly, liberating the free enzyme (E) (eq 1) (37).
| 1 |
To study the kinetics of AChE inhibition by HLA20A, the enzyme was incubated with 1.0, 2.5, 5.0, 10, and 15 μM HLA20A in assay buffer at 37 °C. Aliquots were removed, diluted 100-fold, and assayed for enzyme activity at various preincubation periods. The time course of inhibition of AChE by HLA20A was shown in Figure 1A. The kinetic parameters describing the interaction of HLA20A with AChE were determined from the Kitz−Wilson equations, eqs 2 and 3(38,39):
| 2 |
| 3 |
wherein Ki represents the dissociation constant for EI, k+2 is the carbamylation rate constant, k+3 is the decarbamylation rate constant (k+3 ≪ k+2), kapp is the pseudo-first-order rate constant, and ki is the second-order rate constant, approximated by k+2/Ki, under the experimental condition that [I] ≫ [E0].
Figure 1.

Kinetics of inhibition of AChE by HLA20A. The enzyme was incubated with various concentrations of HLA20A in phosphate buffer, pH 7.4, at 37 °C. At the indicated times, aliquots were removed and assayed for AChE activity as described in Methods. (A) The natural log of the ratio of the remaining activity (E) and the initial activity (E0) is plotted against the time of incubation. The slopes of the lines represent the pseudo-first-order rate constant kapp determined by linear regression analysis. Values represent means ± SEM from three experiments. (B) Plot of 1/kapp versus 1/[HLA20A]. The straight line was generated by linear regression (R = 0.9903). Extrapolating from this line, 1/k+2 was obtained from the intercept on the ordinate and Ki/k+2 from the slope of the line.
The values of kapp were calculated from the slopes in Figure 1A based on eq 2. Double reciprocal plot of 1/kapp versus 1/[I] resulted in reasonably good straight lines (Figure 1B). Extrapolating from this plot, 1/k+2 was obtained from the intercept on the ordinate and Ki/k+2 from the slope of the line. The kinetic parameters calculated for HLA20A were Ki = 9.66 × 10−6 M, k+2 = 0.14 min−1, and ki (approximated by k+2/Ki) = 1.45 × 104 M−1 min−1, comparable to those found for rivastigmine (Ki = 3.66 × 10−5 M, k+2 = 0.26 min−1, and ki = 7.10 × 103 M−1 min−1). The half-time of inactivation (t0.5) of AChE by HLA20A calculated from Figure 1A was 7.9 min (10 μM), 18.6 min (5 μM), 26.8 min (2.5 μM), and 60 min (1 μM, calculated from the time-dependent inhibition curve at 1 μM as published previously (26)).
Inhibition of MAO-A and MAO-B in Vitro
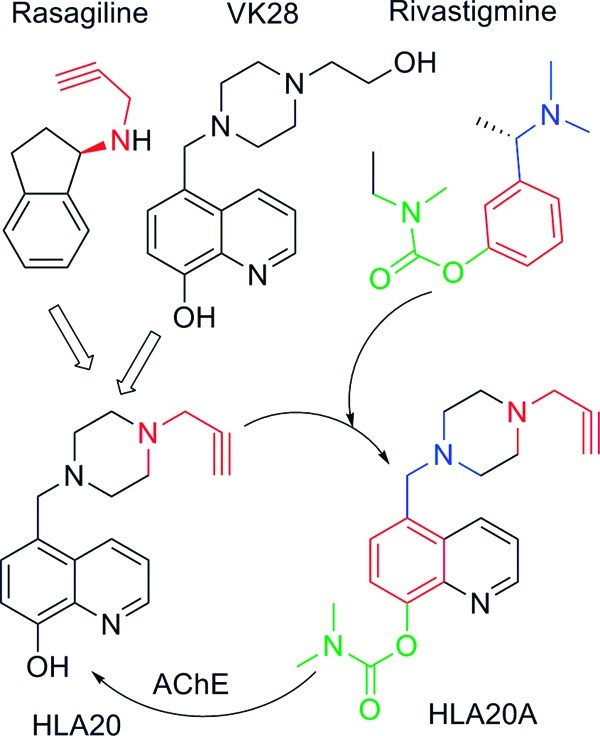
The inhibition of MAO by HLA20A and HLA20 was determined in vitro using rat brain homogenate. It was found that at a concentration of 10 μM HLA20 did not have any significant inhibitory effects on MAO-A and -B but HLA20A showed some activity against MAO-A and -B (about 9% and 5% inhibition, respectively, Figure 2)
Figure 2.
Inhibittion of rat brain MAO-A and MAO-B by HLA20A and HLA20 at 10 μM. Rat brain homogenates were incubated with drugs in the presence of 0.05 μM deprenyl (for the assay of MAO-A) or 0.05 μM clorgyline (for the assay of MAO-B) for1 h. This was followed by 30 min incubation with 14C-5-hydroxytryptamine or 20 min with 14C-β-phenylethylamine, respectively. The results are the mean ± SEM, n = 3 in triplicate.
Formation of Metal Complexes
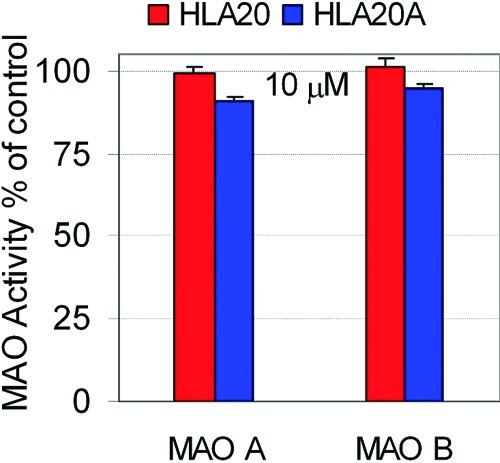
Previous studies have shown that HLA20A has little affinity for metal ions, but its activated chelator HLA20 can chelate iron and copper ions with a higher binding selectivity for iron over copper. HLA20 forms stable metal complexes with the stoichiometry of 3:1 and 2:1 (ligand/metal) for iron and copper, respectively (26). Here we further showed that HLA20 can form a stable complex with Zn(II) in 5% NH4Ac (pH 6.9)] at room temperature (Figure 3A, trace d), while there was no significant complexation between HLA20A and Zn(II) (Figure 3A, trace b). Addition of CuSO4 to the solution of HLA20−Zn complex (Figure 3A, trace e) resulted in disappearance of the characteristic absorbance band of HLA20−Zn complex (362 nm) and appearance of a new band at 372 nm (Figure 3A, trace e) that is the characteristic maximal absorption of Cu(II)−HLA20 complex. The changes in absorption can be explained by the replacement, Zn(II)−HLA20 + Cu(II) → Cu(II)−HLA20 + Zn(II), which suggests that HLA20 has a higher chelating activity for Cu(II) than for Zn(II). The stoichiometry of Zn(II)−HLA20 complex was determined by Job’s method (40), and the results are shown in Figure 3B. The maximum absorbance, ΔA, obtained from Figure 3B is 0.33, which indicates that a complex with 2:1 stoichiometry or (HLA20)2Zn complex was formed in 5% NH4Ac (pH 6.9) at room temperature.
Figure 3.
(A) Changes in absorbance of HLA20A and HLA20 (both 0.1 mM in 5%NH4Ac, pH = 6.9) on addition of metal salts (0.05 mM): (a) HLA20A; (b) HLA20A + ZnCl2; (c) HLA20; (d) HLA20 + ZnCl2; (e) HLA20−Zn complex + CuSO4. (B) Job plots for complexation of HLA20 with Zn2+ determined by UV−vis spectrophotometry in 5% NH4Ac (pH = 6.9) at room temperature, [HLA20] + [Zn2+] = 0.2 mM. Results are values of three assays.
Iron-Binding Potency
The iron-binding capacity of HLA20A and HLA20 was measured by the ferrozine method. In this method, iron-binding capacity of drugs is determined by assessing their ability to compete with ferrozine for Fe(II) ions, resulting in decreased absorbance at 562 nm (41). Because high-affinity iron(III) chelators chelate iron(II) cations and rapidly autoxidize them to the corresponding stable Fe(III)−complexes under aerobic conditions (42), this method actually reflects the Fe(II)/(III)-binding potency. As expected (Figure 4), DFO, a very strong Fe(III) chelator [log β = 30.6 for Fe(III), 7.2 for Fe(II)] (43), had a very high iron chelating effect (99.2%). The iron chelating effect of HLA20A was 1.4%, comparable to that of the FDA-approved anti-Parkinson’s drug rasagiline, which exhibited 1.3% iron chelating effect in the same conditions. These results indicate that both HLA20A and rasagiline have a very weak iron-binding capacity, which is in agreement with the previous results (26). By contrast, the activated chelator HLA20 displayed a strong iron-binding capacity with an iron chelating effect 90.1%, comparable to strong iron chelators clioquinol (91.2%) and VK28 (92.1%), but less than DFO (99.2%) (Figure 4).
Figure 4.

Comparison of the iron chelating effects of HLA20A and HLA20 with other known chelators DFO, VK28, clioquinol (CQ), and anti-Parkinson’s drug rasagiline (R). The iron chelating effects are expressed as percentage of control (50 μM ferrozine, 20 μM FeSO4 in pH 6.9 ammonium acetate buffer). The concentration of test compounds is 50 μM. Values represent the mean ± SEM, n = 3 in duplicate.
Inhibition of Lipid Peroxidation in Rat Brain Homogenates
As shown in Figure 5, HLA20A displayed weak activity against iron(II)-induced LPO at the range of 1−50 μM. By contrast, its activated chelator HLA20 exhibited high potency in inhibiting LPO with an IC50 of about 14 μM. For comparison, the effect on iron(II)-induced LPO by anti-Parkinson’s drug rasagiline was also presented in Figure 5.
Figure 5.
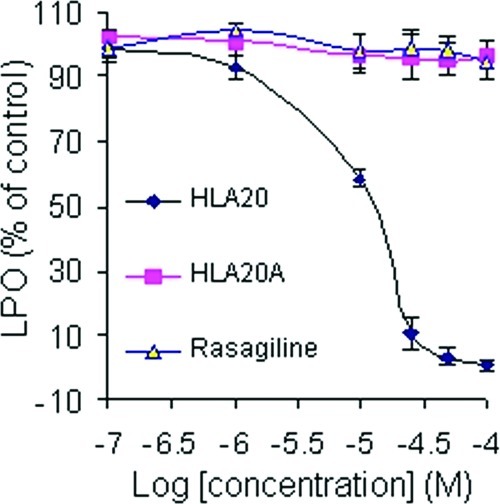
Effects of HLA20A, HLA20, and rasagiline on lipid peroxidation in rat brain homogenates. Lipid peroxidation was induced by 1.5 μM FeSO4/50 μM ascorbic acid. The results represent the means ± SEM, n = 3, p < 0.05.
Previous studies have shown that rasagiline is inactive against iron(II)-induced LPO (34).
Drug-Like Properties
The important pharmacological parameters of HLA20 and HLA20A were calculated online by the computer programs Marvin and ALOGPS 2.1 (44,45). The values were presented in Table 1, in comparison with VK28 and rivastigmine (Exelon).
Table 1. Values (MW, HBD, HBA, NROT, PSA, and LogP)a of VK28, HLA20, HLA20A, and Exelon.
| VK28 | HLA20 | HLA20A | Exelon | |
|---|---|---|---|---|
| MW | 287.36 | 281.35 | 352.43 | 250.34 |
| HBD | 2 | 1 | 0 | 0 |
| HBA | 5 | 4 | 5 | 3 |
| NROT | 4 | 3 | 5 | 5 |
| PSA | 59.83 | 39.6 | 48.91 | 32.78 |
| LogP | 0.45 | 1.37 | 1.79 | 2.41 |
| AClogP | 1.1 | 1.51 | 1.75 | 2.4 |
| ALogP | 1.24 | 2.79 | 3.21 | 2.56 |
Abbreviations: MW, molecular weight; HBD, hydrogen-bond donor atoms; HBA, hydrogen-bond acceptor atoms; PSA, polar surface area; NROT, number of rotatable bonds; logP, AClogP, and ALogP, calculated logarithm of the octanol−water partition coefficient. MW, HBD, HBA, NROT, PSA, and LogP were calculated online by Marvin software (44). AClogP and ALogP were calculated online by the ALOGPS 2.1 program (45).
Discussion
In order to improve the efficacy, the brain targeting, and permeability and minimize potential side effects of chelators for AD therapy, we have recently introduced a new strategy for designing the site-activated chelator−AChE inhibitor HLA20A (26). HLA20A was partly evaluated in the previous paper and was further studied in this paper.
As seen in Figure 1, HLA20A shows time-dependent inhibition of AChE, exhibiting pseudo-first-order kinetics at all the test concentrations. To compare the mode of action of HLA20A to rivastigmine, their kinetic constants (Ki, k+2, and ki) were calculated based on Kitz−Wilson method (38,39). The Ki values (9.66 × 10−6 M for HLA20A versus 3.66 × 10−5 M for rivastigmine) suggest HLA20A has a higher affinity than rivastigmine for AChE. Because the ki (the second-order rate inhibition constant) value can be correlated with inhibitor potency, the ki values (1.45 × 104 M−1 min−1 for HLA20A versus 7.10 × 103 M−1 min−1 for rivastigmine) indicate that HLA20A is more potent than rivastigmine in inhibiting AChE. This result is consistent with our previous studies in which HLA20A is slightly more potent than rivastigmine in inhibiting AChE at 1 μM (26). The carbamylation rate constant, k+2, which governs both the onset of AChE inhibition and the formation of the OH-metabolite HLA20, is low (0.14 min−1), implying that both AChE inhibition and HLA20 formation step proceed slowly. In fact, the t0.5 values (the time required for HLA20A to attain 50% inhibition of AChE) are 7.9 min (10 μM), 18.6 min (5 μM), 26.8 min (2.5 μM), and 60 min (1 μM), indicating that HLA20A inhibits AChE slowly and progressively with increase in inhibition as the incubation time and concentration increase. Previous studies suggest that a rapid onset of inhibition of AChE in the brain is undesirable for AD therapy, as it increases considerably the incidence of side effects like nausea and vomiting (46). A slow onset of inhibition of AChE in the brain has been shown to significantly reduce these side effects (47).
Propargylamine-containing compounds such as rasagiline and pargiline inhibit MAO-A/B irreversibly. Irreversible nonselective MAO-A and -B in liver and intestine is associated with “cheese effect”, a sharp elevation in blood pressure that may be life-threatening (48,49). HLA20, possessing a neuroprotective moiety (a propargylamine) from anti-Parkinson’s drug rasagiline, is a weak MAO-A/B inhibitor in vitro with a 66.21% and 33.21% inhibition for MAO-B and MAO-A, respectively, at a concentration of 100 μM (34). Attachment of a dimethyl carbamyl moiety at the 8-hydroxyl oxygen in HLA20 slightly increases the inhibitory activity as seen with HLA20A (about 9% and 5% inhibition of MAO-B and MAO-A, respectively, at a concentration of 10 μM in vitro). Such poor inhibition of MAO-A/B found in HLA20A is not likely to induce the “cheese effect” in vivo.
Using the ferrozine method, we calculated that the iron-chelating effect of HLA20A is 1.4%, comparable to that of the FDA-approved anti-Parkinson’s drug rasagiline, which exhibited 1.3% iron chelating effect under the same conditions (Figure 4). These results suggest that there may be some complexations between HLA20A or rasagiline and iron(II)/(III) as observed in previous studies (26), but their iron-binding capacity in the test concentrations is too weak to compete with ferrozine to demonstrate significant iron-binding capacity in the ferrozine method. In contrast to HLA20A, HLA20 shows iron-binding potency (90.1%) in the ferrozine method similar to other 8-hydroxyquinoline containing chelators CQ (91.2%) and VK28 (92.1%) but lower than DFO (99.2%, Figure 4). DFO is a very strong iron chelator (log K = 30.6 for Fe(III) and 10.3 for Fe(II)) (43), showing very high iron-chelating potency in the same experiments (99.2%). The very high affinity of DFO for iron is believed to cause abstraction of the metal from endogenous ligands, which can eventually lead to systemic iron depletion (49). 8-Hydroxyquinoline-containing chelators such as CQ were recently termed metal protein attenuating compounds (MPACs), which usually possess intermediate metal affinity and ability to reach specific intracellular compartments to target the harmful “up stream” metal−protein reactions. MPACs are used not primarily for removing a group of metals (with potential systemic toxicity), but for restoring metal dyshomeostasis, blocking free and reactive coordination positions of protein bound metals, and abolishing specific and deleterious metal−protein interactions. 8-Hydroxyquinoline-containing chelators, which largely fit these requirements, have been considered to be very appropriate and promising agents for AD (1,50,51). Recent studies have shown that clioquinol, an 8-hydroxyquinoline-containing chelator like HLA20, is able to chelate Cu and Zn from both amyloid plaques and the synaptic cleft and act as an ionophore to increase intracellular metal bioavailability. The increase in intracellular metal level activates an Akt (protein kinase B) signaling pathway, resulting in inhibition of glycogen synthase kinase 3β (GSK-3β). GSK-3β is a critical kinase contributing to tau hyperphosphorylation, and inhibition of this kinase decreases tau phosphorylation and increases expression of Aβ-degrading proteases in vitro (52).
Previous studies have shown that HLA20 exhibits binding affinity for Fe(II/III) and Cu(I/II) and forms stable metal complexes at a ratio of 3:1 [(HLA20)3Fe] or 2:1 [(HLA20)2Cu] (26,33). Here we show that HLA20 can also chelate Zn(II) and form a stable 2:1 complex or (HLA20)2Zn (Figure 3A,B). The formation of 3:1 complex (HLA20)3Fe indicates all the six iron coordinates have been occupied, which may be part of the explanation for the high potency of HLA20 in inhibiting the Fenton reaction and lipid peroxidation. It is known that occupation of all iron coordinates by a chelator precludes the binding of H2O2 to Fe2+-chelators and the subsequence Fenton reaction. Fe3+−EDTA can be easily reduced to Fention active Fe2+−EDTA by reducing agents like ascorbic acid and superoxide anion radical (O2•¯), thereby enhancing the production of OH• in the presence of H2O253,54. The fact that HLA20 remarkably suppresses iron(II)-induced lipid peroxidation (Figure 5) indicates the inability of Fe3+/2+−HLA20 complexes to catalyze the Fenton reaction even in the presence of strong reducing agents like ascorbic acid.
Oxidative stress (OS) plays an important role in AD pathogenesis and progression, because it occurs significantly before the development of Aβ plaques and NFTs and serves to increase Aβ and tau hyperphosphorylation (8). Besides, OS can damages all components of the cell, including proteins and lipids, and lead to the cell death associated with AD (8). LPO is one of the major outcomes of OS-related cell damage, and AD has been associated with elevated brain levels of LPO products (55). As seen in Figure 5, HLA20A had no significant activity against iron-induced LPO at concentrations of 1−50 μM. This could be explained by the poor iron-chelating potency of HLA20A (Figure 4), which is not able to chelate iron ions and prevent them from catalyzing the production of hydroxyl free radicals via the Fenton reaction. As expected, HLA20 (the activated analog of HLA20A), which shows high iron-chelating potency (Figure 4), exhibits high activity against iron-induced LPO under the same experimental conditions. Previous studies have shown that HLA20 is capable of acting as a strong metal chelator to block the production of ROS, as well as a potent scavenger to scavenge radicals (33,34). It have been shown that the combination treatment of a metal chelator and a radical scavenger protect brain tissue from OS to a greater degree than is achievable with either of them. Moreover a “hybrid” molecule with dual mode of action (chelating and scavenging) is found to be more efficacious in protecting against OS than with the combination treatment (56,57).
For drugs targeted at CNS, oral bioavailability and the BBB permeability are a necessity when administered for prolonged periods of time. In general, increasing lipophilicity may improve a chelator permeability to the BBB, but its uptake into other nonrelated cells may likewise increase with potential increased cytotoxicity as seen with nonspecific lipophilic chelators (58). Attachment of a lipophilic dimethyl carbamyl moiety to the hydrophilic phenolic oxygen in HLA20 yields a new molecule (HLA20A), not only with anti-AChE activity but also with increased lipophilicity as suggested by the logP values (Table 1). Therefore, it can be expected that HLA20A would be more permeable to the brain and the cells in the body than HLA20. However, in contrast to other lipophilic chelators, the improved permeability of HLA20A to the cells would not result in increased cytotoxicity, because HLA20A has little affinity for metal (Fe, Cu, and Zn) ions to interfere with the healthy metal metabolism in the cells. In fact, previous studies have revealed that HLA20A is nontoxic to human SHSY5Y neuroblastoma cells at 10 μM, and even at high concentrations (25 or 50 μM), it exhibits considerably lower cytotoxicity than HLA20 (26). Upon enzymatic cleavage of HLA20A by AChE in the brain, the dimethyl carbamyl moiety is detached from HLA20A with a concurrent release of the active chelator HLA20. HLA20 is more water-soluble than HLA20A, which may help “lock” HLA20 in the brain for prolonged time to exert its activity.
Lipinski’s rules (MW ≤ 450, calculated LogP ≤ 5, HBD ≤ 5, and HBA ≤ 10), PSA ≤ 90 Å2) provide a useful tool for prediction of intestinal absorption and possible brain penetration (59). VK28 and rivastigmine, which have been shown to possess oral bioavailability and brain permeability (46,60), are in good agreement with the prediction of Lipinski’s rules (as shown in Table 1). Both HLA20 and HLA20A are small and simple molecules with PSA < 60 Å2, NROT < 10, and zero number of violations of Lipinski’s rules, suggesting a potential good oral bioavailability and the BBB permeability.
In summary, in an effort to improve the efficacy, brain targeting, and permeability and minimize the potential toxicity of chelators for AD therapy, we have introduced a new strategy for new selective AChE inhibitors with site-activated chelating activity, in which HLA20A exhibits little affinity for metal (Fe, Cu, and Zn) ions until activation by inhibiting AChE to liberate an active chelator HLA20. HLA20 has been shown to possess neuroprotective and neurorescuing activities with the ability to lower APP expression and Aβ generation and to inhibit Aβ aggregation induced by metal (Cu and Zn) ion. HLA20A, like rivastigmine, displays pseudo-first-order kinetics, slowly producing the OH-metabolite HLA20. HLA20A exhibits little iron-binding capacity and activity against LPO at concentrations of 1−50 μM, whereas HLA20 demonstrates high potency in inhibition of iron-induced LPO and in iron-binding ability similar to other 8-hydroxyquinoline-containing chelators (VK28 and CQ) but lower than DFO. HLA20 forms a six-coordinate iron complex with the inability to induce LPO in the presence of the strong reducing agent ascorbic acid. Defined restrictively by Lipinski’s rules, both HLA20A and HLA20 satisfy drug-like criteria and possible oral and brain permeability. But HLA20A is more lipophilic and considerably less toxic than HLA20 in human SHSY5Y neuroblastoma cells at high concentrations (25 or 50 μM). Together HLA20A may represent in itself a potential drug that deserves further development for AD therapy. Furthermore, our new strategy, which generally generates small and simple drug-like molecules, may find application in designing site-activated multifunctional chelators with safer and more efficacious properties for treating other metal-related diseases such as Parkinson’s disease and cancer where specific elimination of metals in cancer cells is required.
Methods
Determination of Inhibition of AChE Activity
Ellman’s method was adapted for determination of AChE and BuAChE activities in rat brain homogenates. Brain tissue from adult Wistar rats was homogenized at 2% w/v in 0.1 M sodium phosphate buffer, pH 7.4, with added NaCl 58.5 g/L and Triton X-100 0.05% v/v. Aliquots of homogenate (20 μL) were incubated with HLA20A for designed time intervals in phosphate buffer, pH 7.4, before addition of 5,5′-dithiobis(2-nitrobenzoic) acid and 1 mM acetylthiocholine iodide (Sigma, St. Louis, MO). The reaction was run at 37 °C in a final volume of 200 μL in 96-well microplates and was followed at 412 nm for 5 min with a plate reader. In every experiment, cholinesterase-independent (nonspecific) substrate hydrolysis was determined by including one experimental group treated with tacrine 30 μM; appropriate tissue and reagent blanks were also included. Reaction velocities were determined in three or four replicates per condition; these were averaged and expressed as percent activity relative to control (solvent), after subtracting the rate of nonspecific hydrolysis. Results are reported as mean ± SEM.
Preparation of Brain Monoamine Oxidase
Rats were decapitated, and the brains were quickly taken into a weighted ice-cold sucrose buffer (10 mM Tris−HCl buffer, pH 7.4, containing 0.32 M sucrose), and their weights were measured. The brains were homogenized at 0−4 °C in 0.32 M sucrose (one part tissue to 20 parts sucrose) in a Teflon glass homogenizer. The resultant homogenates were used to determine MAO activity. Protein concentration was determined with Bradford reagent at 595 nm, using bovine serum albumin as a standard.
Determination of Monoamine Oxidase Activity in Vitro
The activity of MAO-A and MAO-B were determined by the method as described previously (34). Briefly, the tested drug was added to a suitable dilution of the enzyme preparation (70 μg of protein for MAO-B and 150 μg for MAO-A assay) in 0.05 M phosphate buffer (pH 7.4). The mixture was incubated together with 0.05 μM l-deprenyl, a specific inhibitor of MAO-B (for determination of MAO-A), or 0.05 μM clorgyline, a specific inhibitor of MAO-A (for determination of MAO-B). Incubation was carried on 1 h at 37 °C before addition of 14C-5-hydroxytryptamine binoxalate (100 μM) for determination of MAO-A or 14C-phenylethylamine 100 μM for determination of MAO-B activity, and incubation continued for 30 or 20 min, respectively. The reaction was stopped with 2 M ice-cold citric acid, and the radioactive metabolites (5-hydroxytryptaldehyde and phenylacetaldehyde) were extracted by addition of 2 mL of ethylacetate/toluene (1:1 vol/vol). The radioactivity contained in the organic phase was determined by liquid-scintillation counting in cpm units.
Measurement of Iron-Binding Capacity
Iron-binding capacity of drugs was determined by assessing their ability to compete with ferrozine for ferrous ions, resulting in decrease in the absorbance at 562 nm (61). Briefly, the reacted mixture containing ferrozine (50 μM) and a chelator (50 μM) was initiated by the addition of FeSO4 (20 μM). After 20 min incubation at room temperature, the absorbance (at 562 nm) of the resulting solutions was read. Iron-binding capacity was calculated as follows: iron-binding capacity (%) = [1 − (change in absorbance of sample at 562 nm)/(change in absorbance of control at 562 nm)] × 100.
Lipid Peroxidation Assay
LPO was measured in rat brain mitochondrial membrane homogenates as previously described (29). This method is based on the oxidation of polyunsaturated fatty acids in biologic membranes, giving rise to a variety of lipid breakdown products such as malondialdehyde (MDA). Upon reaction of MDA with thiobarbituric acid (TBA), a pink pigment is formed, which can be detected by UV−vis spectroscopy. LPO was induced by 50 μM ascorbic acid and 1.5 μM FeSO4. The absorption of thiobarbituric acid derivatives (TBARS) was measured spectrophotometrically at 532 nm.
Statistical Analysis
All assays were performed at least in triplicate, and the data were expressed as mean ± SEM. Data were analyzed by Student’s t test and ANOVA. Variations were considered to be statistically significant at a p-value of 0.05.
Department of Medicinal Chemistry, Intracellular Therapies Inc. 3960 Broadway, New York, NY 10032.
We thank the Alzheimer Association (USA), Alzheimer Drug Discovery Foundation (New York, USA), Technion-Research and Development, and the Weizmann Institute for generous support of this work.
References
- Biran Y.; Masters C. L.; Barnham K. J.; Bush A. I.; Adlard P. A. (2009) Pharmacotherapeutic targets in Alzheimer’s disease. J. Cell. Mol. Med. 13, 61–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell M. A.; Robertson J. D.; Teesdale W. J.; Campbell J. L.; Markesbery W. R. (1998) Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 158, 47–52. [DOI] [PubMed] [Google Scholar]
- Smith M. A.; Harris P. L.; Sayre L. M.; Perry G. (1997) Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. U.S.A. 94, 9866–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. A.; Zhu X.; Tabaton M.; Liu G.; McKeel D. W. Jr.; Cohen M. L.; Wang X.; Siedlak S. L.; Dwyer B. E.; Hayashi T.; Nakamura M.; Nunomura A.; Perry G. (2010) Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J. Alzheimers Dis. 19, 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S.; Huang X.; Cho H.; Greig N. H.; Youdim M. B.; Rogers J. T. (2006) Metal specificity of an iron-responsive element in Alzheimer’s APP mRNA 5′untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28, and a piperazine chelator. J. Neural Transm. Suppl. 237–247. [DOI] [PubMed] [Google Scholar]
- Rogers J. T.; Randall J. D.; Cahill C. M.; Eder P. S.; Huang X.; Gunshin H.; Leiter L.; McPhee J.; Sarang S. S.; Utsuki T.; Greig N. H.; Lahiri D. K.; Tanzi R. E.; Bush A. I.; Giordano T.; Gullans S. R. (2002) An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 277, 45518–45528. [DOI] [PubMed] [Google Scholar]
- Barnham K. J.; Bush A. I. (2008) Metals in Alzheimer’s and Parkinson’s diseases. Curr. Opin. Chem. Biol. 12, 222–228. [DOI] [PubMed] [Google Scholar]
- Sayre L. M.; Perry G.; Smith M. A. (2008) Oxidative stress and neurotoxicity. Chem. Res. Toxicol. 21, 172–188. [DOI] [PubMed] [Google Scholar]
- Price K. A.; Crouch P. J.; White A. R. (2007) Therapeutic treatment of Alzheimer’s disease using metal complexing agents. Recent Pat. CNS Drug Discovery 2, 180–187. [DOI] [PubMed] [Google Scholar]
- Scott L. E.; Orvig C. (2009) Medicinal inorganic chemistry approaches to passivation and removal of aberrant metal ions in disease. Chem. Rev. 109, 4885–4910. [DOI] [PubMed] [Google Scholar]
- Ritchie C. W.; Bush A. I.; Mackinnon A.; Macfarlane S.; Mastwyk M.; MacGregor L.; Kiers L.; Cherny R.; Li Q. X.; Tammer A.; Carrington D.; Mavros C.; Volitakis I.; Xilinas M.; Ames D.; Davis S.; Beyreuther K.; Tanzi R. E.; Masters C. L. (2003) Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch. Neurol. 60, 1685–1691. [DOI] [PubMed] [Google Scholar]
- Hider R. C.; Hall A. D. (1991) Clinically useful chelators of tripositive elements. Prog. Med. Chem. 28, 41–173. [DOI] [PubMed] [Google Scholar]
- Rottkamp C. A.; Raina A. K.; Zhu X.; Gaier E.; Bush A. I.; Atwood C. S.; Chevion M.; Perry G.; Smith M. A. (2001) Redox-active iron mediates amyloid-beta toxicity. Free Radical Biol. Med. 30, 447–450. [DOI] [PubMed] [Google Scholar]
- Sayre L. M.; Perry G.; Harris P. L.; Liu Y.; Schubert K. A.; Smith M. A. (2000) In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: a central role for bound transition metals. J. Neurochem. 74, 270–279. [DOI] [PubMed] [Google Scholar]
- Shin R. W.; Kruck T. P.; Murayama H.; Kitamoto T. (2003) A novel trivalent cation chelator Feralex dissociates binding of aluminum and iron associated with hyperphosphorylated tau of Alzheimer’s disease. Brain Res. 961, 139–146. [DOI] [PubMed] [Google Scholar]
- Kruck T. P.; Cui J. G.; Percy M. E.; Lukiw W. J. (2004) Molecular shuttle chelation: the use of ascorbate, desferrioxamine and Feralex-G in combination to remove nuclear bound aluminum. Cell. Mol. Neurobiol. 24, 443–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crapper McLachlan D. R.; Dalton A. J.; Kruck T. P.; Bell M. Y.; Smith W. L.; Kalow W.; Andrews D. F. (1991) Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337, 1304–1308. [DOI] [PubMed] [Google Scholar]
- Di Vaira M.; Bazzicalupi C.; Orioli P.; Messori L.; Bruni B.; Zatta P. (2004) Clioquinol, a drug for Alzheimer’s disease specifically interfering with brain metal metabolism: structural characterization of its zinc(II) and copper(II) complexes. Inorg. Chem. 43, 3795–3797. [DOI] [PubMed] [Google Scholar]
- Mancino A. M.; Hindo S. S.; Kochi A.; Lim M. H. (2009) Effects of clioquinol on metal-triggered amyloid-beta aggregation revisited. Inorg. Chem. 48, 9596–9598. [DOI] [PubMed] [Google Scholar]
- Huang X.; Atwood C. S.; Hartshorn M. A.; Multhaup G.; Goldstein L. E.; Scarpa R. C.; Cuajungco M. P.; Gray D. N.; Lim J.; Moir R. D.; Tanzi R. E.; Bush A. I. (1999) The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 38, 7609–7616. [DOI] [PubMed] [Google Scholar]
- Puglielli L.; Friedlich A. L.; Setchell K. D.; Nagano S.; Opazo C.; Cherny R. A.; Barnham K. J.; Wade J. D.; Melov S.; Kovacs D. M.; Bush A. I. (2005) Alzheimer disease beta-amyloid activity mimics cholesterol oxidase. J. Clin. Invest. 115, 2556–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caragounis A.; Du T.; Filiz G.; Laughton K. M.; Volitakis I.; Sharples R. A.; Cherny R. A.; Masters C. L.; Drew S. C.; Hill A. F.; Li Q. X.; Crouch P. J.; Barnham K. J.; White A. R. (2007) Differential modulation of Alzheimer’s disease amyloid beta-peptide accumulation by diverse classes of metal ligands. Biochem. J. 407, 435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherny R. A.; Atwood C. S.; Xilinas M. E.; Gray D. N.; Jones W. D.; McLean C. A.; Barnham K. J.; Volitakis I.; Fraser F. W.; Kim Y.; Huang X.; Goldstein L. E.; Moir R. D.; Lim J. T.; Beyreuther K.; Zheng H.; Tanzi R. E.; Masters C. L.; Bush A. I. (2001) Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30, 665–676. [DOI] [PubMed] [Google Scholar]
- Ritchie C. W.; Bush A. I.; Mackinnon A.; Macfarlane S.; Mastwyk M.; MacGregor L.; Kiers L.; Cherny R.; Li Q. X.; Tammer A.; Carrington D.; Mavros C.; Volitakis I.; Xilinas M.; Ames D.; Davis S.; Beyreuther K.; Tanzi R. E.; Masters C. L. (2003) Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch. Neurol. 60, 1685–1691. [DOI] [PubMed] [Google Scholar]
- Arasaki K.; Nakanishi T. (1989) Selective neurotoxicity of clioquinol on the function of the posterior column nuclei. Neurosci. Lett. 107, 85–88. [DOI] [PubMed] [Google Scholar]
- Zheng H.; Youdim M. B.; Fridkin M. (2009) Site-activated multifunctional chelator with acetylcholinesterase and neuroprotective-neurorestorative moieties for Alzheimer’s therapy. J. Med. Chem. 52, 4095–4098. [DOI] [PubMed] [Google Scholar]
- Schugar H.; Green D. E.; Bowen M. L.; Scott L. E.; Storr T.; Bohmerle K.; Thomas F.; Allen D. D.; Lockman P. R.; Merkel M.; Thompson K. H.; Orvig C. (2007) Combating Alzheimer’s disease with multifunctional molecules designed for metal passivation. Angew. Chem., Int. Ed. 46, 1716–1718. [DOI] [PubMed] [Google Scholar]
- Charkoudian L. K.; Pham D. M.; Franz K. J. (2006) A pro-chelator triggered by hydrogen peroxide inhibits iron-promoted hydroxyl radical formation. J. Am. Chem. Soc. 128, 12424–12425. [DOI] [PubMed] [Google Scholar]
- Zheng H.; Youdim M. B.; Weiner L. M.; Fridkin M. (2005) Novel potential neuroprotective agents with both iron chelating and amino acid-based derivatives targeting central nervous system neurons. Biochem. Pharmacol. 70, 1642–1652. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Rodriguez C.; Sanchez de Groot N.; Rimola A.; Alvarez-Larena A.; Lloveras V.; Vidal-Gancedo J.; Ventura S.; Vendrell J.; Sodupe M.; Gonzalez-Duarte P. (2009) Design, selection, and characterization of thioflavin-based intercalation compounds with metal chelating properties for application in Alzheimer’s disease. J. Am. Chem. Soc. 131, 1436–1451. [DOI] [PubMed] [Google Scholar]
- Liu G.; Men P.; Perry G.; Smith M. A. (2009) Metal chelators coupled with nanoparticles as potential therapeutic agents for Alzheimer’s disease. J. Nanoneurosci. 1, 42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H.; Youdim M. B.; Fridkin M. (2010) Site-activated chelators targeting acetylcholinesterase and monoamine oxidase for Alzheimer’s therapy. ACS Chem. Biol. 5, 603–610. [DOI] [PubMed] [Google Scholar]
- Zheng H.; Weiner L. M.; Bar-Am O.; Epsztejn S.; Cabantchik Z. I.; Warshawsky A.; Youdim M. B.; Fridkin M. (2005) Design, synthesis, and evaluation of novel bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer’s, Parkinson’s, and other neurodegenerative diseases. Bioorg. Med. Chem. 13, 773–783. [DOI] [PubMed] [Google Scholar]
- Zheng H.; Gal S.; Weiner L. M.; Bar-Am O.; Warshawsky A.; Fridkin M.; Youdim M. B. (2005) Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases: in vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J. Neurochem. 95, 68–78. [DOI] [PubMed] [Google Scholar]
- Amit T.; Avramovich-Tirosh Y.; Youdim M. B.; Mandel S. (2008) Targeting multiple Alzheimer’s disease etiologies with multimodal neuroprotective and neurorestorative iron chelators. FASEB J. 22, 1296–1305. [DOI] [PubMed] [Google Scholar]
- Avramovich-Tirosh Y.; Amit T.; Bar-Am O.; Zheng H.; Fridkin M.; Youdim M. B. (2007) Therapeutic targets and potential of the novel brain- permeable multifunctional iron chelator-monoamine oxidase inhibitor drug, M-30, for the treatment of Alzheimer’s disease. J. Neurochem. 100, 490–502. [DOI] [PubMed] [Google Scholar]
- Groner E.; Ashani Y.; Schorer-Apelbaum D.; Sterling J.; Herzig Y.; Weinstock M. (2007) The kinetics of inhibition of human acetylcholinesterase and butyrylcholinesterase by two series of novel carbamates. Mol. Pharmacol. 71, 1610–1617. [DOI] [PubMed] [Google Scholar]
- Maurer T.; Fung H. L. (2000) Comparison of methods for analyzing kinetic data from mechanism-based enzyme inactivation: application to nitric oxide synthase. AAPS PharmSci 2, E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitz R.; Wilson I. B. (1962) Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J. Biol. Chem. 237, 3245–3249. [PubMed] [Google Scholar]
- MacCarthy P. (1978) Simplified experimental route for obtaining Job’s curves. Anal. Chem. 50, 2165. [Google Scholar]
- Carter P. (1971) Spectrophotometric determination of serum iron at the submicrogram level with a new reagent (ferrozine). Anal. Biochem. 40, 450–458. [DOI] [PubMed] [Google Scholar]
- Harris D. C.; Aisen P. (1973) Facilitation of Fe(II) autoxidation by Fe(3) complexing agents. Biochim. Biophys. Acta 329, 156–158. [DOI] [PubMed] [Google Scholar]
- Anderegg G.; L’Eplattenier F.; Schwarzenbach G. (1963) Hydroxamate Complexes. III. Iron(III) Exchange between Sideramines and Complexones. A Discussion of the Formation Constants of the Hydroxamate Complexes. Helv. Chim. Acta 46, 1409–1422. [Google Scholar]
- Marvin, Calculator Plugin and Chemical Terms Demo. http://www.chemaxon.com/marvin/sketch/index.jsp.
- The ALOGPS 2.1 program. http://www.vcclab.org/lab/alogps/start.html.
- Jann M. W. (2000) Rivastigmine, a new-generation cholinesterase inhibitor for the treatment of Alzheimer’s disease. Pharmacotherapy 20, 1–12. [DOI] [PubMed] [Google Scholar]
- Winblad B.; Grossberg G.; Frölich L.; Farlow M.; Zechner S.; Nagel J.; Lane R. (2007) IDEAL: A 6-month, double-blind, placebo-controlled study of the first skin patch for Alzheimer disease. Neurology 69, S14–22. [DOI] [PubMed] [Google Scholar]
- Bortolato M.; Chen K.; Shih J. C. (2008) Monoamine oxidase inactivation: From pathophysiology to therapeutics. Adv. Drug Delivery Rev. 60, 1527–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.; Singh R.; Sundar S. (1995) Failure of ketoconazole in oriental sore in India. J. Chemother. 7(Suppl 4), 202–203. [PubMed] [Google Scholar]
- Ritchie C. W.; Bush A. I.; Masters C. L. (2004) Metal-protein attenuating compounds and Alzheimer’s disease. Expert Opin. Invest. Drugs 13, 1585–1592. [DOI] [PubMed] [Google Scholar]
- Masters C. L.; Beyreuther K. (2006) Alzheimer’s centennial legacy: prospects for rational therapeutic intervention targeting the Abeta amyloid pathway. Brain 129, 2823–2839. [DOI] [PubMed] [Google Scholar]
- White A. R.; Du T.; Laughton K. M.; Volitakis I.; Sharples R. A.; Xilinas M. E.; Hoke D. E.; Holsinger R. M.; Evin G.; Cherny R. A.; Hill A. F.; Barnham K. J.; Li Q. X.; Bush A. I.; Masters C. L. (2006) Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity. J. Biol. Chem. 281, 17670–17680. [DOI] [PubMed] [Google Scholar]
- Graf E.; Mahoney J. R.; Bryant R. G.; Eaton J. W. (1984) Iron-catalyzed hydroxyl radical formation. Stringent requirement for free iron coordination site. J. Biol. Chem. 259, 3620–3624. [PubMed] [Google Scholar]
- Snyrychova I.; Pospisil P.; Naus J. (2006) The effect of metal chelators on the production of hydroxyl radicals in thylakoids. Photosynth. Res. 88, 323–329. [DOI] [PubMed] [Google Scholar]
- Montine T. J.; Neely M. D.; Quinn J. F.; Beal M. F.; Markesbery W. R.; Roberts L. J.; Morrow J. D. (2002) Lipid peroxidation in aging brain and Alzheimer’s disease. Free Radical Biol. Med. 33, 620–626. [DOI] [PubMed] [Google Scholar]
- Bebbington D.; Monck N. J.; Gaur S.; Palmer A. M.; Benwell K.; Harvey V.; Malcolm C. S.; Porter R. H. (2000) 3,5-Disubstituted-4-hydroxyphenyls linked to 3-hydroxy-2-methyl-4(1H)-pyridinone: Potent inhibitors of lipid peroxidation and cell toxicity. J. Med. Chem. 43, 2779–2782. [DOI] [PubMed] [Google Scholar]
- Aftab N.; Vieira A. (2010) Antioxidant activities of curcumin and combinations of this curcuminoid with other phytochemicals. Phytother Res. 24, 500–502. [DOI] [PubMed] [Google Scholar]
- Singh S.; Khodr H.; Taylor M. I.; Hider R. C. (1995) Therapeutic iron chelators and their potential side-effects. Biochem. Soc. Symp. 61, 127–137. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Shachar D. B.; Kahana N.; Kampel V.; Warshawsky A.; Youdim M. B. (2004) Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydroxydopamine lession in rats. Neuropharmacology 46, 254–263. [DOI] [PubMed] [Google Scholar]
- Carter P. (1971) Spectrophotometric determination of serum iron at the submicrogram level with a new reagent (ferrozine). Anal. Biochem. 40, 450–458. [DOI] [PubMed] [Google Scholar]